NUR 308 EXAM #2
1/163
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
164 Terms
AGA measurements in the neonate
head circumference: 33-35 cm
chest circumference: 30.5-33 cm
birth weight: 2700-4000 gm
cephalocaudal
head to toe
measures growth in length
proximodistal
proximodistal to torso outwards
measures chest/abdomen
Nutrition for the first 6 months
breast milk
Infant formula should be iron-fortified
Adding additional fluids (water or juice) in the first 6 months may result in...
failure to thrive due to really low weight and dehydration
nutrition in the second 6 months
Breastmilk/formula is still the primary source of nutrition
If exclusively breastfed still after 6 months → give Vitamin D
Calorie requirements for infants
about 100 cal/kg/day
Commercial formula is about 20 cal/oz
calories to oz conversions
baby weight (kg) x 100 = how many calories they need
ex. 4 kg baby x 100 cal = 400 cals
Then divide 400 cals by 20 cals = 20 Ounces
congenital abnormalities
serious health problems that occur in 2-4% of all live births
Congenital amnomalies usually occur in the ______ trimester
first trimester
What are the 2 classifications of congenital anomalies?
Syndrome → a recognized pattern of malformations due to a single cause
Association → a non-random pattern with no specific cause
What are some etiologies of birth defects
heredity/genetics (20%)
maternal environment → mother taking OTC drugs/street drugs, illness
Combination of both
What is neural tube defects (NTDs)
Defects/failure of neural tube closure
(normally a neural tube closes around 30 days after conception)
neural tube defect etiology (cause)
Maternal nutritional deficiency in folic acid (Mothers should take prior to pregnancy)
Multifactorial → could be heredity
Characteristics of neural tube defects
The neural tube is embryonic beginning for the brain & spinal column
The brain & spinal cord become encased in a protective sheath of bone and meninges
May involve entire length of neural or small portion
Neural tube defects are more common in what gender and race?
Found more in girls than boys
3 times more in whites than African Americans
Treatment/Prevention of neural tube defects
folic acid supplementation of 0.4 mg/day
If history of NTD: 4 mg/day
Foods that contain folic acid
Dark green leafy vegetables → spinach, kale, cabbage, peas
Beans
Peanuts
Sunflower seeds
Fresh fruit
Whole grains → fortified cereals
antenatal diagnosis of NTD
elevated a-fetoprotein in amniotic fluid at 16 to 18 weeks gestation
amniocentesis is how we identify if there is an NTD
Uterine ultrasound
Can check out spine
Why do we want to know if a fetus has a NTD?
So that parents are prepared to know the extent of the lesion (major or not)
to schedule a C-section to manage sac without labor/stress
What are the two most common types of neural tube defects (NTD)?
Anencephaly
Spina bifida/myelomeningocele
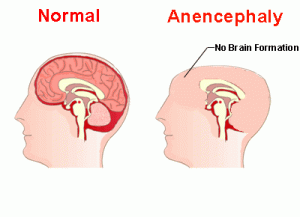
Anencephaly
Absence of cerebral hemispheres
Brainstem function may be intact
Incompatible with life:
survive only a few hours/days and die due to respiratory failure
spina bifida/myelomeningocele
failure of the osseous spine to close
What are the two types of spina bifida/myelomeningocele?
spina bifida occulta: not visible externally
spina bifida cystica: visible defect & saclike protrusion coming from spine
spina bifida occulta
lumbosacral L5-S1 (lower portion of spine)
skin indicators:
sacral dimple
sacral tufts of hair
sacral lipoma (fatty tumor below skin)
abnormal adhesion to bone or fixed structure
Traction on the cord causes:
altered gait
bowel/bladder problems
foot deformities
spina bifida cystica
visible defect with external saclike protrusion
two types:
meningocele
myelomeningocele
meningocele (within spinal bifida cystica)
sac contains meninges and spinal fluid but no neural elements
no neurological deficits (just need surgery to fix)
myelomeningocele (within spinal bifida cystica)
Maybe anywhere along the spinal column
Sac contains:
meninges
spinal fluid
and nerve
Varying and serious degrees of neurologic deficit (depends on where on the spine it’s located)
myelomeningocele sac
may be fine membrane → prone to leakage of CSF; easily ruptured
may be covered with dura, meninges, or skin
Nursing care for Myelomeningocele Sac
Keep moist and intact with a sterile solution
Prone position
myelomeningocele degree
Location and magnitude of defect determine the nature and extent of impairment:
If the defect is below second lumbar vertebra (L2):
Flaccid paralysis of lower extremities → can’t walk, feel legs, or have bowel functions
myelomeningocele initial management
pre op → lay on stomach (prone)
prevent infection → hand hygeine
assess neurological anomalies
Early closure in 12-72 hours after birth
prevent stretching of other nerve roots and further damage
Myelomeningocele is always associated with/linked to...
