Genetic Diseases
1/23
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
24 Terms
polygenic inheritance
Many diseases have a genetic component, albeit without a specific identifiable gene mutation (Ex: coronary artery disease, hypertension, gout, and diabetes mellitus)
- Multiple genes and environment determine one trait
These conditions have a multifactorial inheritance pattern. Most likely follow polygenic inheritance
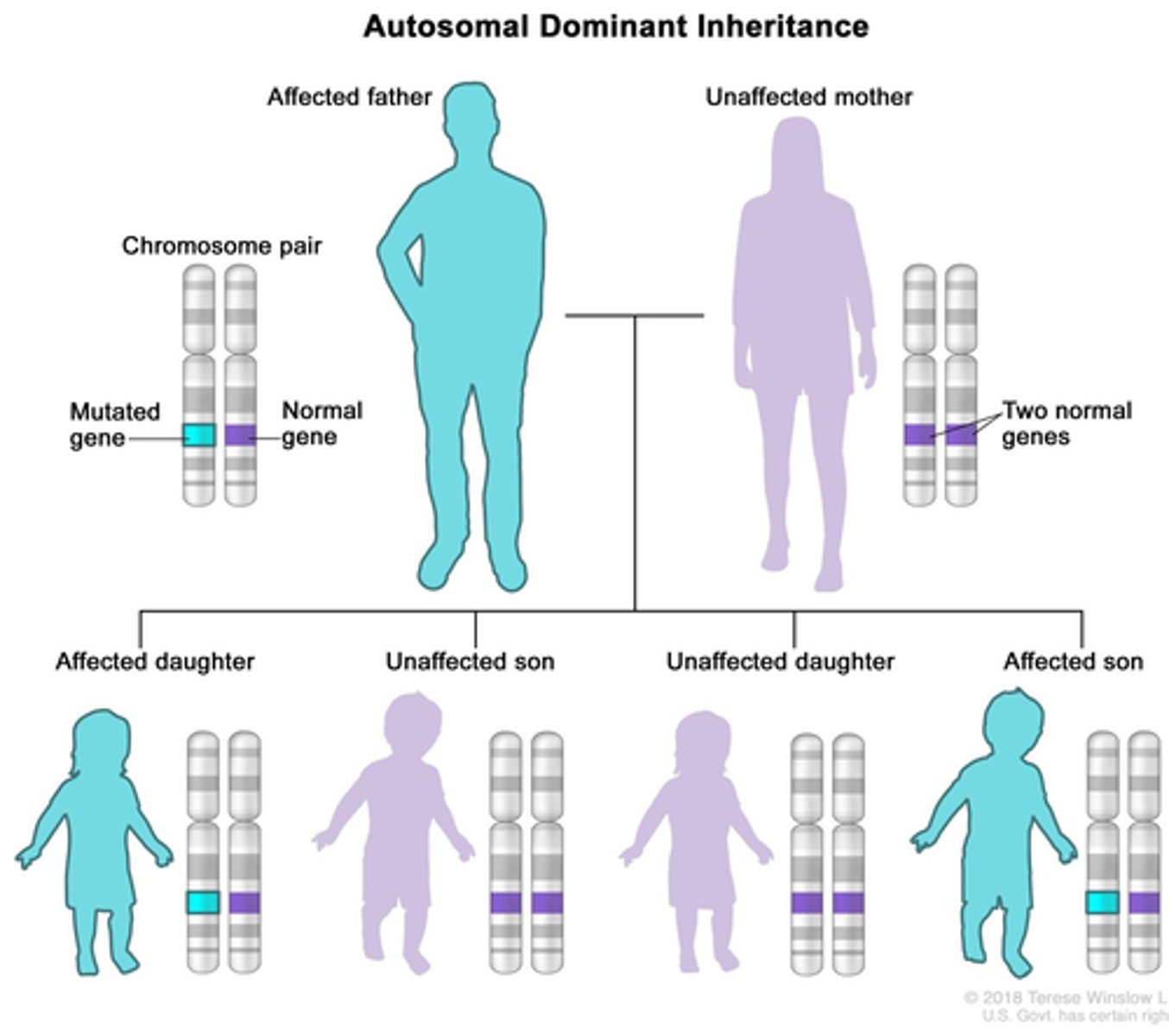
autosomal dominant disorders
Autosomal disorders have reduced penetrance and visible expressivity. They usually do not encode enzymes (a loss of 50% of an enzyme's activity can be compensated for by activity if the enzyme encoded by the normal allele)
- Only one allele needed to show trait.
