Antibacterial Agents
1/78
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
79 Terms
antibiotics: few targets but plenty of resistance
- bacteria doubling time = 30 minutes (single cell organisms, asexual cell division)
- single cell organisms means that they do not have a lot of protections (only from membrane - peptidoglycan layer, periplasmic space is much different than ours)
- gram positive = can retail gram stain (membrane is more accessible, similar to human cell)
- gram negative = tough penetrating outer membrane (do not retain dye, harder to treat) - polar, highly charged external membrane
- chemically modified natural structures (overcome resistance, expands targets)
- we want selective agents (not against our cells)
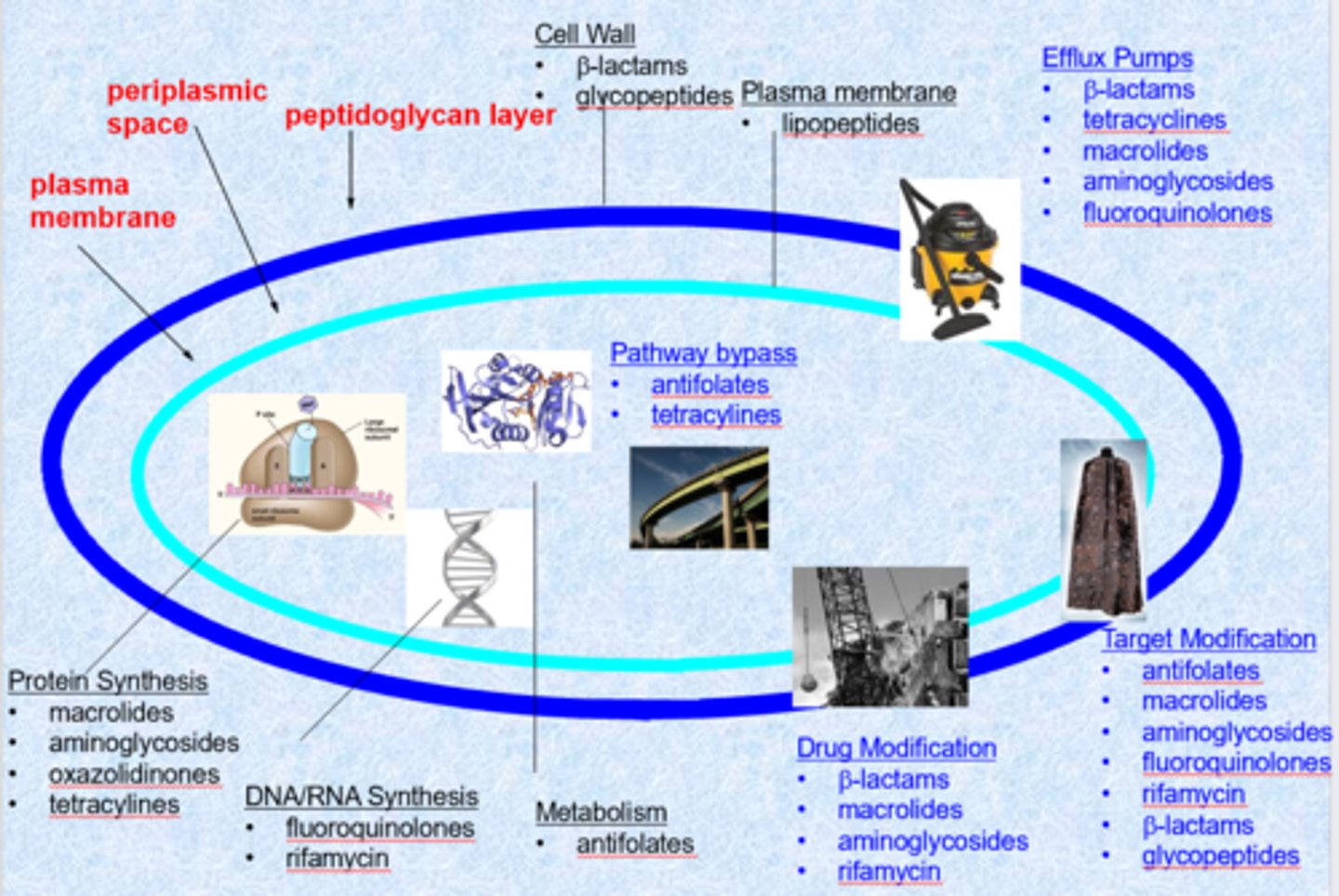
- targets = cell wall, plasma membrane, protein synthesis, DNA/RNA synthesis, metabolism
- cell wall is the biggest target (ex. penicillin)
- protein synthesis is also a large target - ribosome is very different than ours and the drugs are very selective
- changes to the membrane or efflux pumps cause resistance
importance of the bacterial cell wall on antibiotics
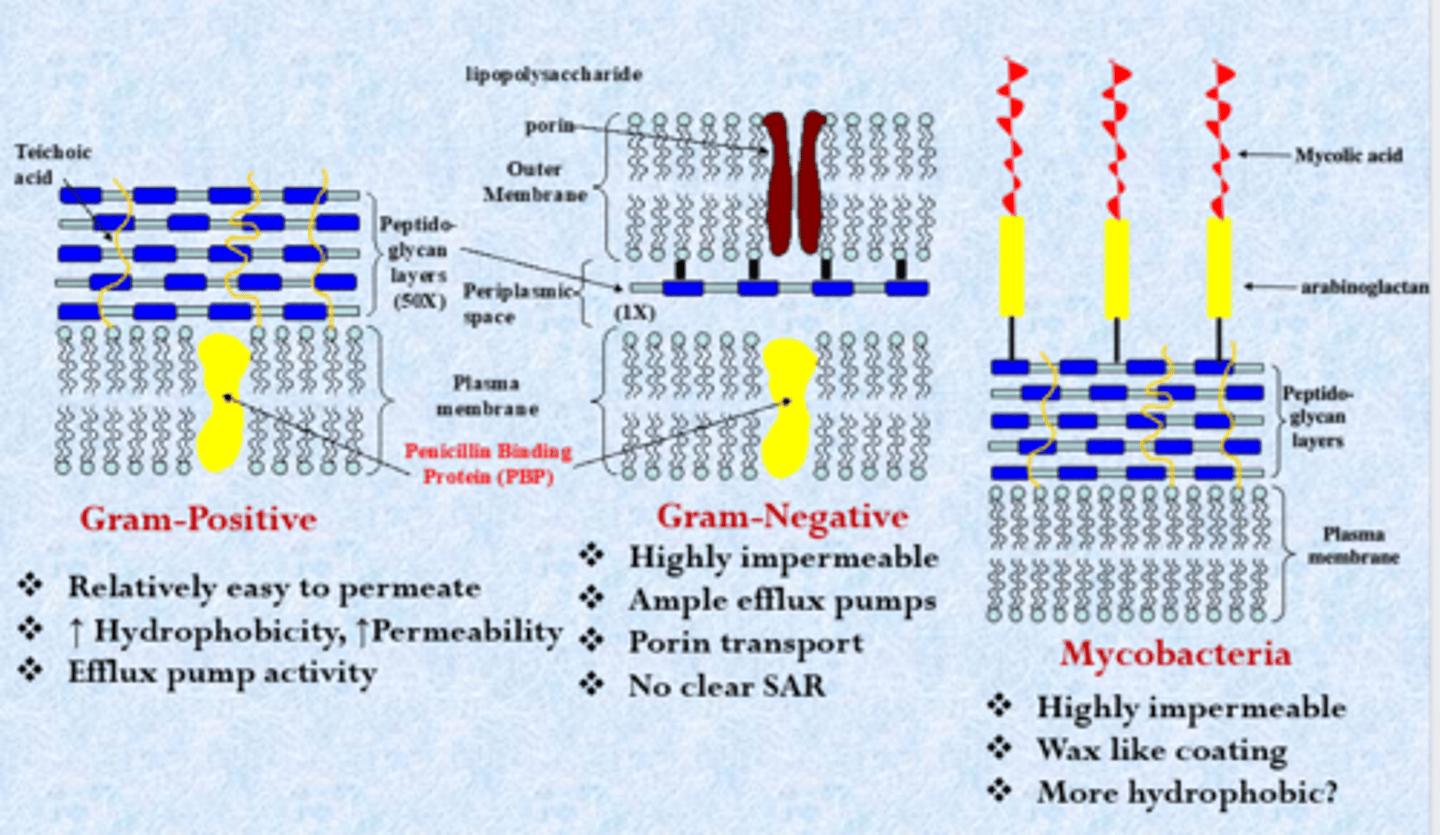
gram-positive
- most closely related to us - classic lipid bilayer but with harder external layer called peptidoglycans (can block some uptake of compounds)
- relatively easy to permeate
- increased hydrophobicity, increased permeability (similar to humans)
- efflux pump activity
gram-negative