latex allergy
cerebrospinal fluid
secreted by choroid plexus
circulates throughout the ventricular system
absorbed within the subarachnoid spaces
hydrocephalus
Extra CSF in the ventricles in the brain ("water on the brain")
Commonly associated with myelomeningocele
80-85% of spinal bifida cases will develop hydrocephalus
May not be apparent after birth
May appear after primary closure of defect (need to do daily head circumferences)
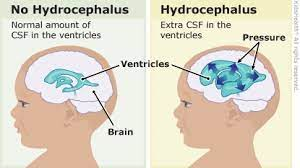
hydrocephalus monitoring
Measure head circumference
fontanel tension (bulging outward on top of head)
serial ultrasounds (shows ventricle size)
hydrocephalus initial management
Treatment of excessive CSF by shunt
surgical intervention: goes into the brain down the neck into the stomach
treatment of complications
Manage problems related to psychomotor development
signs and symptoms of shunt malfunction
SHUNT MLAFUNCTION = EMERGENCY
increased ICP
worsening neurologic status
altered LOC
signs and symptoms of shunt infection
shunt malfunction
fever and inflammation of tract
abdominal pain
Redness or drainage at site
microcephaly
abnormally small head
primary exposure: intrauterine (mother) exposure to toxins
CMV, Rubella (measles), toxoplasmosis, radiation
secondary exposure:
third-trimester, infection, early closure of cranial sutures, metabolic disorders, anoxia
microcephaly effects
mild hyperkinesis (muscle spasms/stiff)
mild motor impairment
complete unresponsiveness: autistic behavior
Impaired brain development
craniosynostosis
premature closure of one or more cranial sutures
some increased ICP
progressive papilledema (swelling in face), optic atrophy, and blindness
generally sagittal suture closes prematurely & results in elongation of skull
craniofacial abnormalities
usually not life-threatening
surgical corrections if possible
ex. Pierre robin syndrome
small chin & feeding problems
congenital clubfoot
AKA talipes equinovarus
Incidence is 1-2 per 1000 live births
more common in males
bilateral clubfeet in 50% of cases
familial tendency (history)
clubfoot categories
positional (mild) - believed due to intrauterine crowding
syndromic (tetralogic) - associated with other congenital abnormalities
congenital (idiopathic) - unknown reason, wide range of severity and prognosis
therapeutic management of clubfoot
correction of the deformity
maintenance of the correction until normal muscle balance is regained
follow-up observation for recurrence
management & treatment of clubfoot
serial casting shortly after birth (progression of frequent casting)
prognosis available
nursing considerations → circulation, education
chromosomal alterations
occur in 1/150 live births
deviation in either structure or number of chromosomes
leading cause of mental delay and spontaneous abortions
can be related to maternal age/prenatal care
trisomy 21
down's syndrome
most common chromosomal abnormality
occurs in 1/600-800 live births
occurs more often in whites
trisomy 21 etiology
extra copy of chromosome 21
cause is largely unknown
statistically greater risk for women over 35
women over 40 have 1/110 chance
only 5% of time, the extra chromosome is from the father
trisomy 21 clinical manifestations
usually diagnosed clinically, but chromosome analysis is always done to confirm outward
outward, upper slant of eyes
small ears
large, protruding tongue, mouth kept open
short, broad neck
short, broad hands, transverse palmar crease
trisomy 21 outstanding features
Intelligence:
varies from severely delayed to low-average intelligence
Social development:
strength in sociability 40-45% have congenital heart defects
Congenital abnormalities
40-45% have congenital heart disease
trisomy 21: other problems
Sensory problems:
cataracts
hearing loss
altered immune systems:
leukemia
prone to respiratory infections
Growth:
reduced height and weight
congenital heart anomalies
Sexual development
delayed or incomplete
trisomy 21: therapeutic management
surgical repair of serious congenital anomalies
evaluation for sight and hearing
special growth charts
trisomy 21: nursing considerations
Family support
Education regarding developmental potential and how uncertain it is
turner syndrome
occurs in 1/10,000 female births
missing or incomplete 2nd X chromosome
normal growth until 3 years old
behavioral problems:
difficulty with social relationships/rules
Usually infertile
most lead productive lives, independent adults
klinefelter syndrome
most common sex chromosome abnormality
have multiple X chromosomes along with Y (MALE)
decreased masculinization with
gynecomastia (enlarged breasts)
hypogonadism (small testicles/not fully distended)
sterility
mental development is usually normal
newborn chromosomal defective gene screenings → important to do as soon as birth to treat early
Screenings exist that are reliable and inexpensive
there is effective treatment or intervention
if untreated, the baby will die or be severely impaired
some babies appear normal at birth
endocrine disorders (in Illinois)
congenital hypothyroidism
congenital adrenal hyperplasia
metabolic disorders (in Illinois)
phenylketonuria (PKU)
galactosemia
cystic fibrosis
hemoglobinopathies (in Illinois)
sickle cell disease
congenital hypothyroidism
autosomal recessive disorder
deficiency of thyroid hormone
occurs 1/3600-5000 live births
more common in females
Cause: thought to be caused by defective embryonic development of thyroid gland
congenital hypothyroidism symptoms
usually appear by 6 weeks
poor feeding
lethargy
prolonged jaundice
decreased metabolism → weight gain
respiratory difficulty
cyanosis
hoarse cry
bradycardia
if congenital hypothyroidism is not treated...