- Close to 50%
familial hypercholesterolemia
- Autosomal dominant
- Mutation: Low-density lipoprotein receptor gene (LDLR); there are more than 900 known mutations
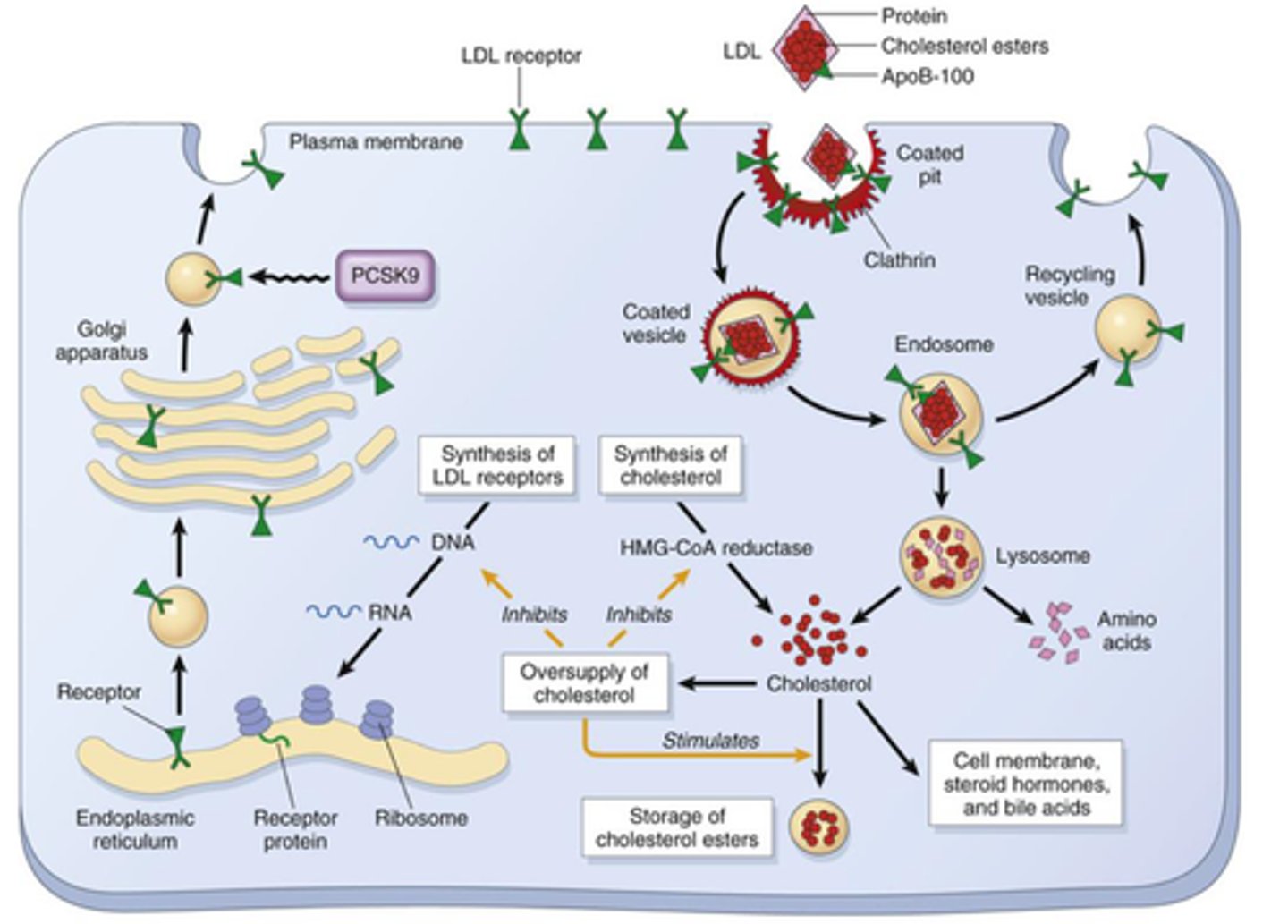
familial hypercholesterolemia mechanism
LDL receptor recognizes apolipoprotein B100 (ApoB100) or apolipoprotein E (ApoE); therefore, a mutation of the receptor results in impaired uptake of cholesterol into cells
- Liver can't take in these proteins
Phenylketonuria (PKU)
Mutation: gene for phenylalanine hydroxylase (PAH)
Phenylketonuria (PKU) mechanism
Hyperphenylalaninemia (inability to convert phenylalanine to tyrosine)
Phenylketonuria (PKU) manifestations
