Clin Med II exam 1
1/321
Earn XP
Description and Tags
gbalsam
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
322 Terms
HLA-B27 disease association
- ankylosing spondylitis
- reiter (reactive arthritis)
- acute anterior uveitis
HLA-DR4 disease association
rheumatoid arthritis
HLA-DR3 disease association
SLE in caucasians
HLA-B*57:01 disease association
abacavir hypersensitivity
what are the 4 cardinal signs of inflammation?
- rubor (redness)
- tumor (swelling)
- calor (heat)
- dolor (pain)
what is sometimes considered a 5th cardinal sign of inflammation?
functio lassae (loss of function)
PG12, PGD2, and PGE2 in acute inflammation
vasodilation and vascular permeability
PGE2 in acute inflammation
mediates pain and fever
LTB4 in acute inflammation
attract and activate neutrophils
LTC4, LTD4, and LTE4 in acute inflammation
slow-reacting substances of anaphylaxis (i.e., vasoconstriction, bronchospasm, and increased vascular permeability)
CD4+ T-cells
MHC class II molecules
CD8+ T-cells
MHC class I molecules
how is pain caused during inflammation?
bradykinin and PGE2 sensitizes sensory nerve endings
deficiency in terminal components of complement
recurrent neisseria infections
deficiency in complement C3
susceptibility to encapsulated bacteria (pyogenic infection)
deficiency in complement C6,7
Raynauds phenomena
deficiency in complement C1 esterase inhibitor
hereditary angioedema
hypersensitivity reaction
allergy to drugs, pollens, foods, bacteria, or any other substance may induce a protective reaction
how are hypersensitivity reactions classified?
on the basis of the principal immunologic mechanism that is responsible for tissue injury and disease
variations of hypersensitivity reaction
mild itching to severe bronchial asthma
Gell-Coombs classification
the mechanisms of immune responses to antigen grouped into 4 distinct types of reactions
type I hypersensitivity reaction
immediate hypersensitivity
associated diseases with type I hypersensitivity reaction
atopy, anaphylaxis, and asthma
mediators of type I hypersensitivity reaction
IgE
type II hypersensitivity reaction
antibody-mediated hypersensitivity
associated diseases with type II hypersensitivity reaction
autoimmune, hemolytic anemia, Goodpasture's disease, and erythroblastosis fetalis
mediators of type II hypersensitivity reaction
IgG or IgM and complement
type III hypersensitivity reaction
immune complex-mediated hypersensitivity
associated diseases with type III hypersensitivity reaction
serum sickness, Arthus' reaction, and lupus nephritis
mediators of type III hypersensitivity reaction
IgG and complement
type IV hypersensitivity reaction
delayed hypersensitivity
associated diseases with type IV hypersensitivity reaction
transplant rejection, contact dermatitis, and tuberculosis
mediators of type IV hypersensitivity reaction
T-cells, macrophages, and histiocytes
pathological lesions of type I hypersensitivity reactions
- vascular changes (vasodilation)
- edema
- contraction of smooth muscle
- production of mucous
- inflammation
effects of type I hypersensitivity reactions
- IgE is produced
- vasoactive amines released from mast cells
- inflammatory cells accumulate later at the site
when do type I hypersensitivity reactions occur?
within minutes after the combination of antigen with antibody bound to mast cells
in what people do type I hypersensitivity reactions occur?
those who have been previously sensitized to the antigen
what are the chief cells in type I hypersensitivity reactions?
mast cells
immediate phase of type I hypersensitivity reaction
seen within 5-30 minutes
late phase of type I hypersensitivity reaction
sets in after 2-24 hours (seen in allergic rhinitis or bronchial asthma)
what can activate mast cells?
- IgE
- anaphylatoxins (C3a and C5a)
- IL-8, drugs, and mellitin (found in bee venom)
histamine
a biogenic amine that causes smooth muscle contraction, increased vascular permeability, and secretions
chymase and tryptase
enzymes that cause tissue damage
heparin and chondroitin sulfate
proteoglycans of type I hypersensitivity that help to store mediators in granules
lipid mediators of type I hypersensitivity reactions
leads to the activation of phospholipase A2 and then to the activation of arachidonic acid metabolism
cytokine mediators of type I hypersensitivity reactions
play in a role in the "late phase" reaction
- TNF
- IL-1, 3, 4, 5, and 6
- GM-CSF
leukotrienes
LTB4 is chemotactic for eosinophils, neutrophils, and monocytes
prostaglandin D2
causes intense bronchospasm
platelet-activating factor (PAF)
platelet aggregation, release histamine, and bronchospasm
local immediate hypersensitivity reaction
known as "atopic allergy;" often caused by pollen, house dust, animal dander, food, etc.
