Metabolic Disorders Exam #2
1/41
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
42 Terms
Congenital Enterokinase Deficiency
cause: low trypsin as a result of a dysfunctional enterokinase
enterokinase is used to active trypsinogen into trypsin
symptoms: failure to thrive, chronic diarrhea, low serum protein, and generalized edema
diagnosed at infancy, grew to adults leading normal lives
treatment: isolated enterokinase is added, trypsin activity returns to normal
some with this deficiency have residual enterokinase activity
thus, trypsinogen can be self-activated at a slow rate. but once it is activated, it leads to a positive feedback loop
How are some adults able to live with congenital enterokinase deficiency, while infants have profound issues related to malabsorption?
the self-activated trypsinogen mechanism is sufficient for adults but not infants because infants have a higher demand for protein digestion
describe how protein in ingested.
adults needs 60-100g of protein a day
protein is digested into amino acids
the amino acids are absorbed by the enterocyte
enter the amino acid pool
the pool is derived from the diet and proteolysis of proteins
describe the characteristics of amino acids
20 amino acids
each differ in their R-group
R-group determines the chemical properties of the amino acid
charged side chain
uncharged side chain
hydrophobic side chains
special cases
what is the difference between synonymous SNP and non-synonymous SNP?
synonymous SNP
change in the base, but not the codon
ex: ACU and ACC both code for Thr
synonymous SNP and non-synonymous SNP
change in the base and the codon
in severe cases, it can result in a stop codon
disruptions can affect enzyme activity, interactions with other proteins, or ligand-binding
ex: arg —> leu
results in a conformational change in the structure of the protein
what are the 6 phases of amino acid digestion and absorption?
mechanical breakdown
the physical breakdown of food in the mouth
digestion that starts in the stomach
gastric hydrolysis of the peptide bonds in proteins
digestion continues in the small intestines
pancreatic proteases further breaks down the proteins into smaller peptides
the proteases are secreted as zymogens and activated in the small intestines
brush border membrane peptidases further hydrolyzes peptide linkages
transport of amino acids
amino acids, dipeptides, and tripeptides cross the brush border via enterocytes
digestion of dipeptides and tripeptides
cytoplasmic peptidases in the enterocyte further breakdown the peptides
transport across the basolateral membrane
describe the gastric phase
location: the stomach
plays a minor role in overall digestion
gastric HCl and pepsins
gastric chief cells release pepsinogens (inactive)
the pepsinogens are activated in the presence of acid into pepsin
pepsin acts as an endopeptidase to target peptide bonds
purpose:
prepare polypeptides for digestion and absorption within the small intestine
describe the small intestinal luminal phase
location: small intestine
mucosal cells release cholecystokinin (CCK)
CCK reaches the pancreas and binds to acinar cells to stimulate release of zymogens via the pancreatic duct and common bile duct
trypsinogen
enterokinase cleaves 8 amino acids off of trypsinogen to produce trypsin
trypsin activates the other zymogens and forms a pool of activated proteases
purpose:
further breakdown peptides into amino acids
describe the mucosal phase
location: small intestines
after pancreatic hydrolysis, free amino acids, dipeptides, tripeptides, and oligopeptides remain
oligopeptides must be hydrolyzed by membrane-bound enzymes in the brush border
dipetides and tripeptides must be hydrolyzed by cystolic aminopeptidases
purpose:
further breakdown peptides into amino acids
what are amino acid transporters?
purpose: amino acids transporters moves protein in and out of the cell
belong to solute carriers superfamily
describe the X- and x- amino acid transporters.
purpose: transports anionic amino acids
X- AG: aspartate and glutamate
x- C: acceptance of cysteine
describe the Y+ and y+ amino acid transporters.
purpose: transports cationic amino acids
ex: arginine and lysine
describe the B and b amino acid transporters.
purpose: transports neutral amino acids
describe the T amino acid transporters.
purpose: transports aromatic amino acids
such as tyrosine and tryptophan
describe the N amino acid transporters.
purpose: transports neutral amino acids with a selectivity for amino acids with N in the side chain
describe the IMINO amino acid transporters.
purpose: transports proline and hydroxyproline
which amino acid transporters are sodium-dependent and sodium-independent?