Clinical features of untreated PKU may include severe intellectual disabilities, seizures, and decreased pigmentation of skin (due to not producing tyrosine), which can be avoided by restricting the intake of phenylalanine in the diet
- Female patients with PKU who discontinue dietary treatment can give birth to children with malformations and neurologic impairment resulting from transplacental passage of phenylalanine metabolites
Gaucher disease
Mutation: gene for B-glucocerebrosidase (GBA)
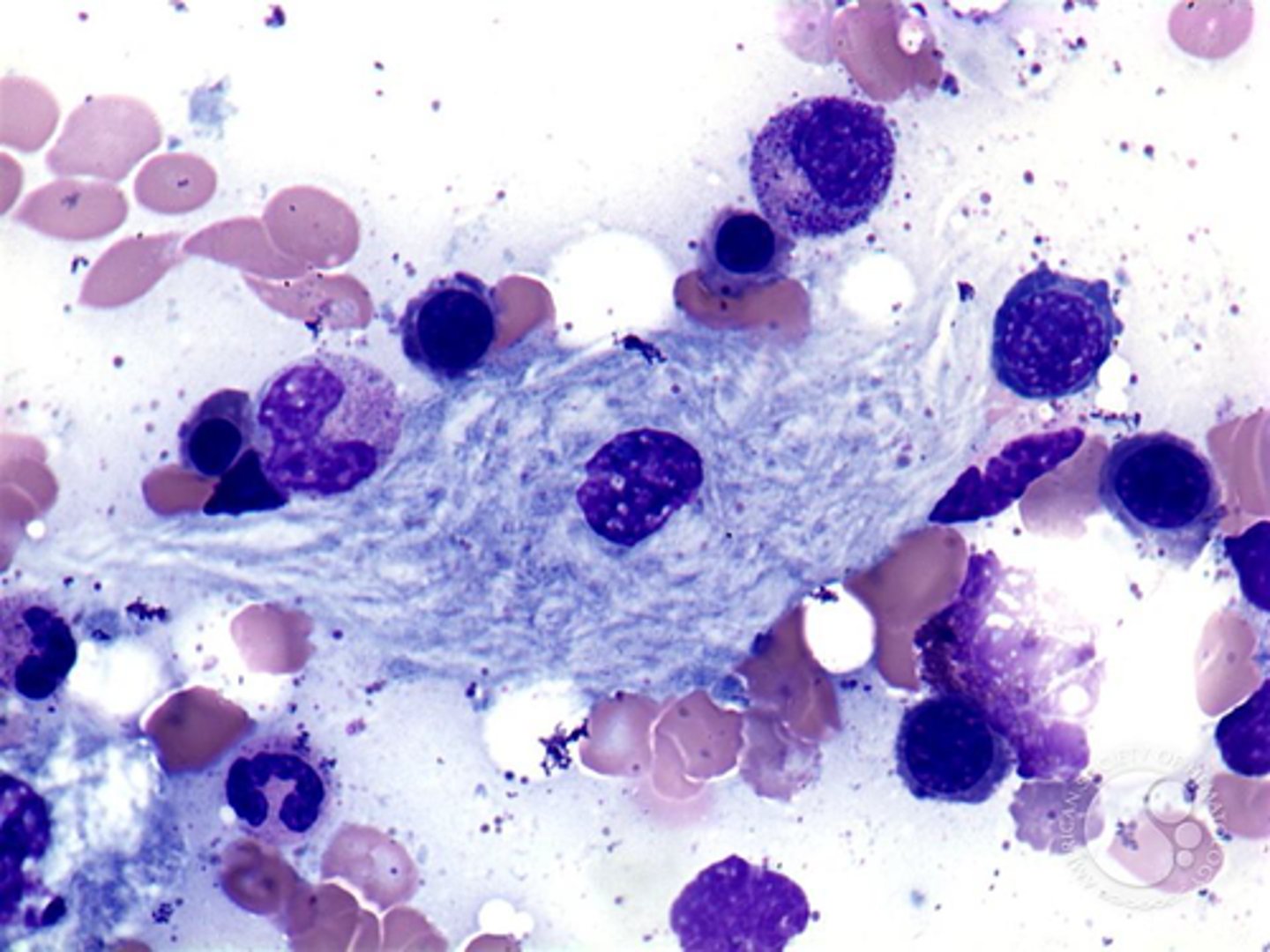
Gaucher disease mechanism
Accumulation of glucocerebroside in organs with high concentration of phagocytic cells (ex: spleen, liver, lymph nodes)
Type I (3 total): Chronic non-neuropathic form represent 99% of cases of Gaucher disease. Presents later in childhood or early adulthood
- Puffy macrophages
Gaucher disease manifestations