- thinner peptidoglycan layer, lipopolysaccharides highly negative charge (tough exterior, hydrophobic molecules bounce off this layer - can live in tough evolutionary environments and harder to kill)
- polar compounds on drug molecule (could not get in gram-positive)
- highly impermeable
- ample efflux pumps
- porin transport
- no clear SAR
mycobacteria
- mycolic acid
- highly impermeable (just as bad as gram-negatives)
- wax like coating
- more hydrophobic?
- classified as gram-positive bacteria
agents targeting the cell wall
- b-lactam family (ex. penicillin derivatives) - largest, oldest, most reliant on these derivatives
- glycopeptides (ex. vancomycin)
- lipopeptides (ex. daptomycin)
- fosfomycin
penicillin - a b-lactam
- isolated from the Penicillium fungi
- Fleming reports seeing antibiotic effect from a mold growing on a Petri plate covered with Staphylococcus (Sept. 28, 1928)
- March, 1942 – Merck treated the first patient
- cantaloupe from Peoria, IL best source in 1943
- US produced 2.3 million doses in time for Normandy invasion
- structure determined by Dorothy Crawfoot Hodgkin in 1945
- subject to rapid clearance
SAR
- 4 membered amid is beta-lactam and is most important
- beta = 4 carbon ring (molecule is really strained)
- side chain gets changed
- multiple families within the b-lactams but what we call them changes based on ring to the right
- thiazolidine ring = 5 membered ring in penicillin - sulfur and nitrogen
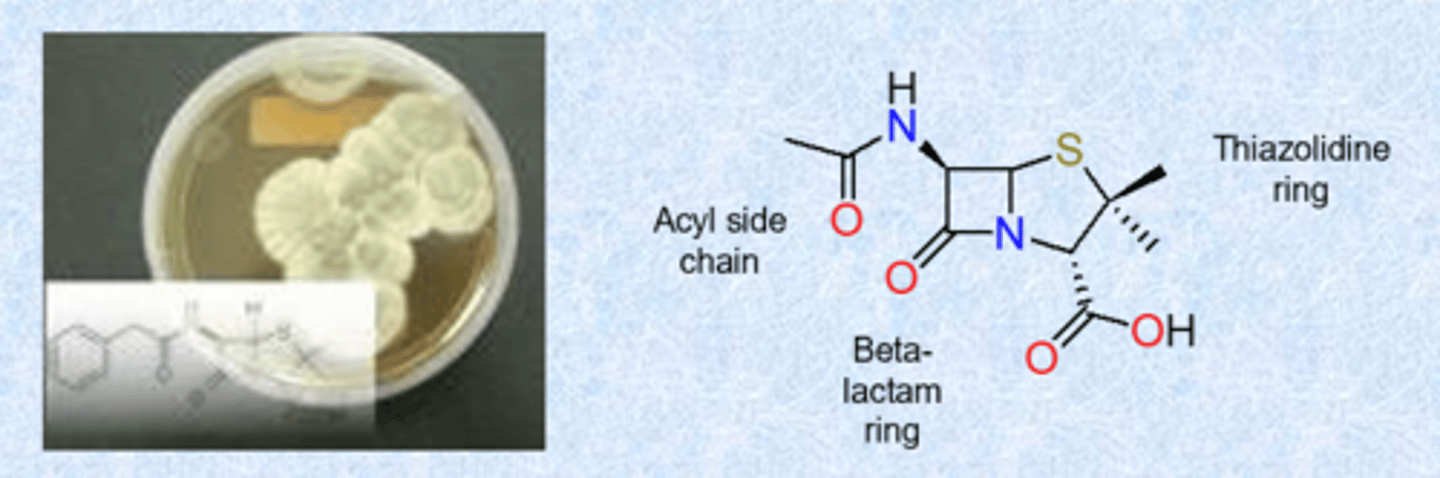
What tells you that it is a penicillin?
