BMM Module 6: Potential Energy surface
1/18
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No study sessions yet.
19 Terms
Relation PES and FF
PES= full energy landscape of a moleucle, showing how energy changes with atomic positions
Force field= simplified mathemeatical model (using equations and paramaters) to aporximate that landscape
Relation: FF provide an approximate PES that simulations can explore
Relation to MD
Gradient of PES= negative slope of PES at a certain postion = force acting on atoms
MD simulations use FF to calculate these force to navigate the PES
What is the PES?
it maps every possible conformation to it’s energy, allowing us to predict which conformations are the most stable and/or the most populated
Boltzmann distribution determines the populations
the deeper the well depth the more populated
transitions occur through saddle point by structural changes
It gives a 3N dimensional landscape (N= number of atoms)
it can’t be visualised
you can visualise a 2D slice when varying two coordinates (like torsion angles) while keeping all other others fixed
at minima:derivative of the energy is 0, so the force acting on it is 0 and it’s in a stable conformation
Goal molecular modelling
Identiy, characterize and navigate between the minima of the PES
How to navigate the PES
Simply minimizing E (goin downhill) is not enough for complex processes like protein folding or ligand docking where many local minima exist so you need sampling techniques to explore multiple minima and find the global one:
Monte carlo: randomly generates a new conformation, accepts or rejects based on energy and repeats this a lot of times which allows broad exploration of the landscape quickly
Simulated annealing= type of monte carlo with gradually decreasing temperature, this allows the system to escope local minima in the beginning and later settle into low energy conformations
Molecular dynamics: solves newtonian equations of motion for all atoms, the system continuously moves over the PES over time, this captures the thermally most accessible position
Genetic algorithms
PES of propane
it has 11 atoms so a total of 33 coordinates = 33 variables that define the PES
Total number of paramaters
10 covalent bonds
18 bond angles
each carbon atom is connected to 4 atoms, and these four atoms form bonds with each other
atom 4 forms bond with 3 others, atom 3 with 2, atom2 with 1 = 3+2+1 = 6 or (N*(N-1)/2)
6×3 carbon centers
18 torsion angles
27 non bonded terms
11×10/2 = 55 atom pairs
10 are bonded
18 are connected via bond angles and are not counted
55-10-18 = 27
non bonded terms are counted twice for coulomb AND vdw
=27×2+18+18+10= 100 parameters
XYZ parameters are the independent positional degrees of freedom of the atoms, while bonded and non-bonded parameters are force-field energy terms that depend on and are coupled through those Cartesian coordinates.