Affected phagocytes become enlarged and accumulate in the liver, spleen and bone marrow causing hepatosplenomegaly (lysosomes can't function) and bone erosion
Tay Sachs disease
Mutation: Deficiency of one of the genes encoding for lysosomal enzymes that catabolize gangliosides (glycolipid), hexosaminidase (HEXA)
- Unable to break down glycolipids and hexolipids
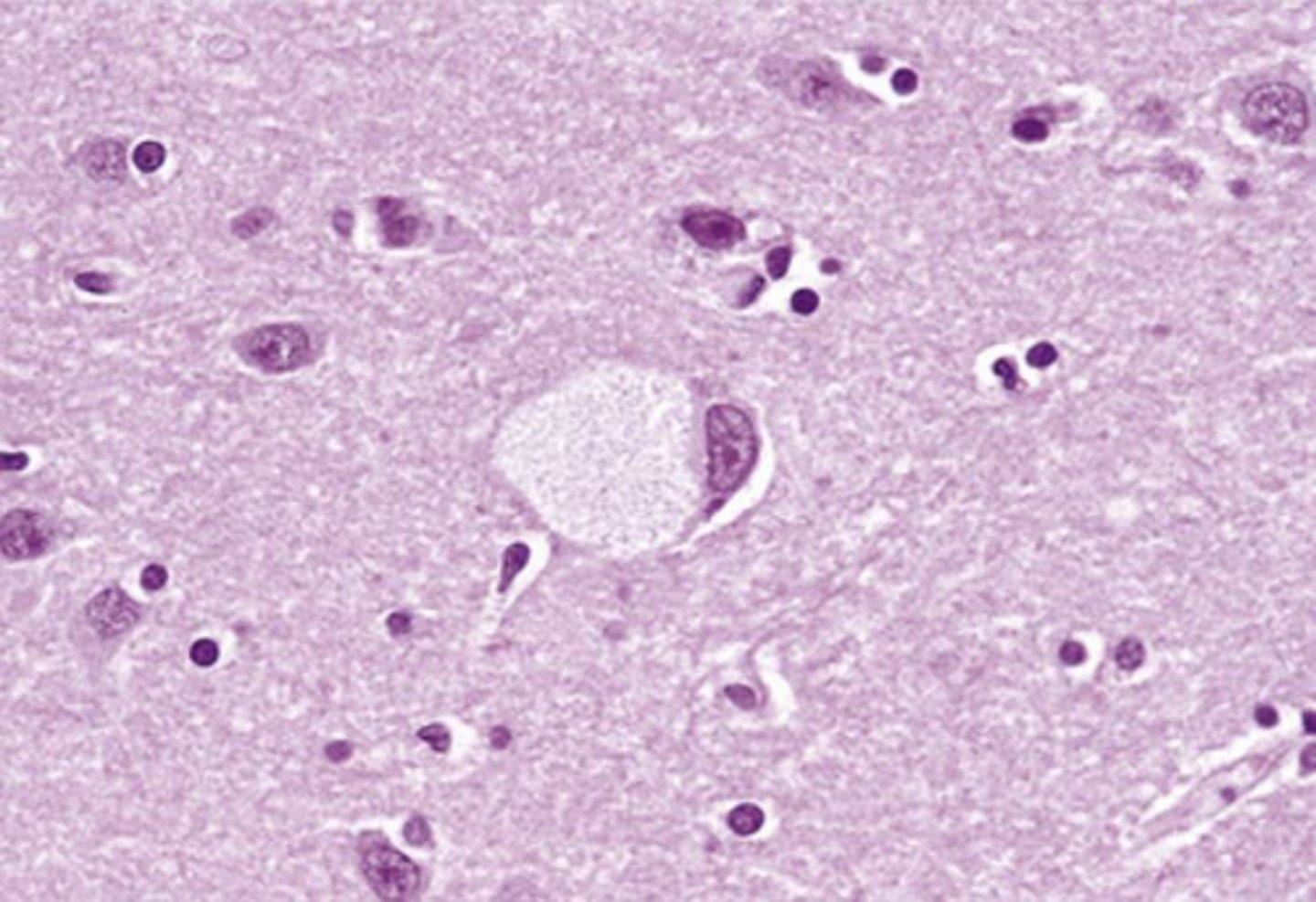
Tay Sachs disease manifestations
Ballooned neurons leading to progressive neurological deterioration (motor and mental) and involvement of ganglion cells in the retina causing a cherry-red spot. Signs and symptoms develop by 6 months; death usually occurs by the age of 2 to 3 years
Duchenne muscular dystrophy mechanism