b-lactam and thiazolidine
medicinal chemistry of penicillin
- used to treat infections caused by Gram-positive bacteria (S. aureus, S. pneumoniae, S. pyogenes, E. faecalis)
- mechanism of action:
inhibit formation of peptidoglycan crosslinks in bacterial cell wall
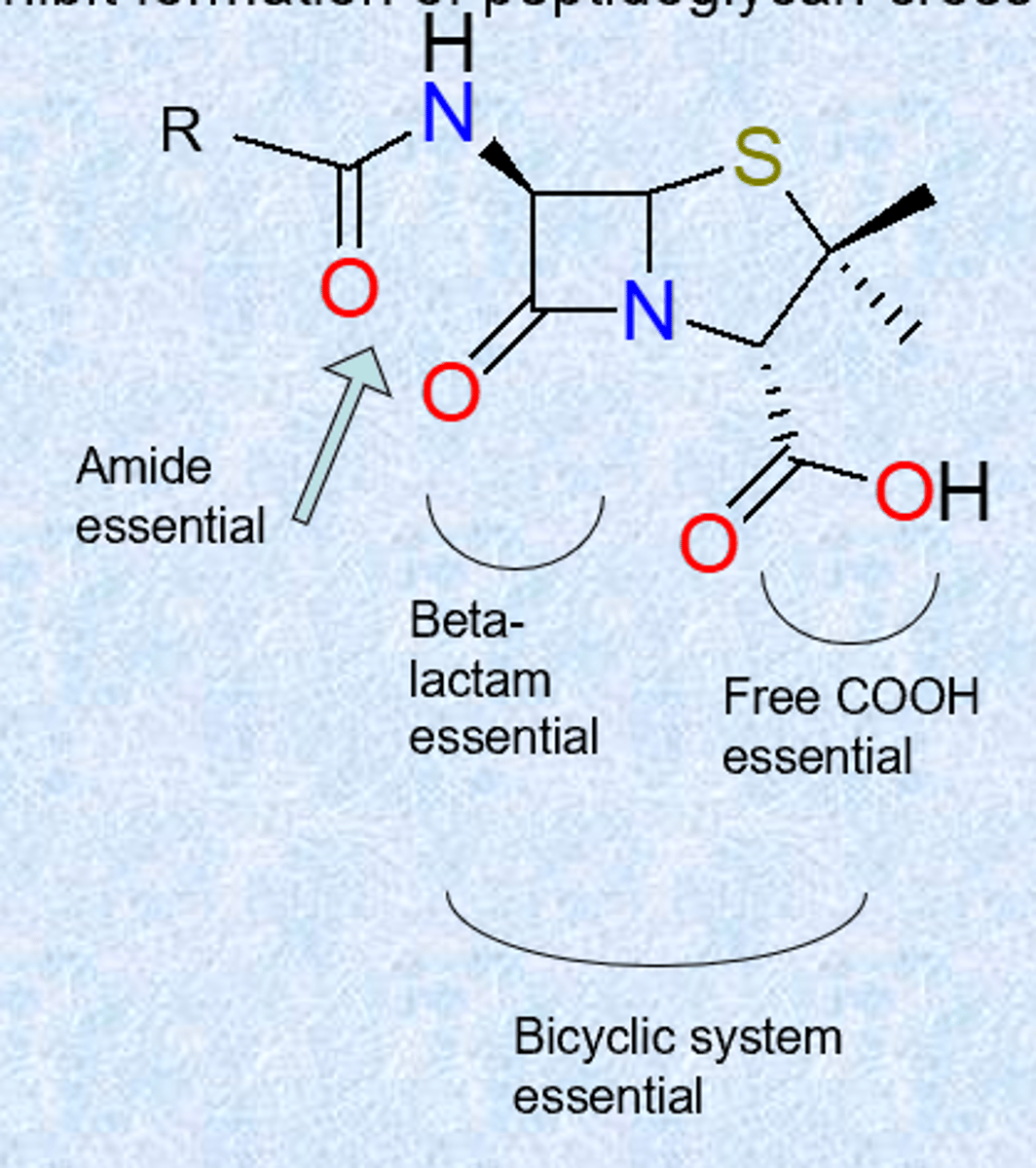
medicinal chemistry of penicillin - SAR
- acyl chain: only location where variation can occur (R=benzyl: Penicillin G)
- strained beta-lactam - strain, resonance of amid is broken (far more reactive, can be attacked by many things)
- free COOH ionized – allows administration as Na+ or K+ salt
- acylamino chain essential
- sulfur is usual but not essential
medicinal chemistry of penicillin - acid sensitivity
- ring strain
- highly reactive beta-lactam carbonyl group
- influence of acyl side chain – incorporate electron-withdrawing groups to reduce sensitivity
medicinal chemistry of penicillin - biosynthesis
- biosynthesis: derives from cysteine and valine
- acyl side chain varies depending on components of fermentation medium (most analoged)
