Purification and Primary Sequence Determination
1/18
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
19 Terms
Formation of Peptides
peptides are small condensation products of amino acids
Peptides
linear sequence of amino acids with one free N terminus and one free C terminus held together by peptide bonds
peptide bonds have partial double bond characteristic due to resonance
sequences are written from the N to C terminus
Covalent Structures of Proteins
gene sequence - each amino acid is coded for by 3 nucleotides; DNA sequence can be translated into protein sequence
chemical and enzymatic sequencing of purified protein, mass spectrometry sequencing
Protein Purification
two options:
know gene sequence, protein sequence, size + charge + maybe something about the structure and function, design a purification scheme based on those properties
know protein function, don’t know sequence, don’t know size + charge + something about the structure, try different techniques and monitor protein concentration and function
separation relies on differences in physio-chemical properties
size
charge
affinity for a ligand
solubility
hydrophobicity
thermal stability
chromatography is commonly used for preparative separation
Protein purification needs…
an assay for function and an assay for protein
UV absorbance by the Beer-Lambert Law for the presence of protein
Separation Based on Solubility
ammonium sulfate precipitation: as the ionic strength of the solution increases, water molecules get “tied up” with hydrating the ions and no longer hydrate the dissolved protein → precipitation
larger protein surface = lower ammonium sulfate concentration at which it will precipitate
small polar molecules will have stronger ionic strength
frequently the first step in purification because it removes large fractions of contaminating proteins
not very specific (only solubility matters), proteins require further purification
at pH = pI proteins will usually precipitate because loss of charge-charge repulsion (less solubility)
Separation Based on Size
gel filtration chromatography
size exclusion chromatography
each size exclusion resin has a specific range of sizes (300-15000, 50000-500000, 10000-100000)
protein below the limit will all elute together at the bed volume and proteins greater than the limit will elute together at the void volume
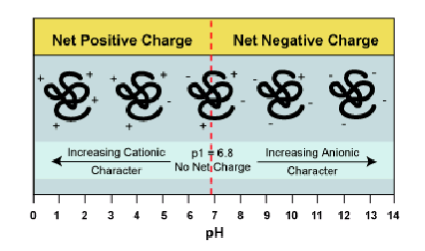
Charge
pI of a protein is the pH at which the protein had no net charge → can be determined experimentally
if pI is less than pH the protein is NEGATIVELY charged
if pI is more than pH the protein is POSITIVELY charged
charge on protein depends on pI and pH of the buffer in ion exchange resins
anion exchange resins: DEAE sepharose, Q sepharose, bead-linker quaternary amine
cation exchange resin: CM CH2-COOH, SP CH2SO3H
Affinity
ATP columns are used to purify any protein that binds to ATP or any of the nucleotide cofactors
metal binding proteins or His rich proteins bind to Ni columns
ATP is covalently linked to the column - any protein that binds to ATP will bind to the column and will constantly come off + rebind
add ATP to the column buffer to get the protein off the column
the tighter the protein binds to the column the more free ATP in the buffer it takes to get protein off
Polyacrylamide Gel Electrophoresis
separates proteins based on size
samples are boiler in detergent SDS containing DTT to denature protein and break disulfide bonds → multi-subunit proteins are separated into individual chains
all proteins are coated in SDS so they are negatively charged regardless of amino acid composition
SDS PAGE: Molecular Weight
SDS = sodium dodecyl sulfate - a detergent
SDS micelles bind to and unfold all the proteins, gives all proteins a uniformly negative charge (native shape doesn’t matter)
rate of movement will only depend on size
SMALL PROTEINS MOVE FASTER - too thick = nothing moves, too thin = everything moves too much
Quantitative Amino Acid Analysis
gives basically all the amino acids in the protein but no sequence information
hydrolyze all amide bonds for 12-36 hours in sealed tube
run the mixture on an ion exchange column to isolate
amino acids are derivatized with a chromophore (DABS) before or after the column is run
asn and gln will not show up because they have amide bonds that get hydrolyzed to glu and asp
End-Group Analysis
to determine the identity of the amino acid at the N or C-terminal of a peptide/protein and the # of polypeptide chains in the protein
dansyl chloride and FDNB react with free amino groups, also lys side chain
standards can be used to identify dansyl or DNP labelled amino acids
2+ amino acids indicate heterogenieity (one peak) or more than one polypeptide chain in the protein (multiple peaks)S
Disulfide Bond Cleavage
cysteine residues can interfere with sequence determination due to disulfide bonds
beta-mercaptoethanol or DTT (makes cyclic disulfide) break disulfide bonds by disulfide exchange - making intermolecular bonds within molecule to break the ones from protein
carboxymethylation with iodoacetate prevents disulfide bonds from reforming
Protein and Peptide Sequencing
only reliable for 30-50 amino acids in a sequence; method has to break big proteins into smaller, specific peptides for sequencing
chymotrypsin and trypsin are specific proteases to make smaller pieces
reduce and alkylate disulfide bonds (hydrolyze + reduce)
cleave polypeptide to smaller fragments by 2 or more methods e.g. trypsin and cyanogen bromide (cleaves at MET)
sequence smaller fragments e.g. Edman degradation
overlap sequences to determine overall sequence
repeat without reduction of disulfides to locate position of bridges
Edman Degradation Method
makes amino acid sequence from the N-terminal end but each step is only 99% efficient
after 20-50 you get too much noise and can’t differentiate between what you just cut
cannot sequence through disulfide bonds → need to reduce disulfide bonds to Cys first
Putting Everything Together
purify protein
treat with beta-mercaptoethanol or DTT and iodoacetate if needed
treat separately with trypsin, chymotrypsin and/or CNBr
separate the peptides by ion exchange or reverse phase HPLC
sequence each peptide by the Edman degradation method
analyze data to generate intact peptide
Protein Sequencing by Mass Spectrometry
protein is cleaved into peptides, usually with trypsin since it generates and fragments
fragments are subjected to tandem MS-MS by electrospray ionization
patterns of fragments are analyzed and compared to known sequences
sequence is generated
What can sequences do for you?
identify the target protein by comparison to a similar, known protein to predict new protein’s function
allows to get a crystal structure to get a hypothetical structure
can show how related a particular protein is to the same protein from other organisms by looking at primary sequences