In skeletal and cardiac muscles, dystrophin is part of a protein complex that strengthens muscle fibers and protects them from injury as muscles contract and relax. In dystrophin deficiency, the cell membrane of the muscle fiber is damaged when the muscle contracts
Duchenne muscular dystrophy signs and symptoms
Weakness of pelvis first, with delayed ability to walk; pseudohypertrophy (calf muscles enlarge due to fat replacement), and muscle atrophy
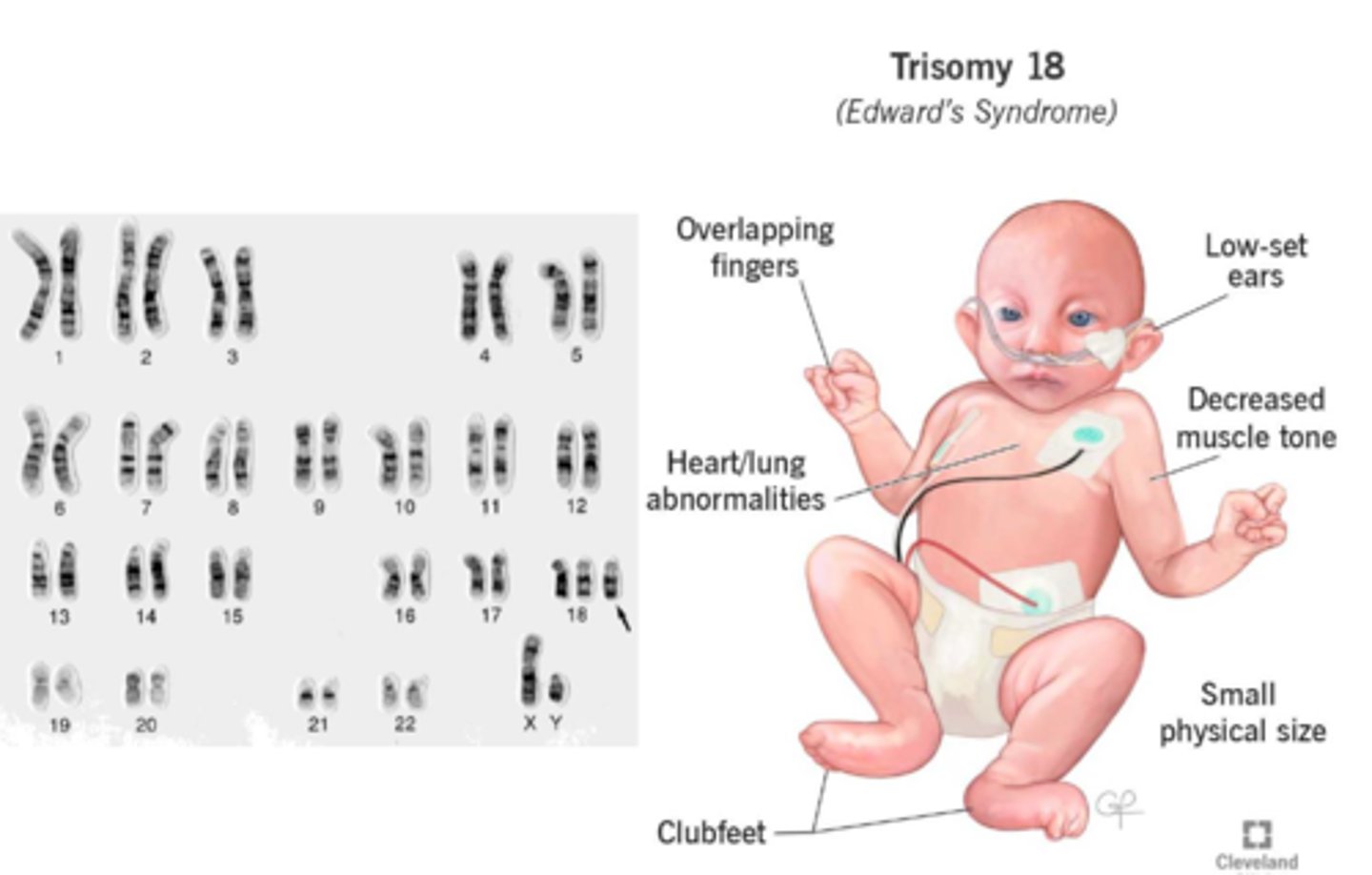
trisomy 18 (Edwards syndrome)
Incidence: 1 in 8,000 live births
Manifestations: Prominent occiput, low-set ears, small jaw, intellectual disabilities, cerebellar and brainstem abnormalities, congenital heart disease, rocker bottom feet
Almost all patients die of apnea in the first year of life
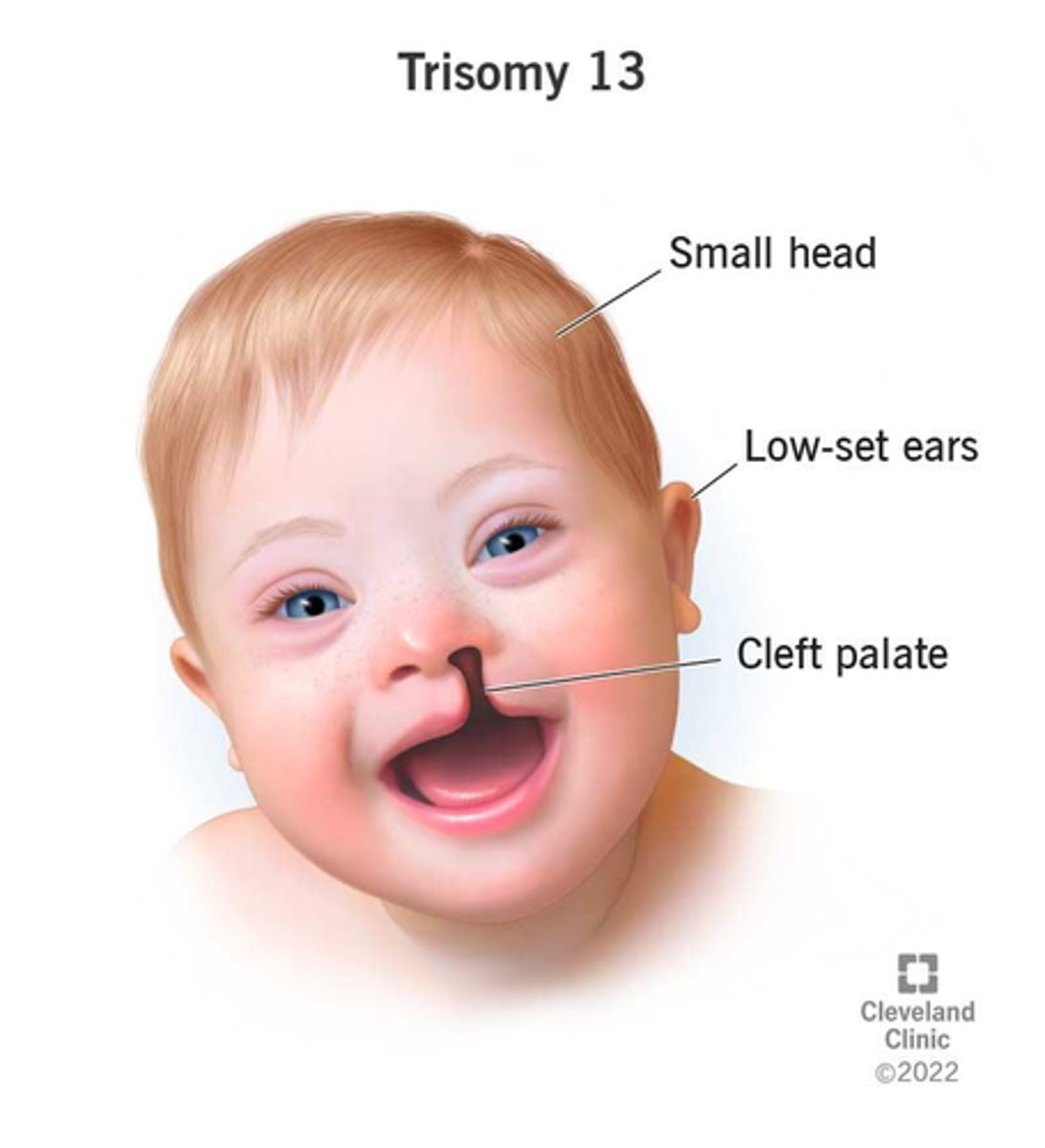
