Lecture 11: Phylogenetics II
1/22
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
23 Terms
Clade
group of organisms that share a common ancestor and includes all descendants
Sister Taxa
taxa derived from the same common ancestor (node)
Polytomy
unresolved relationship between 3+ taxa
Monophyletic
includes the most recent common ancestor and all descendants
ideally, organisms are grouped according to pattern of descent, and only monophyletic groups should be named
BUT not all groups in practice are monophyletic
Paraphyletic
includes the most recent common ancestor but not all descendants
most traditional groups are paraphyletic
ex. dinosaurs; Dinosauria should include birds
ex. reptiles: Reptilia should also include birds
Polyphyletic
does not include the most recent common ancestor of all members of a group
group has at least 2 separate evolutionary origins
Rooted Tree
common lineage indicated from base of tree
direction indicates the passage of time
in theory, it is possible to root a tree along any branch
different roots provide different sequences of branching events
one tree will therefore be more correct than others
Unrooted Tree
does not indicate (fully) the direction of the passage of time
unclear which internal node is the most ancestral
Root
point on a tree representing the earliest time in the evolutionary history of the taxa included
hypothetical common ancestor to all taxa in the tree
root trees via outgroups
Model Based Phylogeny
parsimony analysis does not incorporate statistical model or evolutionary change
best tree = one with the fewest changes
model based methods incorporate probability models of how characters change over time
calculates probability of a change occuring in a given branch
maximum likelihood is the most common method of tree-building now
Maximum Likelihood
idea:
find the tree that maximizes the probability of obtaining the observed data
need:
original data
an underlying model of evolution
trees and their branch lengths
then:
search possible trees and find most likely tree; the tree that maximizes the probability of observing the actual data
Cladogram
branch length has no meaning
Phylogram
branch length indicates the amount of evolutionary change
corresponds to model of subsitution rate from one nucleotide to another
ML methods use phylograms to find the tree most consistent with observed sequence data
Statistical Confidence
we can rarely be certain an entire tree is correct
instead, we can assign confidence values to parts of the tree
how strongly does data support a given clade?
Bootstrap Resampling
build replicate trees by creating new datasets from original dataset (repeated sampling)
pick with replacements
create 100s of replicates, and a tree for each
generate consensus tree indicating hat percentage of the trees each particular branch occurred
how often do we obtain the same clade?
bootstrap values: 0-100% (bad-good)
if results are being biased by a few nucleotide sites, branch values will be low
Bootstrap Resampling Results
0% (bad) - 100% (good)
>70% suggest a particular grouping is reliable
50-70% considered suggestive but not conclusive
<50% suggests a particular grouping is not strongly supported by the dataset
Molecular Clocks
molecular traits change at a steady rate, in a ‘clock-like’ fashion
recall neutral theory:
if mutation rate is constant and generation times are similar,
# neutral molecular differences between two taxa should be proportional to the age of their most recent common ancestor
can this be used to date when and how rapidly major events occurred?
yes, # substitutions proportional to divergence time
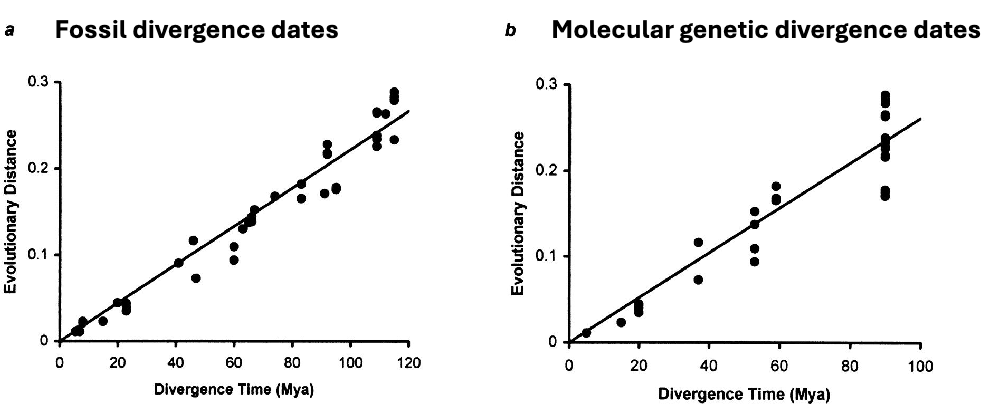
Calibrating Molecular Clocks
measure genetic distance bewteen two taxa whose divergence is known from:
fossil record
geological record
however, we now know that molecular clock runs at different rates in different species
even in different regions of the genome within a species
rates calibrated for particular gene and lineages might not work for other groups, but can still be useful within a clade
calibration with fossil record or geological events are only estimates of actual divergence dates
WHIPPO Phylogeny Background
who are the whales’ closest living relatives?
whales, dolphins, and porpoises form a monophyletic group: Cetacea
relationship between cetaceans and ungulates (horse, hippo, deer, etc.) suggested by skeletal characters
proposed as a sister group to artiodactyls (cows, hippos, pigs)
outgroup is perissodactyls (horses, rhinos)
are whales ungulates? or are ungulates paraphyletic?
WHIPPO Synapomorphy
the astragulus, a bone in the ankle, unites the Artiodactyls
whales dont have the Artiodactyl-type of astragulus
does this imply that whales are outside of artiodactyla?
on the other hand, whales dont have an astragulus at all!
need another kind of data
DNA Synapomorphy WHIPPO
Beta-casein gene sequencing data suggest that whales are a sister taxa to hippos
SINE and LINE elements
Short of Long INterspersed Elements (transposons)
transposition events are rare, and therefore VERY unlikely that 2 homologous SINEs would insert themselves into 2 independent host lineages at exactly the same location
reversal is also very unlikely, as these can be detected
much more likely to be synapomorphies suggesting that whales are a sister taxa to hippos
Fossil Evidence WHIPPO
fossils discovered after SINE/LINE analysis have:
whalelike characteristic
pulley-shaped astragulus
WHIPPO Conclusion
suggests that traits of hippos and whales that were thought to be convergent adaptations for aquatic life might actually be synapomorphies
modern taxonomic classification refers to this group as Cetartiodactyla