features of local immediate hypersensitivity reaction
- urticaria
- angioedema
- allergic rhinitis
- asthma
local lesion
wheal-and-flare reaction confined to one region of the body
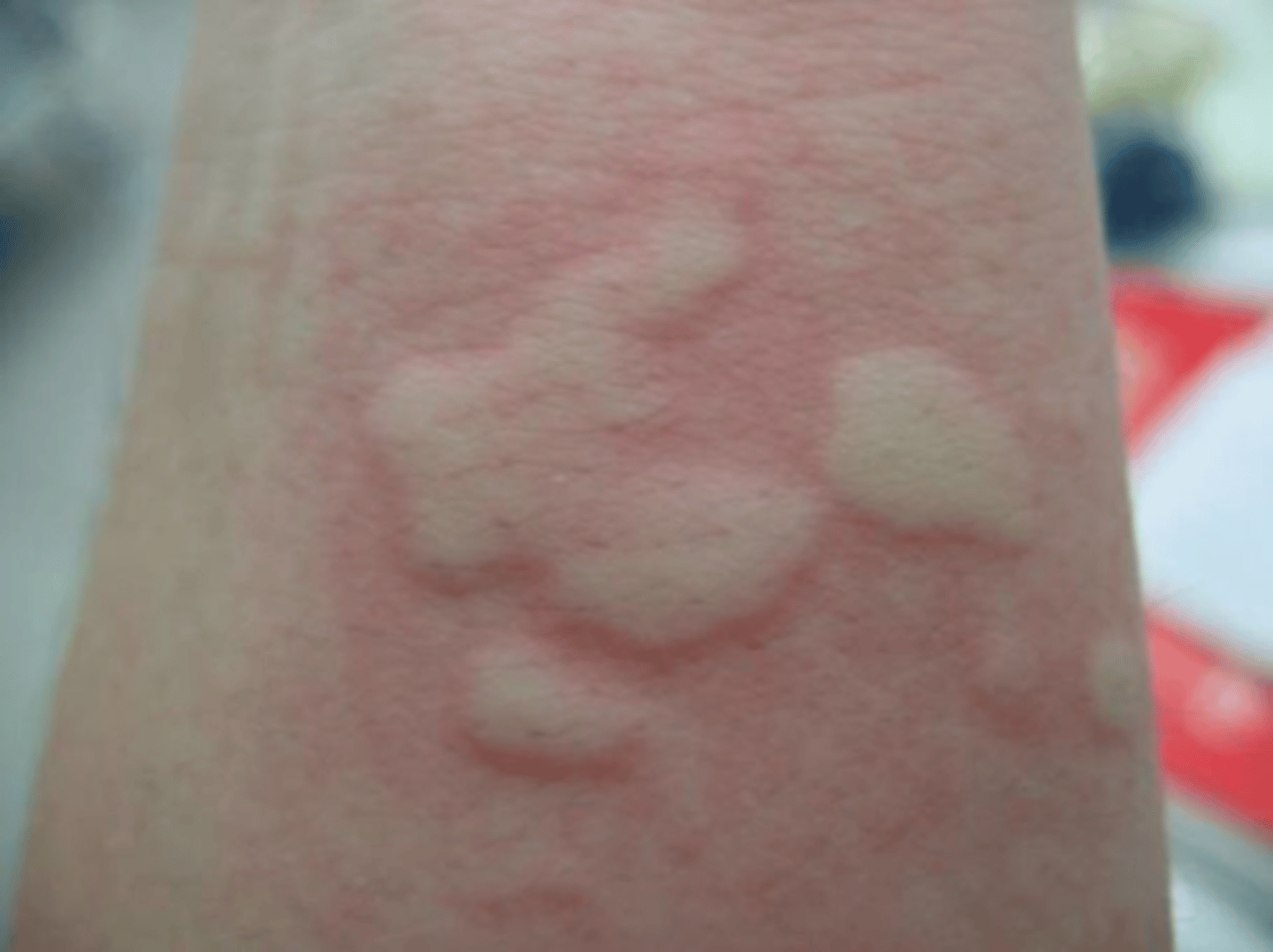
urticaria
disseminated form of a wheal-and-flare reaction
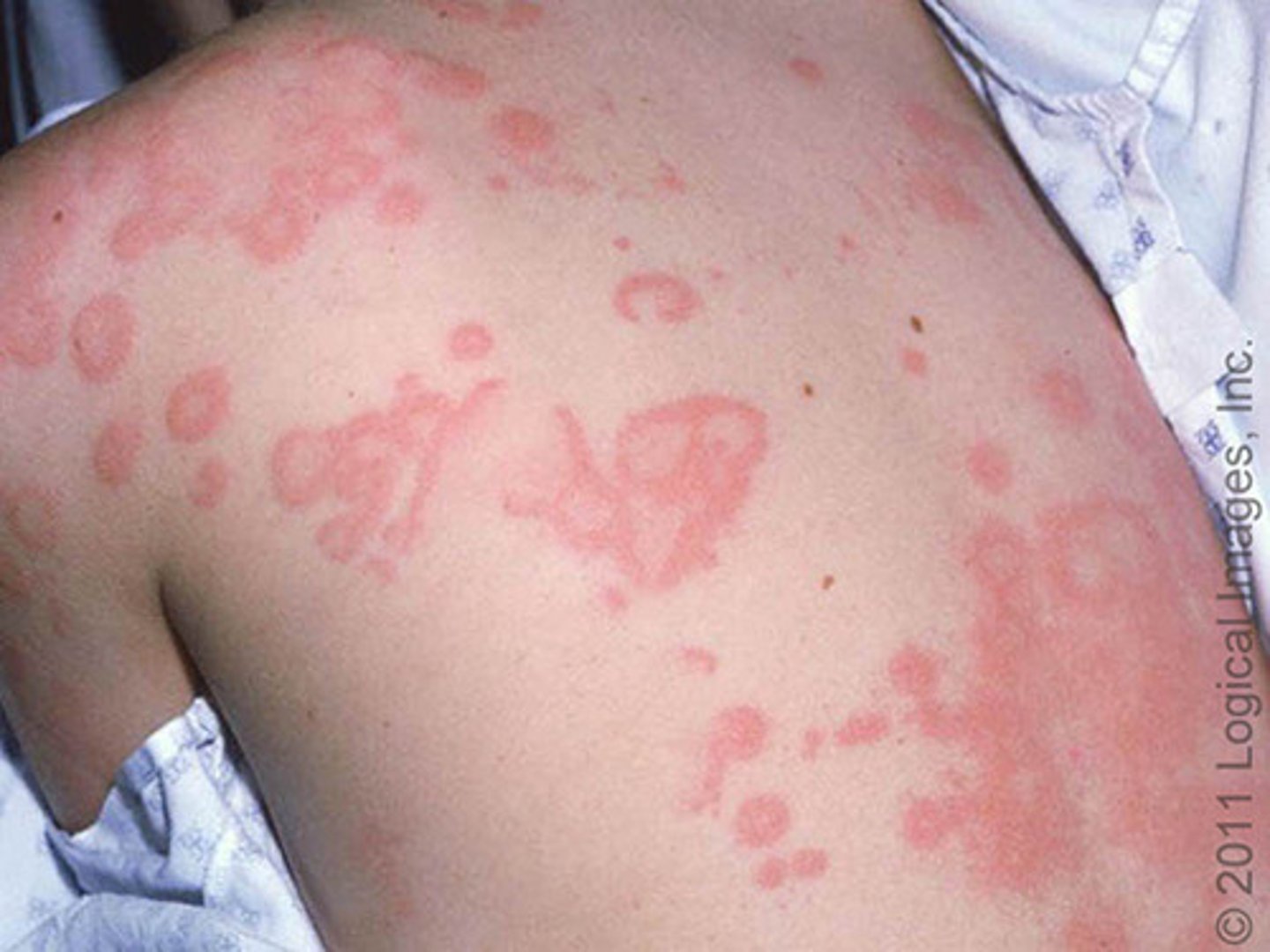
angioedema
localized areas of swelling beneath the skin, often around the eyes and lips, but it can also involve other body areas as well
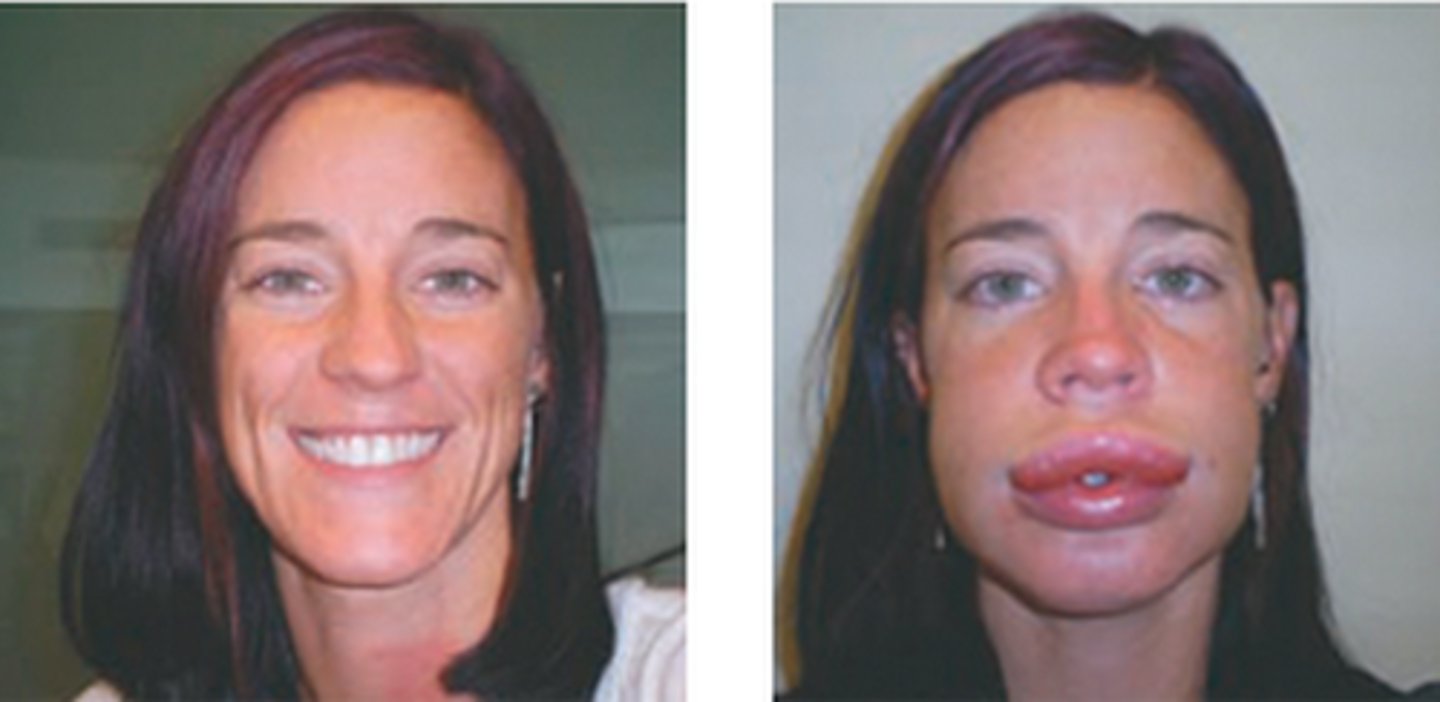
hereditary angioedema
autosomal dominant deficiency or malfunction of C1 esterase inhibitor (C1-INH)
symptoms of hereditary angioedema
- recurrent episodes of severe non-pruritic swelling of the face, limbs, GI tracts, and airways
- recurrent abdominal pain
- no urticaria
- begins in childhood and worsens during puberty
- death from laryngeal edema may occur
prevalence of family history in hereditary angioedema
positive in 80% of patients
diagnosis of hereditary angioedema
- low C4 level (due to exaggerated cleavage of C4 by C1 complex)
- low C2 level
- low C1 inhibitor protein or function
acute treatment of hereditary angioedema
FFP or C1-INH concentrate
chronic treatment of hereditary angioedema
ecallantide and androgens (i.e., danazol or stanazol)
commonly understood patho of acquired angioedema
ACE inhibitors
anaphylaxis
a life-threatening manifestation of immediate hypersensitivity
when do symptoms of anaphylaxis develop?