Bonded and non-bonded interaction terms are functions of the Cartesian (xyz) coordinates and are fully determined by them.
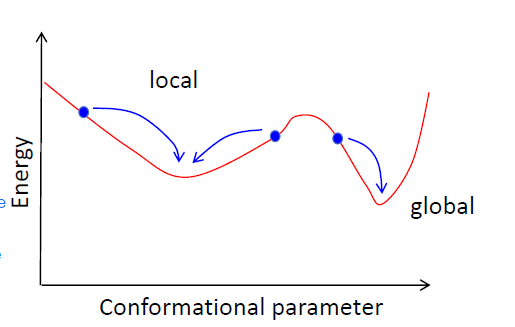
Minima
LMEC= local minimum energy conformation
GMEC= global minimum engergy conformation
gmec is not always the most populated: a wide valley that is higher in energy might be more populated than a steep lower energy conformation
prion disease = normal protein misfolds into deep low energy conformation, which is stable, resists degradation and induces other proteins to misfold leading to aggregates that can dammage neurons
EM workflow
EM vs forcefields
(not really in slides but just for clarity)
1)build molecule with molecule editior or importing coord,
geometry is a rough guess, you rarely start from a stabble conformation
2)assign a MM force field that describes how atoms interact and provides the PES
3) EM algorithms use the FF to calculate forces on each atom and iteratively move the coordinates to lower the energy
4)output = new xyz coordinates of atoms and the minimized structure is then used for tasks like
ligand binding
docking
MD simulations
EM methods
non-derivative methods
simplex method
sequential univariate method (bad)
derivative methods
steepest descent
conjugated gradient
simplex method
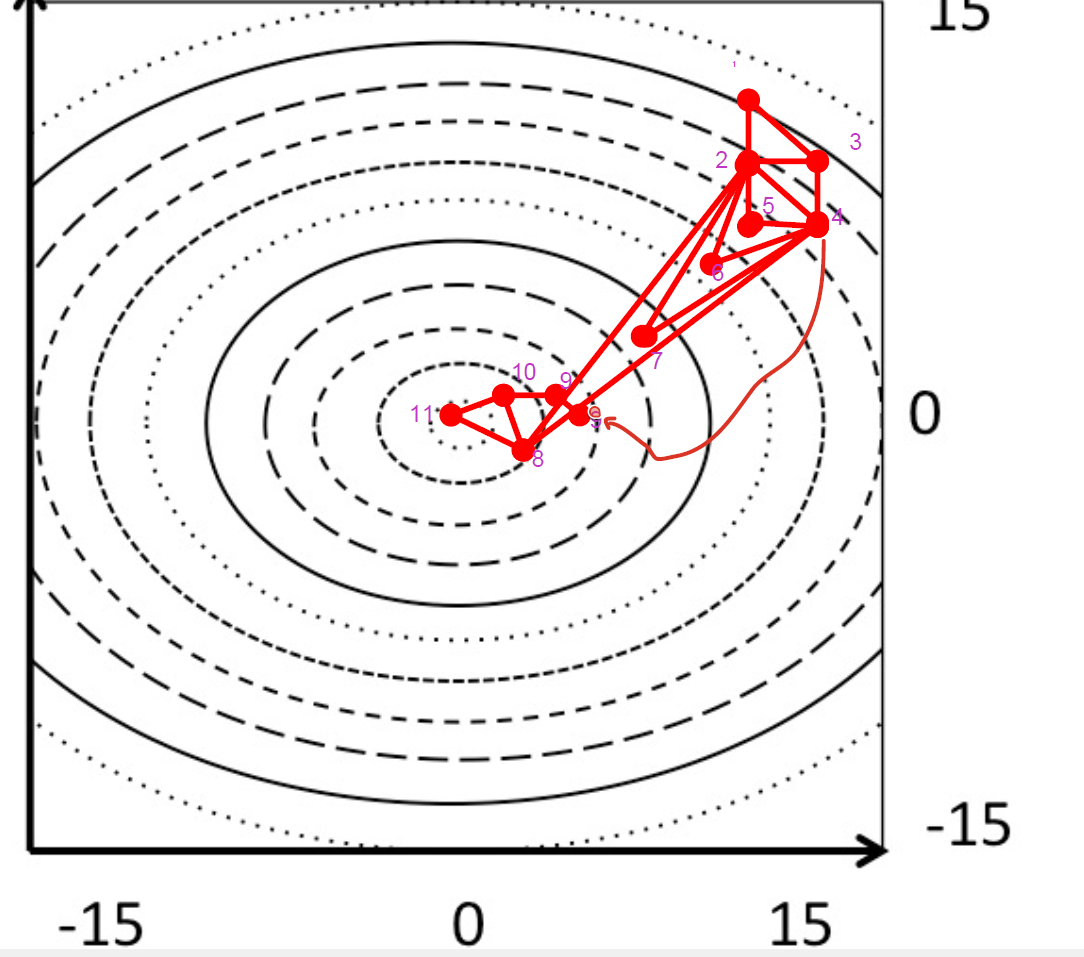
EM algorithm that finds local minima in N-dimensional space for small molecules
simplex= geometric figure with N+1 vertices (corners) each with their own energy
1)start with random poin on PES with N neighbouring points
2) ID highest E point and reflect through the centroid of the lower energy points
3) if reflected point:
improves the situation: stretch further along the reflection direction
doesn’t improve: move point partially back towards the centroid and then contract the the vertices of higher E towards the lowest E one
4) repeat until a local minimum is found
useful when inital energy is very high and good for intial steps
but costly in comp time,
needs many conformations and E’s before it can start
can get trapped in local minima and converges slowly once close to minimum, so better to swithch to other methods at that point
sequential univariate method
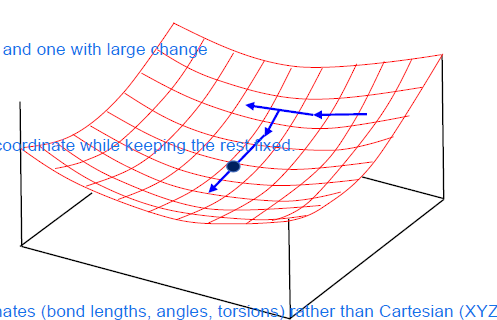
= EM method by optimizing one internal coordinate at a time: xi= bondlengths, bond angles and torsion angles rather than cartesion coordinates
1) start wiht conformation xi
2) create two new structures xi+deltaxi and xi+2deltaxi and compute the energy
one wiht big change and one with small
3)fit parabole through the three points and determine the minimum xi+1 = xi+a*delta*xi
4) continue with xi+1 keeping the rest fixed until minium is reached
But changing one coordinate can affect others, so each coordinate has to be revisited multiple times - so this method is slow, inefficient and limited to small molecules
Derivative methods
Principle = move in direction parallel to the force (first derivative of energy) until minimum of the line
When to stop along the line?