trisomy 13 (Patau syndrome)
Incidence: 1 in 15,000 live births
Manifestations: Microphthalmia, holoprosencephaly, intellectual disability, polydactyly, cleft lip and palate, congenital heart disease, rocker bottom feet
Almost all patients die in the first year of life
endocrine system events for Klinefelter syndrome

Fragile X syndrome notes
- Normal individuals have about 30 repeats; affected individuals have 230+ repeats
- Individuals with 50 to 230 repeats are referred to as premutations
- Premutations can expands the number of repeats during oogenesis, but not during spermatogenesis. Therefore, fathers with premutations cannot pass the disease to sons, but repeats will amplify in his daughters and affect his grandsons and granddaughters. Understanding of this potential by parents produces the anticipation effect
- Fragile X is the most common inherited genetic cause of intellectual disability
genomic imprinting
Epigenetic process whereby certain homologous genes are differentially "inactivated" during paternal and maternal gametogenesis
Imprinting occurs in ova or sperm and is then transmitted to all somatic cells derived from the zygote
- Functionally delete a gene
Prader-Willi syndrome
Deletion of paternal-derived 15q12
- Must receive paternal-derived gene at 15q12 to not develop
Angelman syndrome
Deletion of maternal-derived 15q12
- Must receive maternal-derived gene at 15q12 to not develop
Female pseudohermaphrodite mechanism
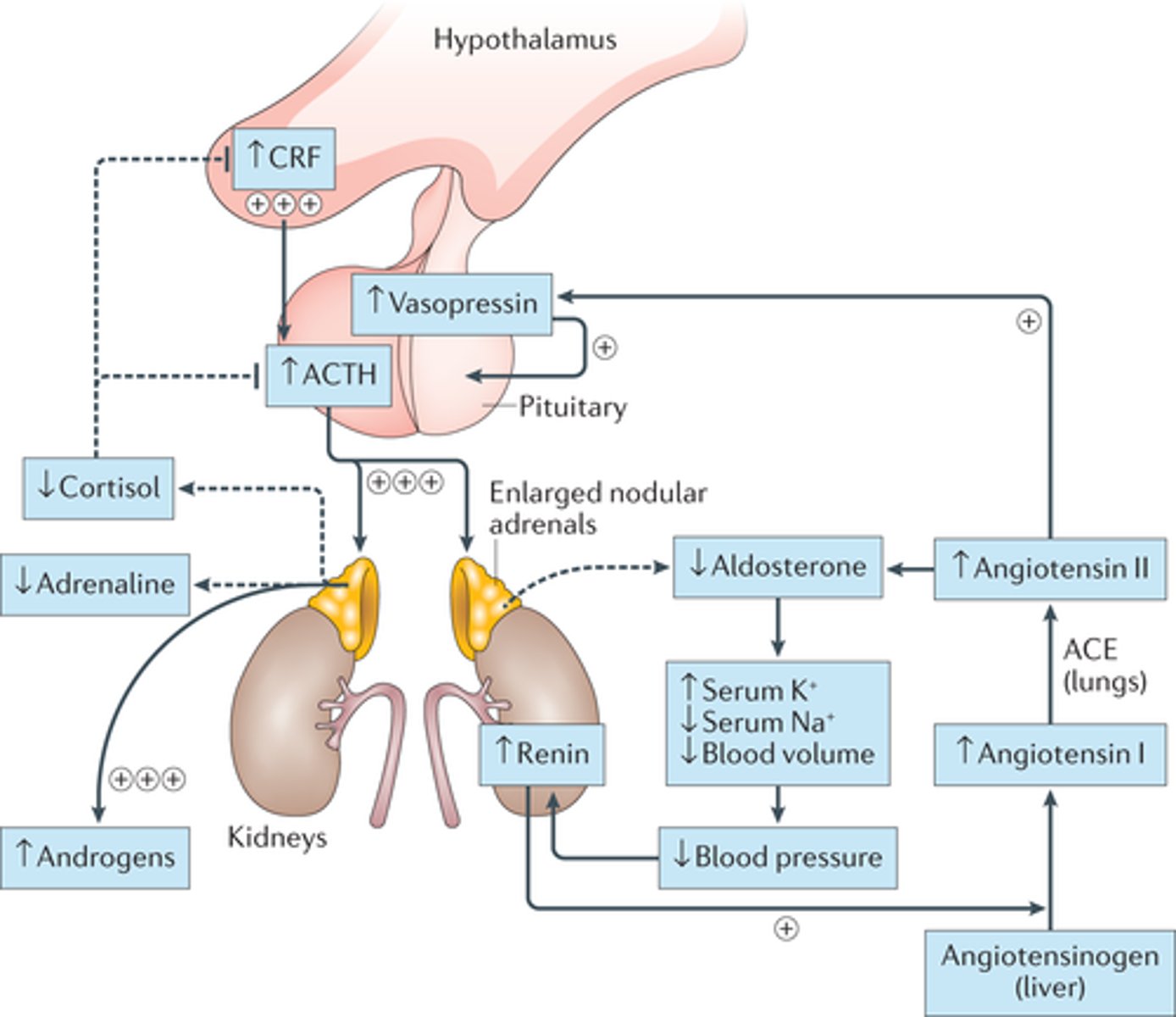