within 30 minutes of exposure to the inciting agent
symptoms of anaphylaxis
- urticaria or severe upper airway obstruction resulting from edema of the larynx, epiglottis, and surrounding structures
- hypotension, secondary to profound vasodilation
- possible seizures
management of anaphylaxis
maintenance of airway and oxygenation (i.e., attach a pulse oximeter and cardiac monitor)
pharmacologic management of anaphylaxis
epinephrine 1:1000 solution (0.01 mL/kg with max of 0.5 mL IM repeated every 15 minutes as needed)
other pharmacologic management of anaphylaxis
- for bronchospasm, administer albuterol by metered-dose inhaler (MDI) with spacer device or nebulizer
- rapid IV fluid if patient is hypotensive
- diphenhydramine 50mg IV/IM/PO
- corticosteroids (such as prednisone 60 mg IV/IM/PO) to reduce late-phase recurrence of symptoms 4-8 hours later
- consider glucagon and/or atropine for patients on beta blockers whose symptoms are refractory to therapy
- injectable epinephrine and antihistamine on discharge
how long should children be monitored following anaphylaxis with airway involvement?
at least 24 hours
effects of type II hypersensitivity reactions
IgG and IgM produced are bound to the target cells that are then lysed by activated complements
two major changes observed in tissues during a type II hypersensitivity reaction
cell lysis and inflammation
mechanism of type III hypersensitivity reactions
deposition of antigen-antibody complexes leading to activation of complements
pathological changes observed in type III hypersensitivity reaction
necrotizing vasculitis and inflammation
examples of type III hypersensitivity reactions