line search method
arbitrary step method
These are two different strategies that tell you how far to move along a certain direction once the derivative has told you which way to move.
these are used in other algorithms and are not complete optimization methods themselves
second derivative
minimum = where curvature changes direction

line search method
1)start with point 1 with E1 move along direction of the gradient and chooses two now point such that they bracket a minimum (E2<E3)
2) fit a parable and compute the minimum= next point
3) continue with the new 3 lowest points and repeat until minimum is found
High accuracy but computationally expensive
difference line search method and sequential univariate method
Line search method optimizes along derivative guided direction in multidimensional space, (based on cartesion coordinates)
non derivative sequential univaritate methods only update one paramater at a time (holding others fixed)without derivatives (based on internal coordinates)
during an energy minimization, cartesian coordinates are updated as you move to a low energy conformation, what happens to parameters like bondlenghts, bond angles, strength constants etc.?
During energy minimization, the Cartesian coordinates of atoms are updated to move the system toward a lower-energy conformation. Force field parameters like bond lengths, bond angles, and force constants remain fixed. However, the observed geometric values in the molecule—actual bond lengths, angles, and dihedrals—adjust toward the parameter-defined “ideal” values.
Force field parameters only define ideal values (like bond lengths r0r_0r0) and how strongly deviations are penalized (force constants). The observed values are the actual bond lengths and angles in the current structure. During minimization, the atoms move to balance all forces, so the observed geometry naturally relaxes toward the ideal values—it emerges from minimizing the total energy, not from the parameters directly.
Arbitrary step approach
1) start from initial conformation and compute gradient (direction of steepest descent)
2) atomic coordinates are moved along the gradient by a predetermined stepsize
3)recompute the gradient
4) repeat until no descent in E anymore
if lambda
to big: it overshoots the minimum, so next iteration lower it
to small = slow convergence, so next itteration increase its value
although it may require more itterations than line search method it will always be faster because no repeated E evaluations are done (no optimal lambda)
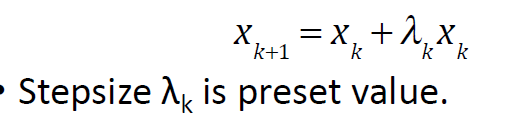
Steepest descent
1)Start with an intial structure and calculate the gradient
2) move coordinates in this direction of steepest descent by a certain stepsize
arbitrary step: fixed
line search: optimized every time
3) recompute the gradient at the new geometry
this itteration will move orthogonal to the previous direction because it now has no component along that direction (if minimization in previous direction is exact with line search, not with aribitrary step)
4) repeat until convergence
simple and good for early stage minimization
not for final refinement
can oscillate in narrow valleys
slow convergence near minima due to zig zag behavior
Conjugated gradient
start with intial structure and calculate the gradiant
move atom coordinates in the direction of the gradient
The first step is a steepest descent
Choose a stepsize via line search
usually nog arbitrary step
The next step will move in a direction which is a weighted ocmination of the current (orthogonal) gradient and the previous search direction
continue unitl convergens
good for fine tuning
higher efficiency because it needs fewer steps to reach the minimum (no zig-zag)
But more complex because you do a line search at every step