sodium-dependent
capital letter abbreviations
sodium-independent
lower letter abbreviations
system L, system T, and proton-coupled AATs
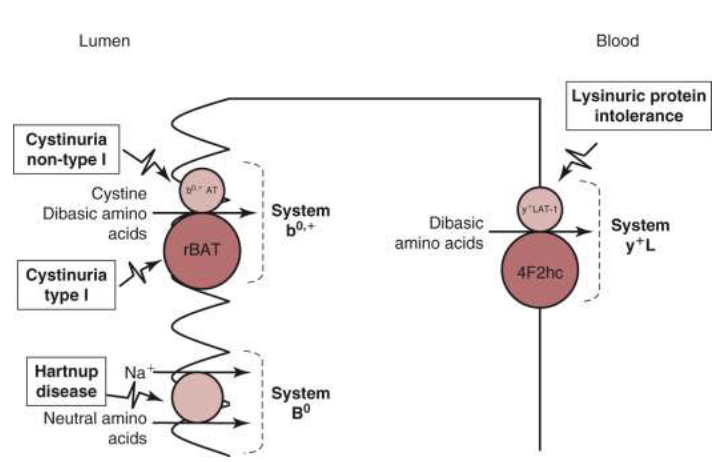
Cystinuria
cause: defective amino acid transporter on the apical membrane of the small intestines and renal proximal tubules
results in amino acids such as L-cystine, lysine, arginine, and ornithine not being transported across the membrane
leads to hyperexcretion of these amino acids in the urine because they are unable to be reabsorbed
this results in high concentrations of cystine, which forms crystals in the kidneys and bladder and causes kidney stones
mutations in either the amino acid transport or the functional unit can lead to cystinuria
symptoms: nausea, hematuria, flank pain, kidney stones, and frequent urinary tract infections
screening can be due to (1) family history of cystinuria (2) recurrent kidney stones (3) development of stones at an early age
testing stone and determining that it consists of cystine
treatment:
initial approach -- prevention of stone formation
drinking more fluids or decreasing sodium intake
reducing animal protein rich in methionine
methionine is broken down into cystine in the body
drug therapies -- thiol agents to form mixed disulfides that are more soluble than cystine, lowering free cystine levels
penicillamine & tiopronin
Cystinosis
cause:
cysteine enters the lysosome, forming cystine
cystinosin (CTNS) is a membrane transporter that removes cystine from the lysosome
BUT, in cystinosis, CTNS is not functional, leading to a build up of cystine and causes cystine crystals inside the lysosome
this can cause damage to the kidneys, and other organs as it progresses
ex: upper GI tract, liver, and lower GI tract
symptoms:
dehydration, polyuria, polydipsia, metabolic acidosis, hypokalemia, etc.
treatment:
cystine-depleting therapy -- breaks cystine into cysteine and cysteine-cysteamine -- creates mixed disulfides
these products are able to leave the lysosome independently of the cystinosin transporter
cysteine leaves via the cysteine transporter and cysteine-cysteamine leaves via the PQLC2 transporter
what are the 3 types of amino acids?
3 types of amino acids:
essential
cannot be synthesized in the body, and must be obtained from the diet
non-essential
can be synthesized in the body
nitrogen is required
conditionally essential
only synthesized if essential amino acids precursors are provided
what are the 3 major metabolic fates of amino acids?
protein synthesis
precursors
catabolism
where does the nitrogen come from when synthesizing non-essential amino acids?
nitrogen is provided by the diet in the form of alpha-amino groups
however, the body has a limited ability to incorporate inorganic nitrogen into amino acids
what is transamination?