irreversible mental delays
Develop classic features such as:
depressed nasal bridge, short forehead, puffy eyelids, large tongue
congenital hypothyroidism treatment
Lifelong thyroid hormone replacement
synthetic levothyroxine sodium → given in morning before feeding
education to parents:
need for evaluation of growth,
routine monitoring of thyroxine levels (frequent blood draws)
strict adherence to treatment (needs to occur everyday for rest of life)
congenital adrenal hyperplasia
occurs in 1/12000-15000 births
causes overproduction of adrenal androgens
results in male hormones in female fetus:
excess androgen
body hair, deep voice, muscle mass
“masculine features”
congenital adrenal hyperplasia: clinical manifestations
varying degrees of ambiguous genitalia
girls:
enlarged clitoris that resembles penis
fused labia which resembles a scrotum
internal sexual organs are normal
boys:
do not display genital abnormalities
fast genital development
congenital adrenal hyperplasia: therapeutic management
with early dx: cortisone is used Iif started early, very effective)
if not diagnosed in infancy, early sexual maturation occurs
and both sexes appear outwardly male
girls can develop normally with medication and surgery
phenylketonuria (PKU)
An inherited genetic disease caused by an absence of the liver enzyme needed to convert phenylalanine to tyrosine
(missing the liver enzyme to break down protein)
Seen in 1/4000-12000 live births
Mostly affects whites
phenylketonuria (PKU) clinical manifestations
failure to thrive
frequent vomiting
irritability
hyperactivity
in older children-
bizarre behavior, screaming, head banging, seizures
phenylketonuria (PKU) screening test
Guthrie test:
detects increased levels of serum phenylalanine
ideal if the test is done 3-4 days after ingesting formula/breast milk
goal of screening and treatment is to prevent mental delays
WANT TO CATCH EARLY TO TREAT EARLY
phenylketonuria (PKU) treatment
restriction of dietary protein
beginning ASAP after birth and maintained throughout life
Avoid all meat and dairy products, including
breads, pasta, legumes, flour, nuts, and eggs
frequent blood phenylalanine monitoring
Special formula
periodic monitoring of intellectual, neurological, and behavioral parameters
phenylketonuria prognosis
With early detection and treatment, may achieve normal cognitive development
Important to screen after birth before leaving the hospital
But outcomes vary
Even with an adequate diet, a high % exhibit some degree of intellectual impairment
galactosemia
inborn error of carbohydrate metabolism (problems digesting carbohydrates)
lack of a liver enzyme that converts galactose into glucose
High levels of galactose results in…
hepatomegaly, cirrhosis, jaundice
enlarged spleen
kidney failure
cataracts
lethargy
hypotonia
brain damage, death if untreated
galactosemia treatment
Eliminate/avoid all milk and lactose-containing foods, including breast milk
lactose-free or soy protein formulas used
galactosemia nursing considerations
Education on:
Diet → emphasize the need to read food labels and recognize early developmental delay
Support:
Of mothers because they feel a great loss due to the fact they cannot feed their baby
sickle cell anemia
most common genetic disease in the US autosomal recessive disorder
Auto recessive gene (both parents have to have the gene)
Pathophysiology of sickle cell anemia
obstruction caused by sickled red blood cells
cells stick together = get stuck/clump & can’t pass arteries
increased hemolysis
sickle cell anemia clinical manifestations
swollen hands/feet in newborn
susceptibility to infection
possible growth restriction
chronic anemia
jaundice
abdominal tenderness
Vasoocclusive crisis - clinical manifestations
painful swelling of:
hands & feet
joints
severe abdominal pain
sickle cell therapeutic management
Early treatment is significant
Hydration → prevents clumping of RBCs
Prompt treatment of sickle cell crisis
hydroxurea
The only drug approved by the FDA to reduce incidence of pain crises in children with sickle cell
sickle cell anemia nursing considerations
adequate hydration
no contact sports with enlarged spleen
avoiding environments with low O2 concentration
avoid sources of infection
strict adherence to medication
prompt report of fever or infection
even mild (get sick very quickly)
cleft lip/palate
the most common craniofacial malformation
more common in males
results from incomplete fusion of the embryonic structures surrounding the oral cavity
cleft palate
Occurs when the primary and secondary palates fail to fuse during embryonic development
can involve only soft palate or extend into hard palate
cleft lip/palate diagnosis
readily apparent at birth
can be diagnosed in utero by sonogram at 14 weeks
thorough examination of hard and soft palate with gloved finger at birth is essential
cleft lip surgery
usually involves no long term interventions after surgery
cleft palate surgery
Management after surgery usually involves a multidisciplinary team including plastic surgery, orthodontics, speech, etc.