- today: fermented from two strains of penicillium
- dipeptide amino acid mimics D amino acid configuration (most are L configuration) - gives the compound low toxicity because we do not have a lot of things that recognize D amino acids (safe)
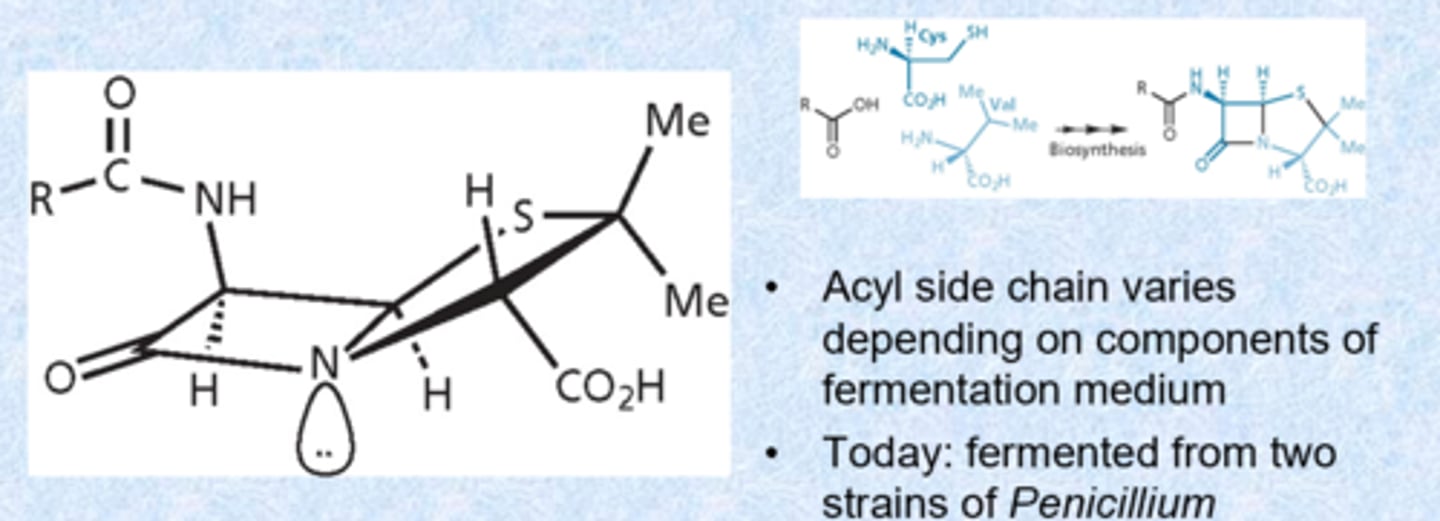
penicillin blocks the transpeptidase necessary for crosslinking
- process is specific to the bacterial (not occurring in patients)
- bacteria has D-alanine branching off of peptidoglycan
- want to cross-link them to make them strong via enzyme transpeptidase (penicillin binding proteins - PBPs is the molecular target)
- enzyme looks for 2 D amino acids - penicillin is a D amino acid
- enzyme binds via covalent bond temporarily
- glycine that branches forms bond with D-alanine and turns over the enzyme - stronger membrane (internal pressure swells bacteria if not and it can explode)
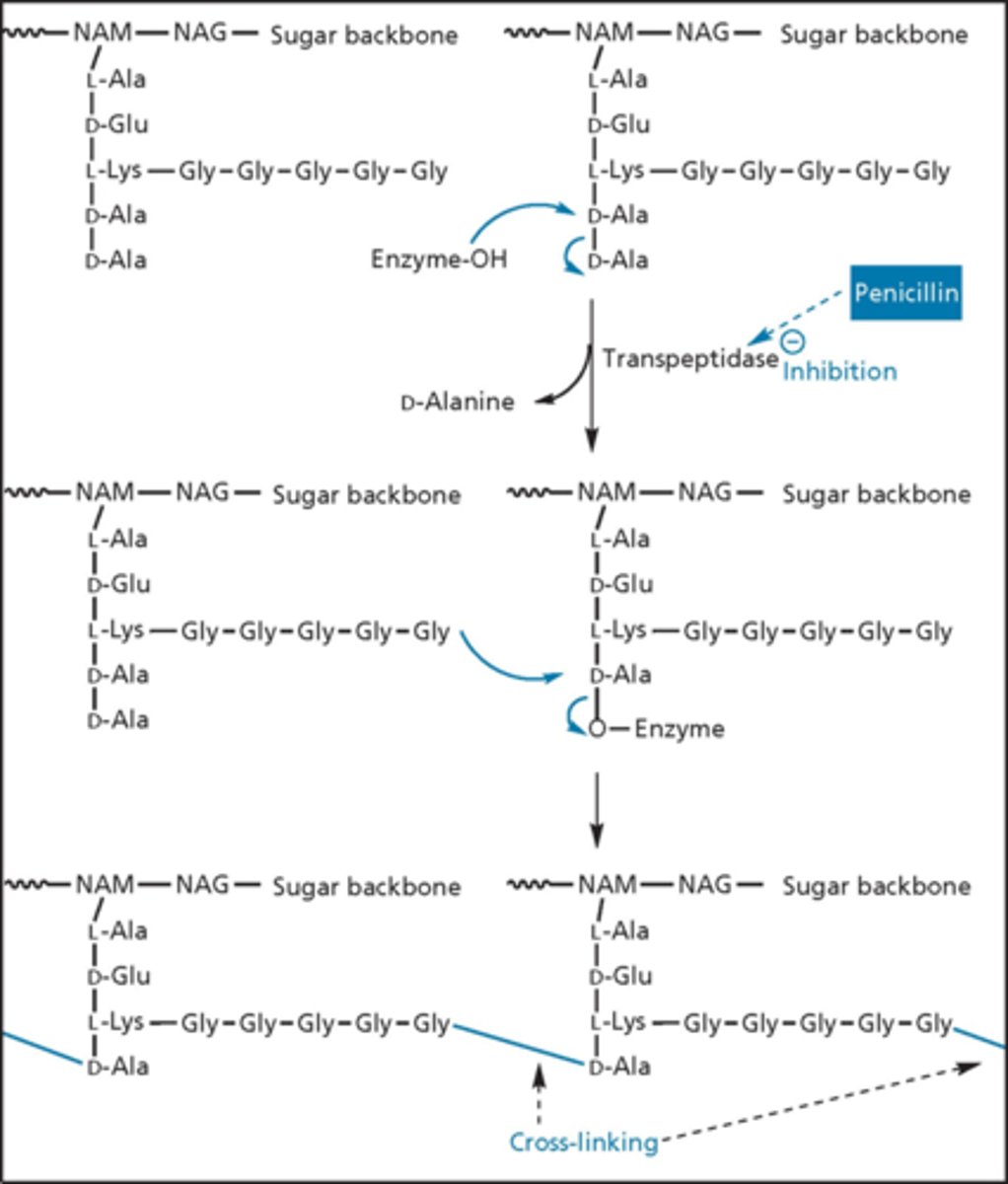
transpeptidase cross-linking
- penicillin gets into site and mimics D-ala D-ala unit