Transamination:
removing an amino group and adding it to a keto-acid to form a different amino acids
catalyzed by transaminase enzymes
these enzymes are particularly found in the liver, heart muscle, skeletal muscle, and kidney
transaminase requires vitamin B6 as a cofactor
a vitamin B6 deficiency can decrease the rate of transamination in tissues
the preferred amino acid/keto-acid is:
alpha-ketoglutarate/glutamate
what amino acids do not participate in transamination?
threonine
lysine
proline
Hypervalinemia & Hyperleucine-isoleucinemia (HVLI)
cause:
elevated levels of valine, leucine, and isoleucine in the blood and urine
results from a transaminase enzyme deficiency - specifically the BCAT enzyme
the BCAT enzyme is the 1st step to break down valine, leucine, and isoleucine
if not broken down, it can lead to build up of these amino acids in the brain and blood
BCAT 1 - cystolic form of the enzyme
rarely found
BCAT 2 - mitochondrial form of the enzyme
most common cause of hypervalinemia
symptoms:
present at birth
protein intolerance, metabolic acidosis, vomiting, failure to thrive, coma
delayed mental and physical development
treatment:
diet low in valine, leucine, and isoleucine
returns valine levels to normal
vitamin B6 supplementation
restore normal valine levels in patients with BCAT2 mutations
what is deamination?
Deamination:
the process by which the amino group is removed to form a keto-acid and ammonia
the ammonia is excreted as urea
the most common deamination is the release of ammonia by glutamate dehydrogenase (GDH)
converting glutamate into alpha-ketoglutarate, ammonia, and NAD(P)H
found in the mitochondria
present at high levels in the liver, kidney cortex, and brain
examples of deamination reactions:
glutamate dehydrogenase deficiency
histidinemia
Glutamate Dehydrogenase Deficiency
cause:
glutamate is converted into alpha-ketoglutarate by glutamate dehydrogenase
alpha-ketoglutarate is important for energy production (krebs cycle) and protein metabolism (transamination & deamination)
during a deficiency, an build-up of ammonia and excess of glutamate
build-up of ammonia: if no more alpha-ketoglutarate, this stalls the process that clears nitrogen from the body
leads to a build-up of ammonia
excess glutamate: unable to regulate the glutamine and glutamate cycle
leads to a build-up of glutamate, and overexcites the brain
glutamate is an important neurotransmitter
symptoms:
mimics Parkinson’s disease
seizures, brain fog, memory issues
Histidemia
cause:
deficiency in histidase prevents the conversion of histidine into trans-urocanic acid, resulting in ammonia not being released
elevated levels of histidine levels in the blood and urine
symptoms:
mostly asymptomatic, but some clinical symptoms were reported in patients
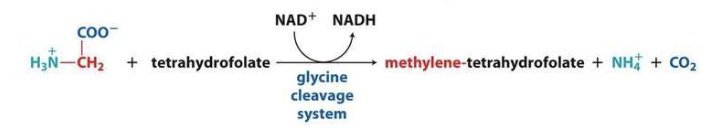
describe the glycine cleavage system.
purpose: uses glycine to donate a methyl to tetrahydrofolate
this cleavage system is important because it continues to methylate tetrahydrofolate in order for the folate cycle to continue running
4 different enzymes in the cleavage system:
GCSH, GLDC, DLD, and AMT
GLDC: needed for the release of CO2 and the transfer of glycine to GCSH
Nonketotic Hyperglycinemia (NKH)
cause:
results for a GLDC not functioning in the glycine cleavage system
nonfunctional GLDC accounts for most cases in NKH
leads to a build-up of glycine and accounts for most cases of NKH
symptoms:
high levels of glycine in the brain
neurological problems & global development delay
coma & death
treatment:
sodium benzoate
combines with glycine in the liver to promote excretion
dextromethorphan
stops glycine receptors from overfiring
diet
limiting gelatin -- high in glycine
coordination for swallowing and breathing may be needed
oral feeding by bottles, etc.
nasogastric tube or gastronomy tube
low glycine diet, ketogenic diet
what are the 3 types of NKH?
classic NKH
severe mutations in the core GCS genes
variant NKH
mutations in the genes outside of the GCS system
transient NKH
short-term and resolves eventually
what is deamidation?