SLE, polyarteritis nodosa, poststreptococcal glomerulonephritis, acute glomerulonephritis, Arthus reaction, and serum sickness
serum sickness
reaction to certain medications or antiserum (snake) that develops 7-10 days after initial exposure
symptoms of serum sickness
- redness & itching at injection site
- fever
- joint pain
- adenopathy
- wheezing
- diarrhea & nausea
causes of serum sickness
- PCN (most common cause)
- Prozac
- barbiturates
Arthus reaction
acute immune complex vasculitis causes a localized area of tissue necrosis in the skin developing 4-10 hours after the injection
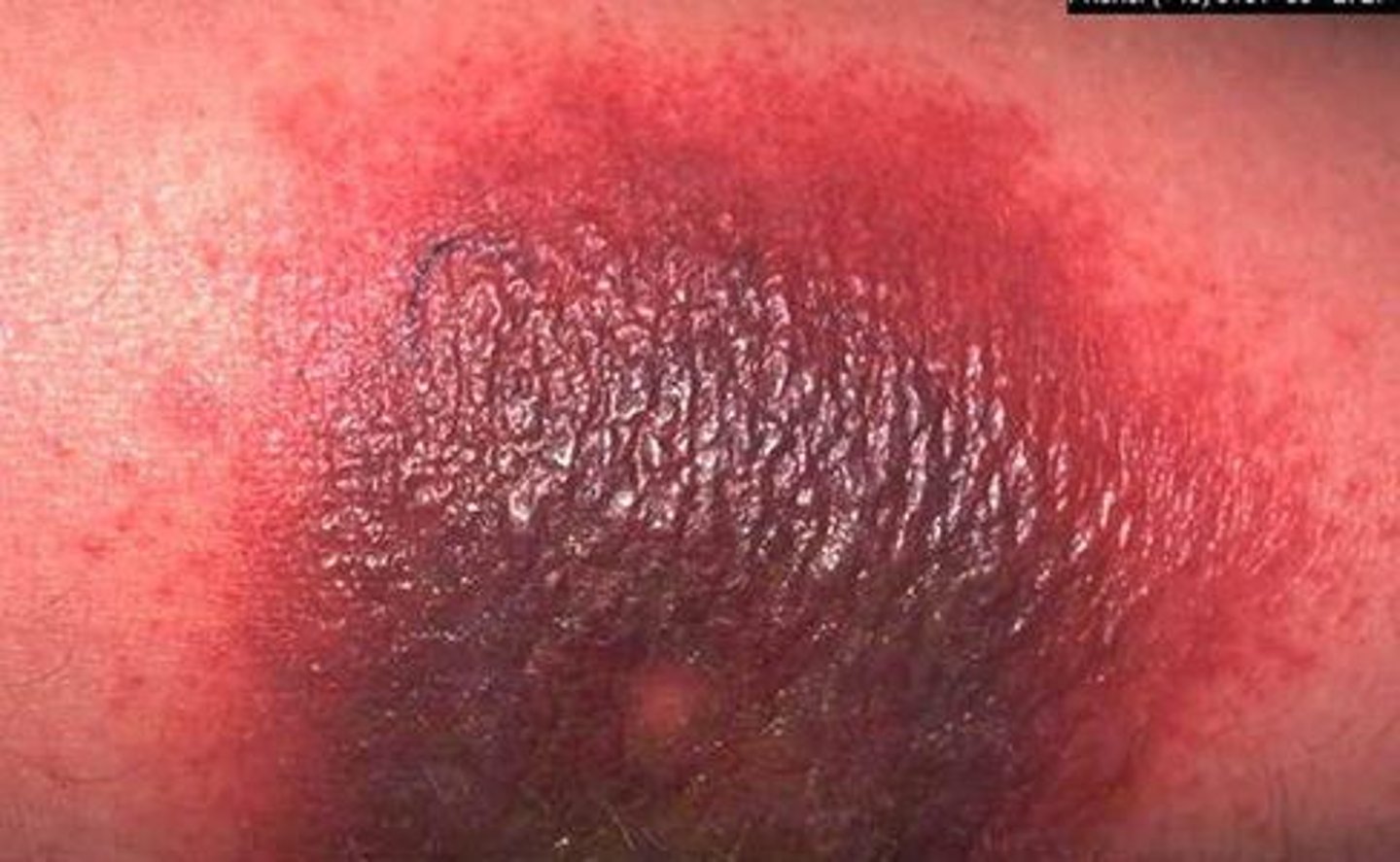
patho of Arthus reaction
as the antigen diffuses into the vascular wall in a previously sensitized person, large immune complexes are formed producing an inflammatory response
mechanism of type IV hypersensitivity reactions
- activation of T-lymphocytes and macrophages
- T-cell mediated cytotoxicity
pathological changes seen in type IV hypersensitivity reactions
- granuloma formation
- edema
- perivascular inflammation
- cell destruction
delayed type hypersensitivity
mediated by CD4+ T-cells as seen in tuberculosis, fungal, viral, and parasitic diseases as well as some autoimmune diseases
T-cell mediated cytotoxicity
sensitized CD8+ T-cells kill antigen bearing target cells
what is the classic example of delayed type hypersensitivity?
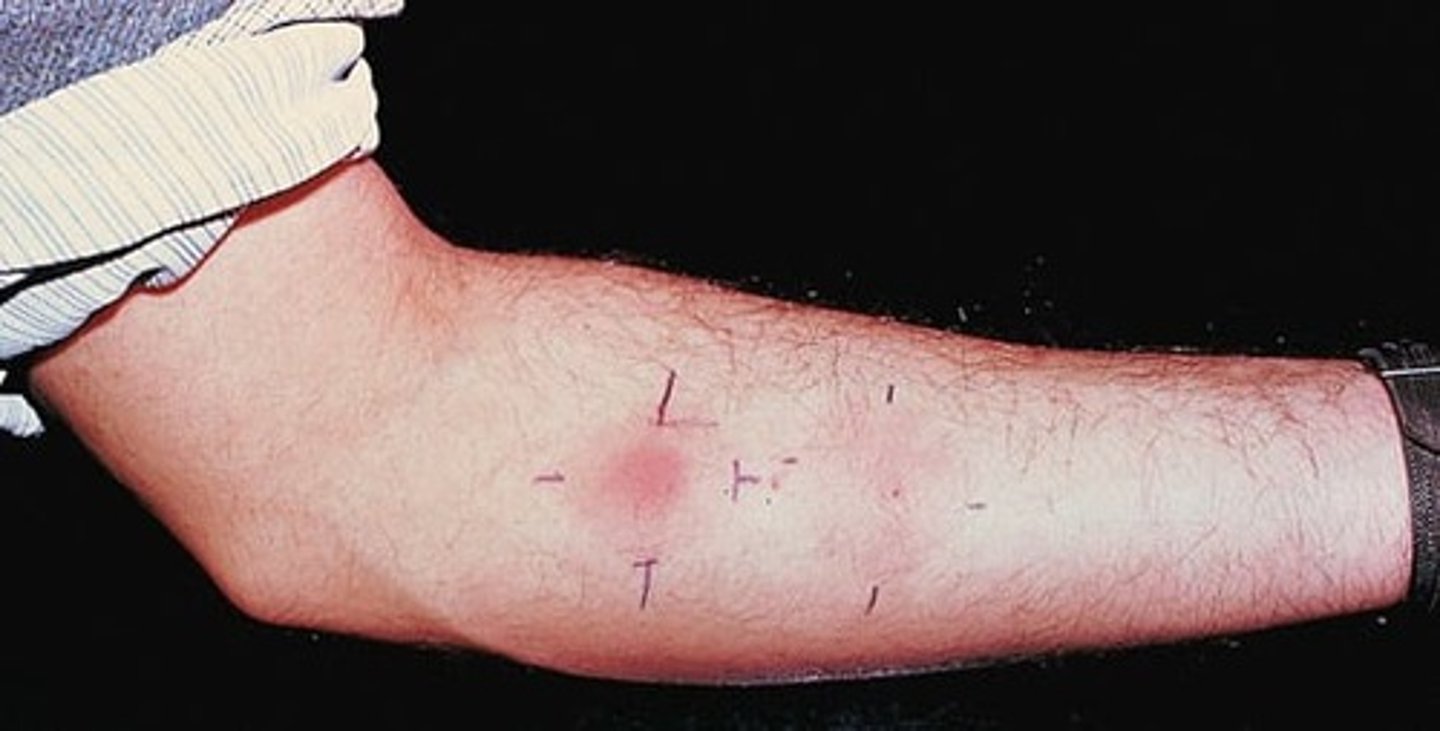
tuberculin reaction (TB skin test)
positive tuberculin reaction
reddening and induration appear within 8-12 hours
what happens to lymphocytes with long-standing infections?
they are replaced by macrophages that are then transformed to look like epithelial cells
granuloma