- ring open penicillin cannot do the next step
- eventually falls off (quasi-irreversible inhibitors)
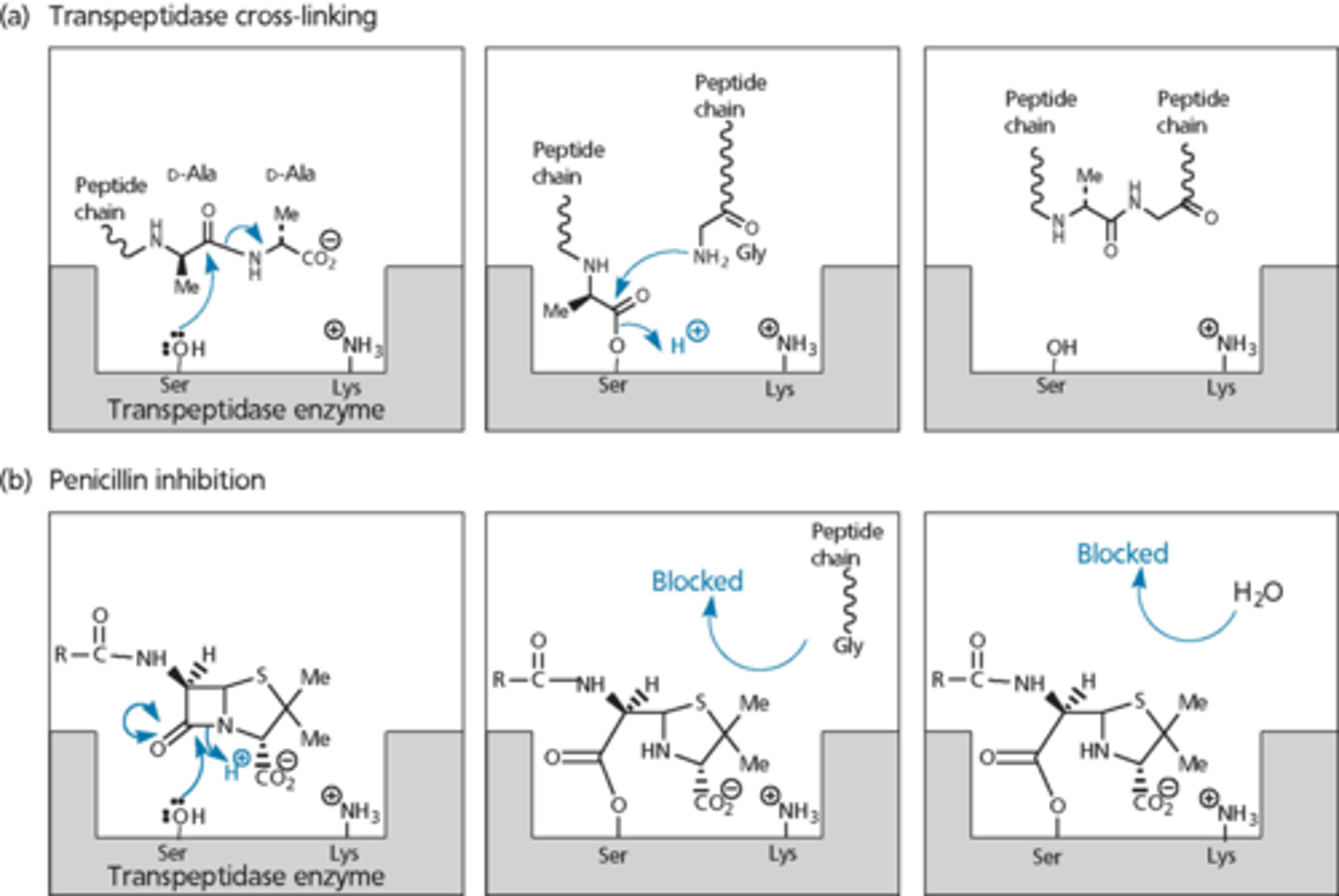
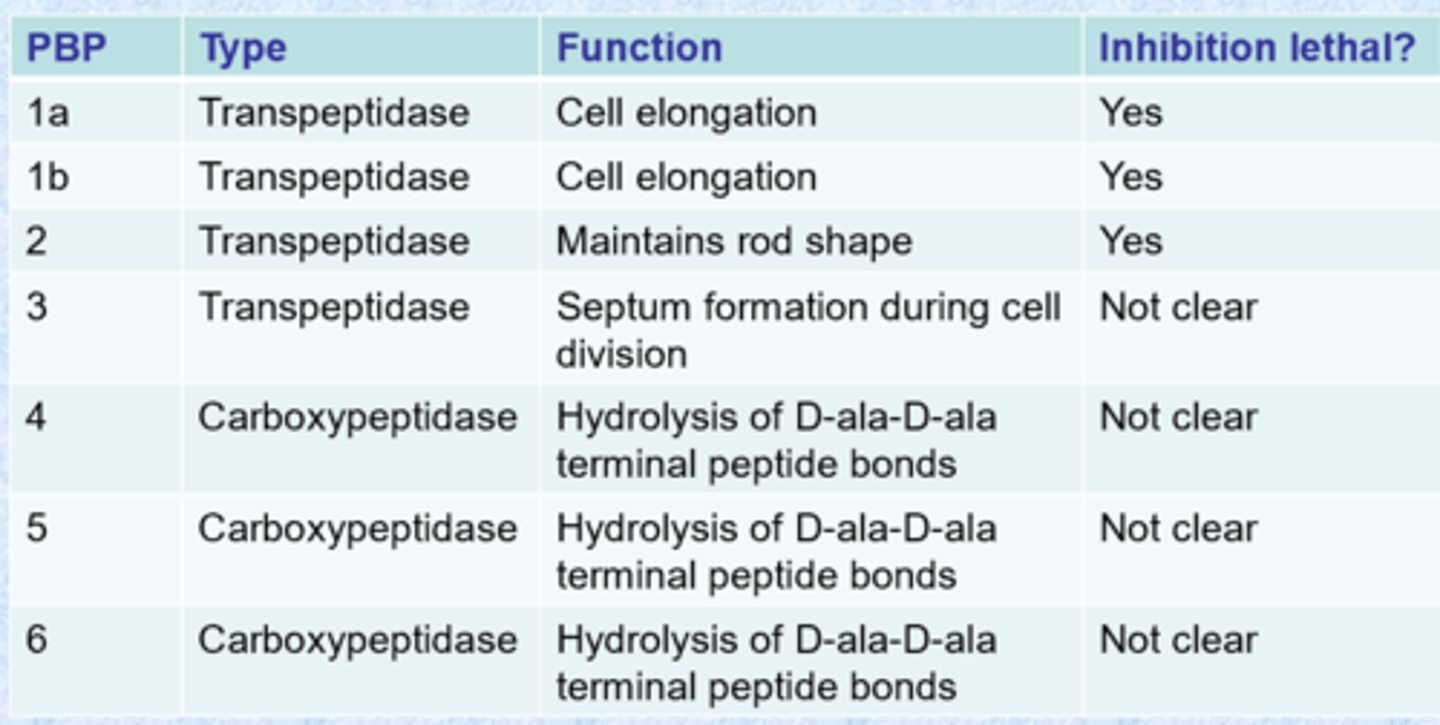
there are many PBPs
- tend to have highly conserved active sites (nucleophilic serine)
- chemists can inhibit all of them
- PBPs vary from one bacteria to another
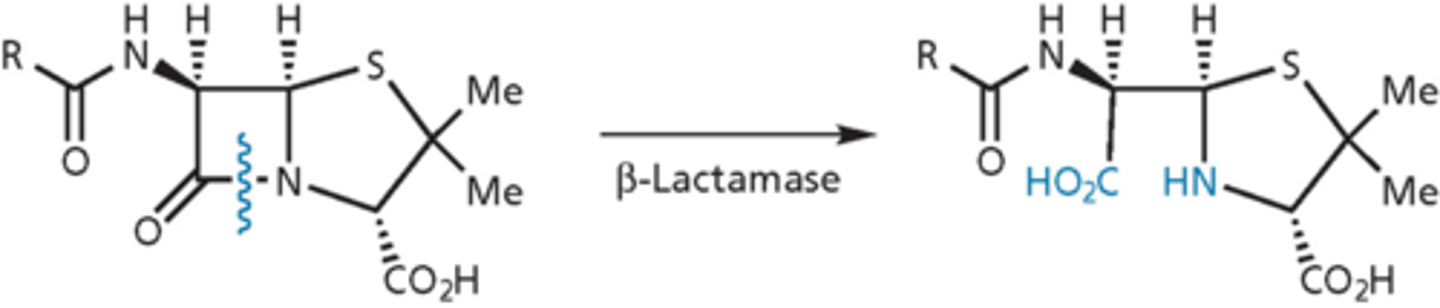
beta-lactamase enzyme confer resistance
- mutated from transpeptidases - do not have transpeptidase activity, no cross-linking
- very efficient: hydrolyze 1000 molecules per second
- contain an active site serine that forms an ester link that is then cleaved to release product
- turnover number is very high, enzyme protects the bug
- antibacterial activity when b-lactam ring is open is gone
- gram-negative (more deadly because more concentrated) and gram-positive
improving penicillins
- reduce acid-catalyzed degradation in stomach
- incorporate electron withdrawing groups to reduce nucleophilicity of side chain carbonyl oxygen, reducing its participation in the ring opening reaction