Deamidation:
removing the amide group to release the ammonia
glutamine -- metabolized by glutaminase
asparagine -- metabolized by asparaginase
what is incorporation of ammonia?
most reactions utilize nitrogen in the amino or amide groups. however, some reactions can use ammonia
ex: glutamate dehydrogenase -- can function in the reverse direction, and can incorporate ammonia into glutamate
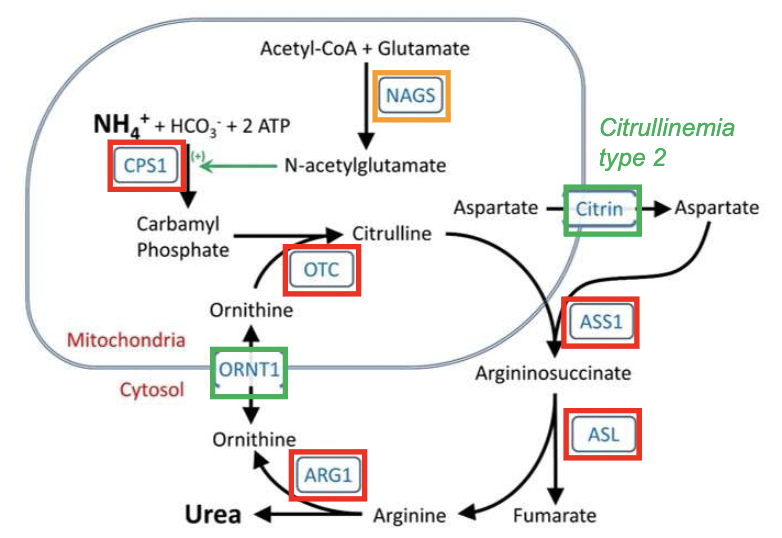
Describe the urea cycle.
Purpose: clearing the waste nitrogen from protein turnover
composed of 6 enzymes and 2 transporters
enzymes:
5 catalytic enzymes
CPS1, OTC, ASS1, ASL, and ARG1
1 cofactor-producing enzyme
NAGS
2 amino acid transporters
ORNT1, citrin
*KNOW: location of the enzymes/transporters -- mitochondria or cytoplasm
Urea Cycle Disorders
cause:
any genetic defects in the enzymes or transporters
leads to high levels of ammonia which increases glutamate and depletes ATP
can cause brain damage
build-up of ammonia, which is a toxin
enzymes at the top of the diagram are more lethal if non-functional compared to those at the bottom
symptoms:
lethargy, anorexia, seizures, coma, etc.
1st signs: drowsiness and failure to feed
hyperammonemia
loss of appetite, lethargy, vomiting, behavioral abnormalities
treatment:
low-protein diet
medications to remove ammonia from the blood
sodium benzoate
sodium phenylacetate
supplements
arginine and citrulline
what is the outcome of the urea cycle testing?
ARG1
increases arginine
ORNT1
increase in ornithine, and decrease in citrulline
ASS1
increase in citrulline, and decrease in arginine
what is an organic acid?
organic compounds with acidic properties -- many are carboxylic acids
can be found as physiological intermediates in other metabolic pathways
ex: sulfonic acids
produced from the breakdown of amino acids
describe organic acidemias
group of disorders characterized by increased excretion of non-amino acids in the urine
this means the body is able to remove the nitrogen from the amino acid, but cannot process the carbon skeleton to form the organic acid
organic acidemias disturb mitochondrial energy metabolism
this means that they have a large range of clinical and biochemical ways to manifest in the TCA cycle
classic organic acidemias
systemic effects
cerebral organic acidemias
brain effects
how are organic acidemias detected during newborn screening?
if there is a metabolic defect in the TCA cycle, this leads to acyl-CoA build-up
the accumulated acyl-CoA compounds are esterified with free carnitine and leads to an increase in acylcarnitines
the acylcarnitines can be detected during newborn screening
what are the clinical manifestations of organic acidemias?
energy deficiency
targets brain, liver, kidney, pancreas, retina, and other organs
describe clinical signs of the 3 types of enzyme activity for organic acidemias.
complete absence of enzyme activity
first few weeks of life
severe, acute-life threatening attacks
provoked by a stress such as an infection, trauma, etc.
an attack is . . . metabolic acidosis, hyperammonemia, and hypoglycemia
caused by: accumulation of neurotoxic organic acids and their acyl-CoA esters
residual enzyme activity
symptom-free between attacks
provoked typically by an infection
neurological signs, movement disorder, epilepsy, feeding difficulties, and intellectual disability
insidious, late-onset forms
progressive neurological and GI symptoms
no apparent crisis during disease progression