collection of macrophages (epithelioid cells)
primary immunodeficiency disorders
genetic defects that can lead to abnormalities in immunocompetence
effects of any immunopathogenic mechanism that impairs T-lymphocyte function or cell-mediated immunity
predisposes the host to the development of serious chronic and potentially life-threatening opportunistic infections with viruses, mycobacteria, fungi, or protozoa involving any or all organ systems
effects of immunopathogenic dysfunction of B-lymphocytes
results in antibody deficiency that can predispose the host to the pyogenic sinopulmonary and mucosal infections
clinical features of adaptive primary immunodeficiency disorders (B-cell defects)
- recurrent bacterial sinopulmonary infections or sepsis, particularly with polysaccharide encapsulated organisms
- unexplained bronchiectasis
- chronic or recurrent gastroenteritis (often with Giardia or enterovirus)
- failure to thrive
- chronic enteroviral meningoencephalitis
- arthritis
clinical features of adaptive primary immunodeficiency disorders (T-cell defects)
- recurrent, severe, or unusual viral infections (VZV, CMV, HSV)
- failure to thrive
- chronic candidiasis
- chronic diarrhea
- lymphopenia during the neonatal period or in infancy
- pneumocystis pneumonia
- graft-versus-host disease
- severe/neonatal eczematoid or seborrheic rashes
symptoms of graft-versus-host disease
maculopapular and/or desquamating skin, abnormal liver function tests, and/or chronic diarrhea
clinical features of innate primary immunodeficiency disorders (phagocytic defects)
- poor wound healing
- delayed separation of the umbilical cord
- lymphadenitis or soft tissue abscesses