Haberman feeders & Dr. Brown Valve/Disk
special nipple/bottle that can be squeezed in sequence with the babies feeding pattern
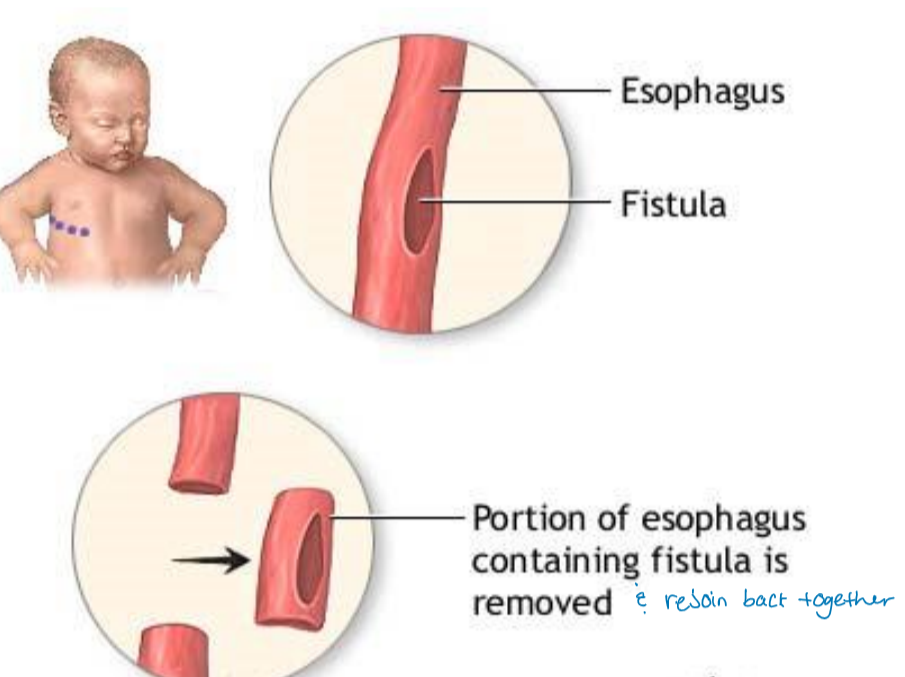
esophageal atresia
failure of the esophagus to develop as a continuous passage
Esophagus doesn’t connect correctly to the stomach → food cannot reach the stomach
Surgical intervention is needed right away
tracheoesophageal fistula
failure of esophagus and trachea to separate into distinct structures
food will go into the trachea to the lungs
Clinical manifestations of esophageal atresia/tracheoesophageal fistula
frothy saliva in mouth → choking and coughing
with feeding → formula from nose and mouth
cyanosis and cessation of breathing with aspiration of feeding (if choking)
Diagnostic evaluation of esophageal atresia/tracheoesophageal fistula
Attempt to pass NG tube → inability warrants further investigation
radiopaque catheter and x-rays
bronchoscopy to visualize fistula
Therapeutic management of esophageal atresia/tracheoesophageal fistula
maintenance of patent airway
prevention of aspiration pneumonia
surgical repair of abnormal structures
crucial that they are NPO
Preoperative care for esophageal atresia/tracheoesophageal fistula
Maintain NPO status
IV fluids/parenteral nutrition
careful suctioning of nose/mouth
proper positioning
Supportive care/Education to parents
Surgical repair: esophageal atresia/tracheoesophageal fistula
usually one stage
Done after adequate hydration, treatment of pneumonia proactively, and patient is well
thoracotomy with division of TEF and end-to-end anastomosis of esophagus
Post-op care: esophageal atresia/tracheoesophageal fistula
First feeding after surgery should be sterile water
If the suture isn’t correct only water will go into body
No formula/breast milk in case surgery is unsuccessful
pyloric stenosis
Severe narrowing of the pyloric canal due to pylorus thickening
Develops in the first few weeks of life
won’t gain weight & failure to thrive
pyloric stenosis clinical manifestations
non-bilious vomiting in early stages → projectile vomit
Infant is hungry, irritable
prolonged vomiting leads to dehydration
olive-shaped mass may be palpated (hypertrophy of pylorus)