- acts as a buffer and prevents from acid catalyzation and opening (decreases electron density)
- medications that are oral
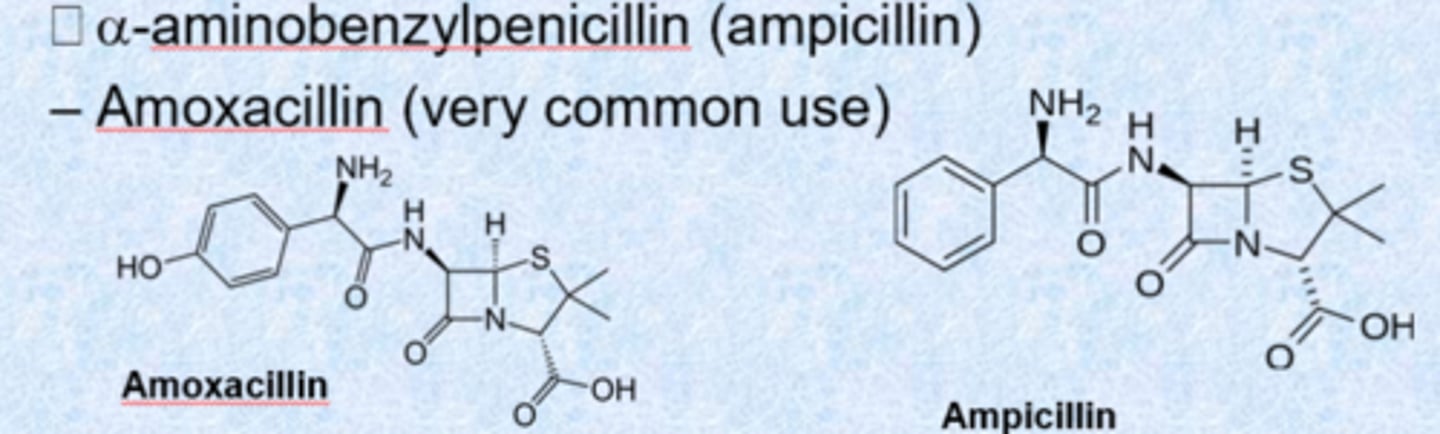
– a-aminobenzylpenicillin (ampicillin)
– amoxacillin (very common use)
- Pen-G (iv only)
- do not memorize which is which, just recognize that they are penicillins
gram-positive and gram-negative effects - penicillins
- gram-positive bacteria generally sensitive
- gram-positive just secrete b-lactamases so they are not as concentrated
- gram-negative bacteria: some resistant, some sensitive (hard to get a drug in there)
– depends on structure of porin and structure of penicillin
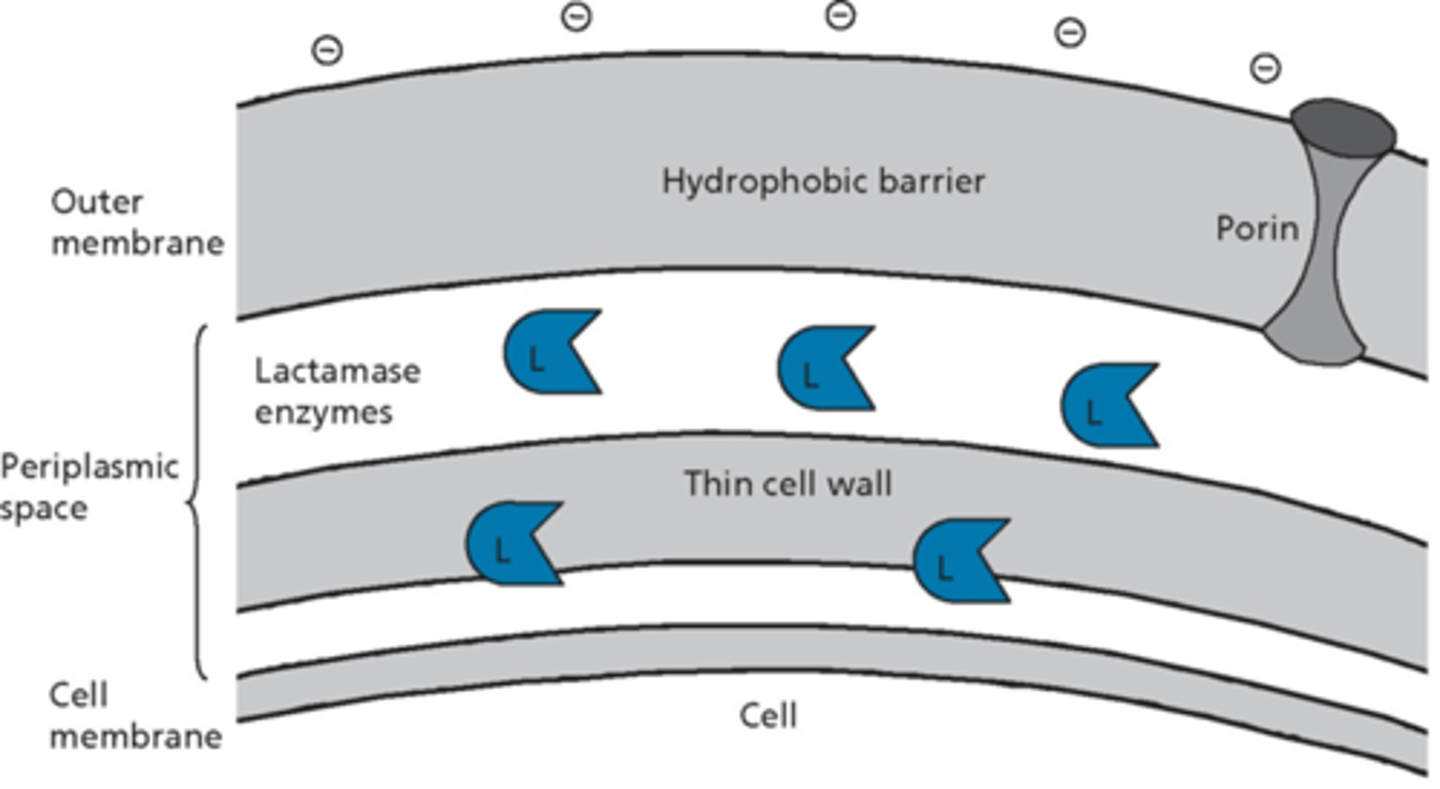
gram-positive and gram-negative effects - b-lactamase
- some gram-positive bacteria release b-lactamases into surrounding environment
- most gram-negative bacteria produce b-lactamases, usually trapped in periplasmic space
- different b-lactamases vary in substrate specificity