- hepatosplenomegaly
- chronic gingivitis and periodontal disease/oral mucosal ulcerations
- infection with catalase positive bacteria and fungi
- recurrent GI and GU tract obstruction
clinical features of innate primary immunodeficiency disorders (complement defects)
- angioedema of face, hands, feet, and GI tract
- autoimmune disease or lupus-like symptoms
- pyogenic bacterial infections (i.e., Neisseria meningitidis)
- history suggestive of autosomal dominant inheritance (should have an affected parent)
clinical features of innate primary immunodeficiency disorders (other defects)
- herpes simplex meningoencephalitis in infancy
- papilloma virus infections of skin, including extensive warts
- ectodermal dysplasia
- pyogenic infections (i.e., sepsis, meningitis)
ataxia-telangiectasia syndrome
autosomal recessive disorder due to a defect in DNA damage repair leading to defective B- and T-cell functions
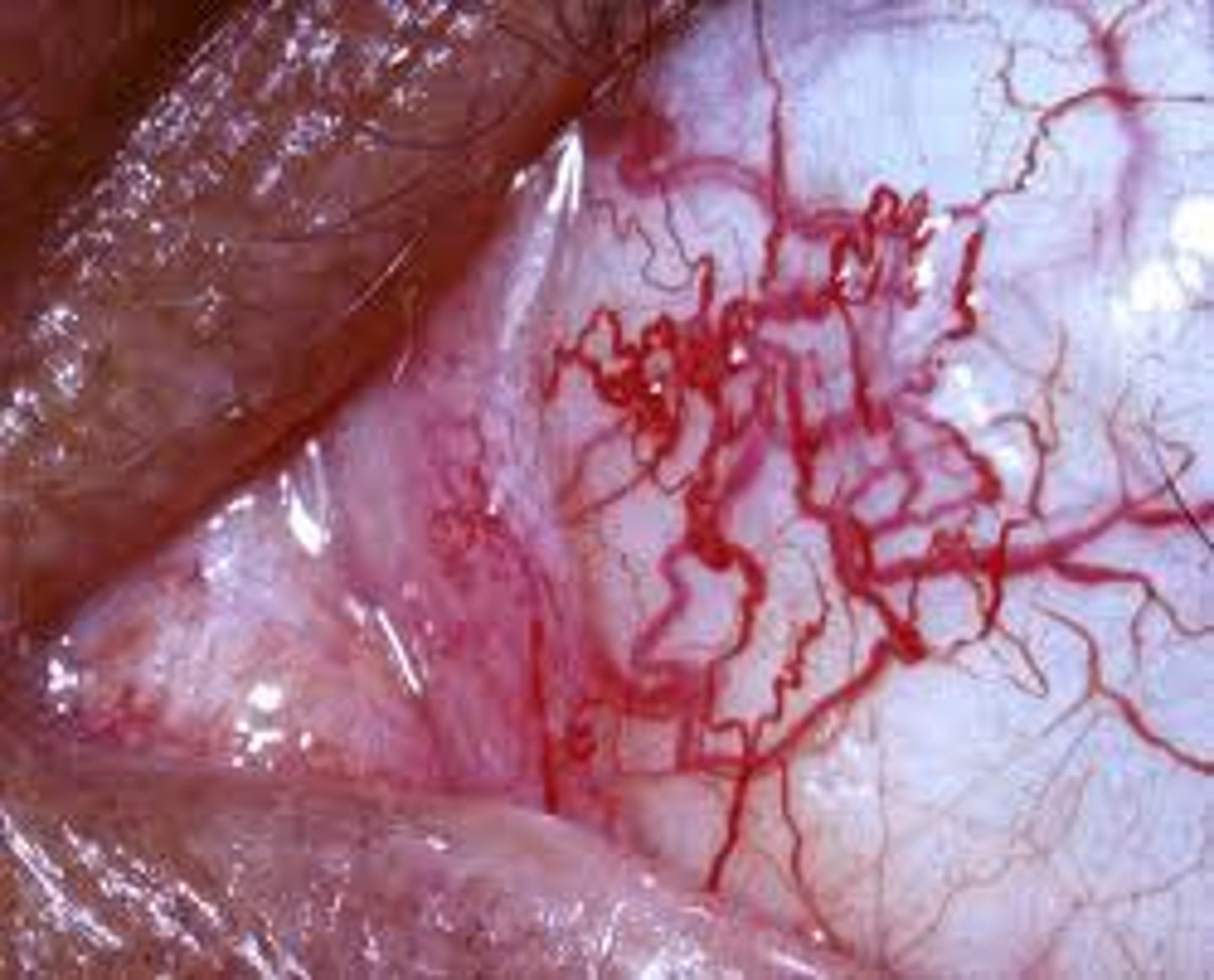
symptoms of ataxia-telangiectasia syndrome
- delay in walking due to atrophy of the cerebellum
- uncoordinated head and eye movements
- speech failure
- characteristic ocular and cutaneous telangiectasias
- decreased IgE and IgA levels
- increased serum alpha-fetoprotein
prognosis of ataxia-telangiectasia syndrome
increased risk for lymphomas and leukemias
treatment of ataxia-telangiectasia syndrome
none specific but is directed at specific symptoms
severe combined immunodeficiency diseases (SCID)
- genetic defect in stem cells resulting in the absence of the thymus, T- and B- cells
- half autosomal recessive; half X-linked leading to prevention of DNA synthesis and problems with interleukin signaling