Excessive androgens during gestation; common cause is congenital adrenal hyperplasia (CAH)
- Born with it, hyperplasia in adrenal cortex (Aldosterone - salt, Cortisol - sugar, Androgens - Sex, DAGA)
Androgen insensitivity syndrome
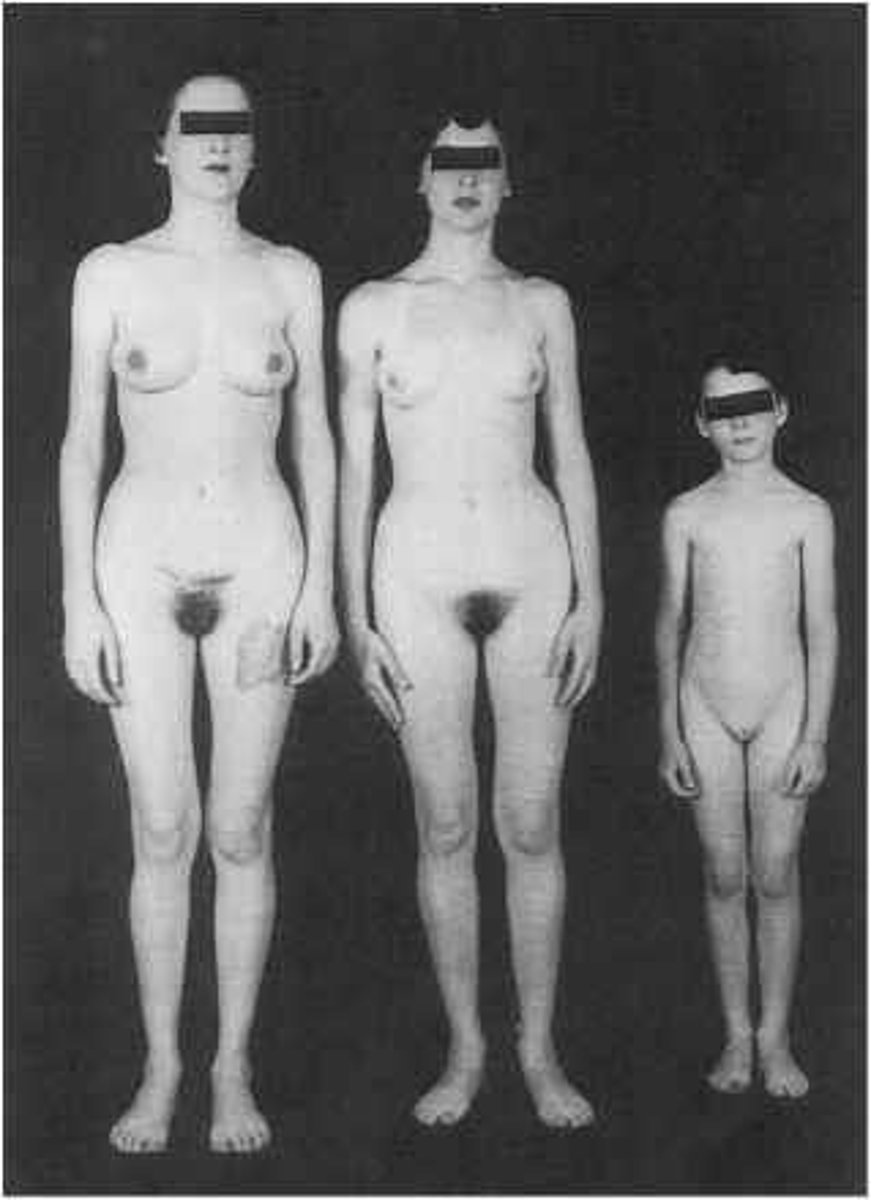
Individuals are genetically male (XY), but are resistant to androgens during embryonic development. The external genitalia and secondary sex characteristics are feminine, but there are no ovaries, uterus, or vagina.
Androgen insensitivity syndrome mechanism
Due to a mutation in the androgen receptor (AR) gene on Xq11-12. Laboratory findings include increased levels of testosterone, estrogen, and luteinizing hormone (LH).
- Mechanism: 5a-reductase is normally required for conversion of testosterone to dihydrotestosterone (DHT)