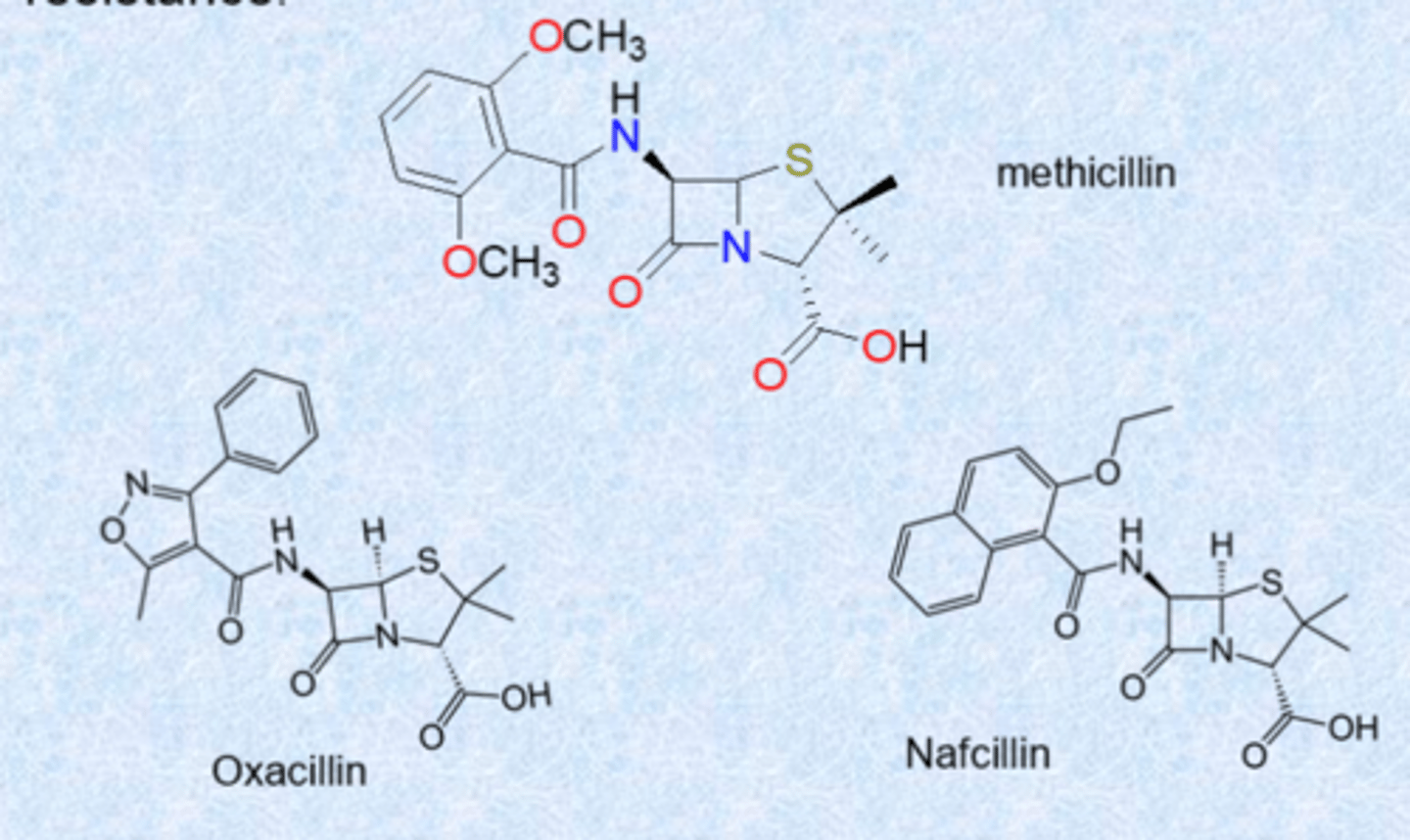
second generation of analogs of penicillin overcome resistance
- analogs present a steric shield to block access to beta-lactamases on amid (large bulky groups slow down beta-lactamases)
- narrow spectrum (gram positive)
- IV delivery
- today: MRSA (methicillin-resistant S. aureus)
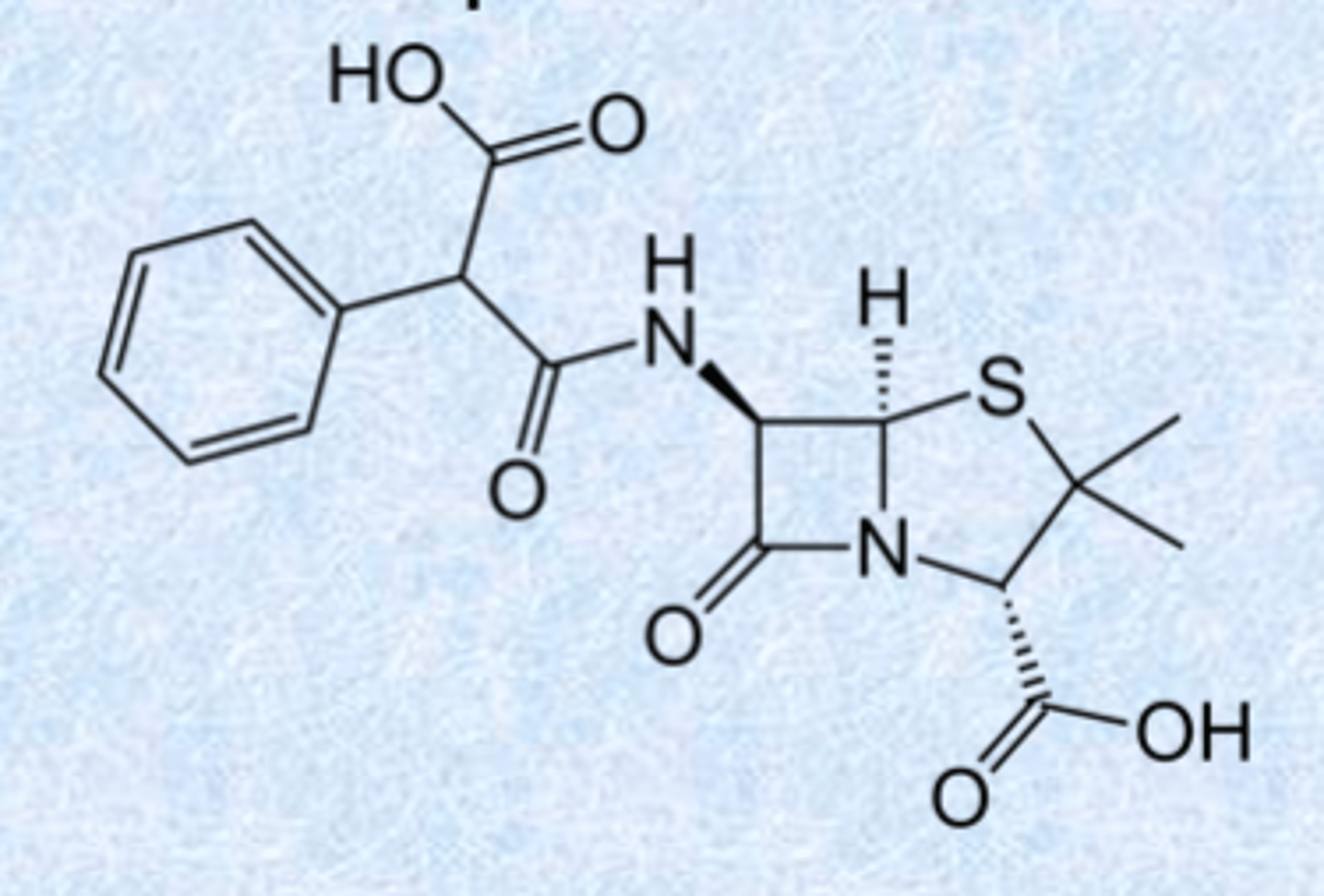
carboxypenicillins - broad spectrum
carbenicillin: first example
– COOH gives broad spectrum activity
– increased negative charge (2 charge)
– gram negative active (ilucing P. aeruginosa) but little gram positive
uriedopenicillins - broad spectrum
- derivatives of ampicillin
- more polar side chain enhances gram-negative penetration
- very susceptible to b-lactamases (especially in gram-positive pathogens)
- given iv or im
- good pseudomonial activity
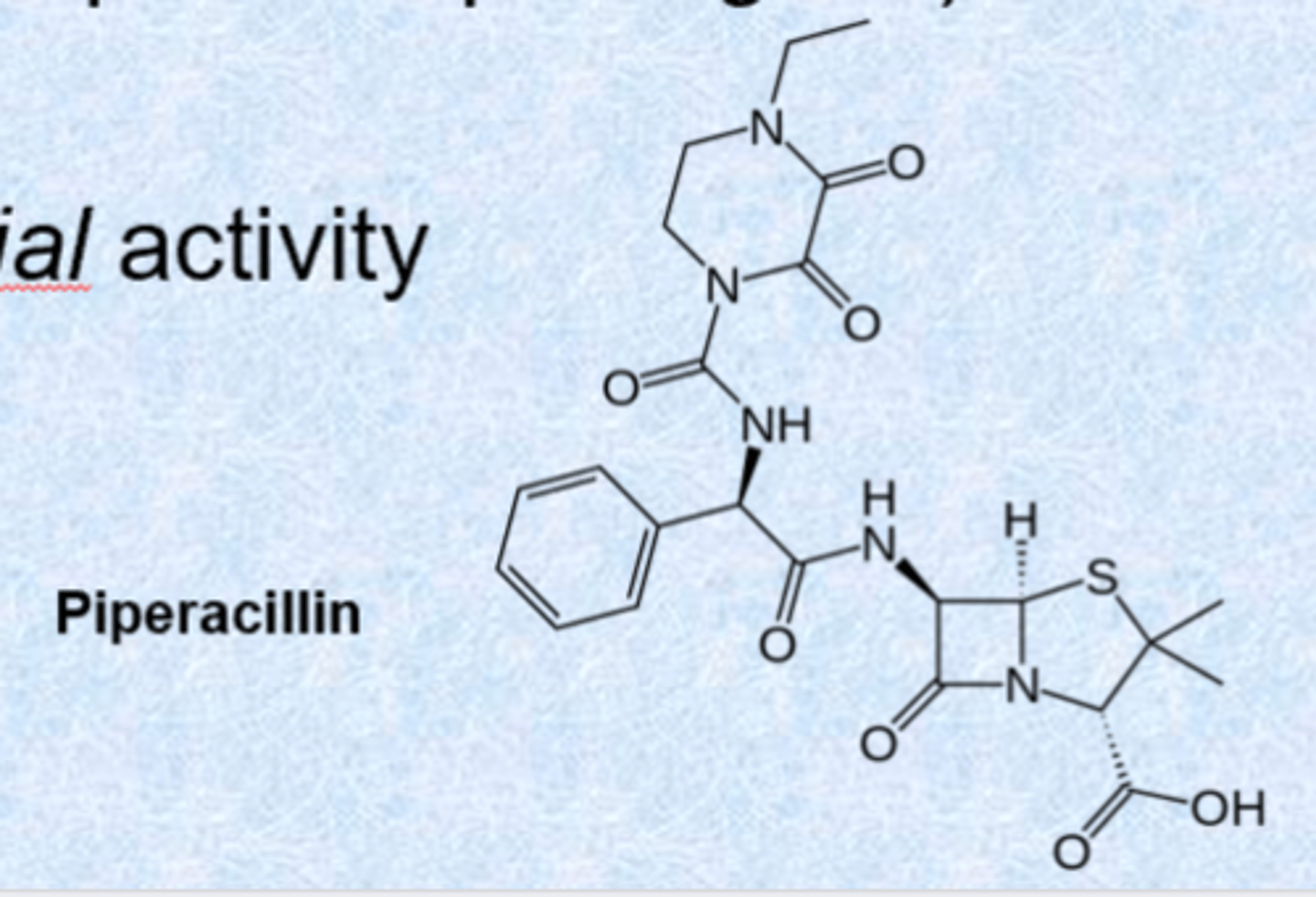
cephalosporins
- still b-lactams but 6 membered dihydrothiazine ring instead of penicillin 5 membered ring
- discovered in mid 1940s, structure established 1961
- isolated from the fungus Acremonium
- has 6-membered dihydrothiazine ring (new scaffold)
- most active against gram(+) but newer versions increased coverage
- developed to have activity against both gram-positive and gram-negative bacteria (H. influenzae, N. gonorrhea, E. coli)
- also has activity in presence of beta-lactamase enzymes, more acid stable
- variations tolerated at 7-acylamino side chain, 3-acetoxylmethyl side chain (unstable) and extra substitution at C7
- 3 acetoxylmethyl side chain unstable because of double bond - swapped out in some medications
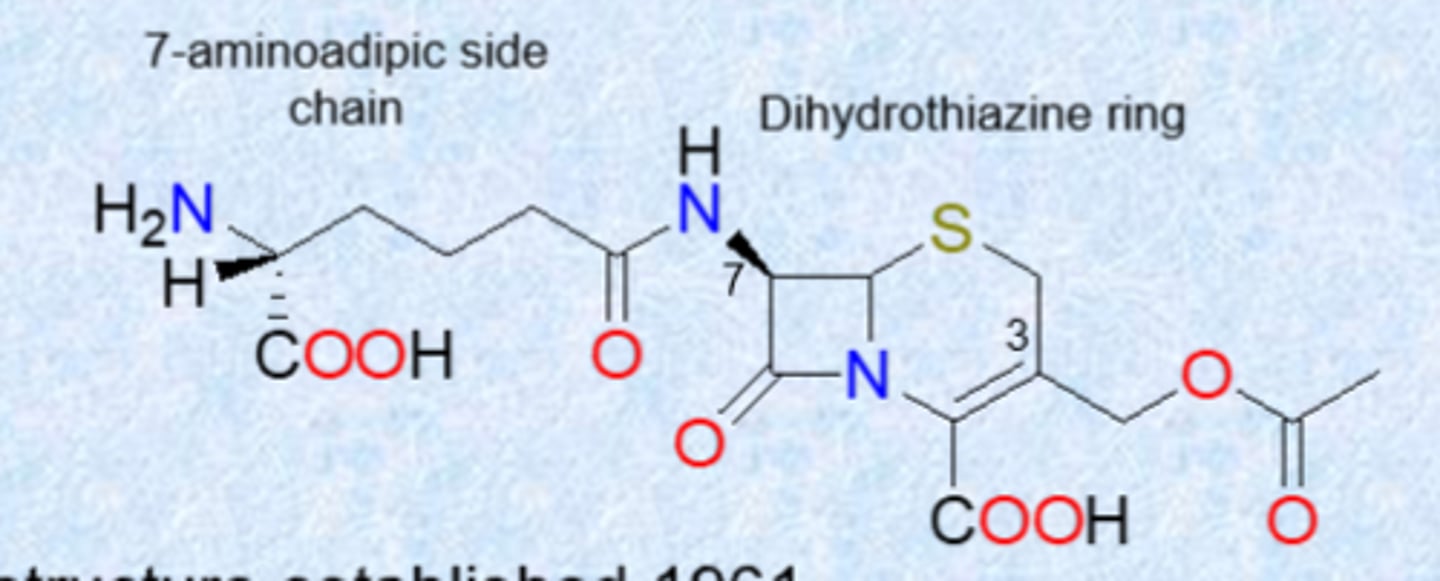
first generation cephalosporins (same strategy as penicillins)
- cefalexin (Keflex) both Gram (+) and some Gram (-) but not MRSA
- cefadroxil both Gram (+) and better Gram (-)
- cefazolin good for UTIs, effective for MSSA but not MRSA
- got rid of acetoxy group to simple methyl or ring to increase stability and increase coverage
- change side chain to the left
- MRSA picked up its own PBP
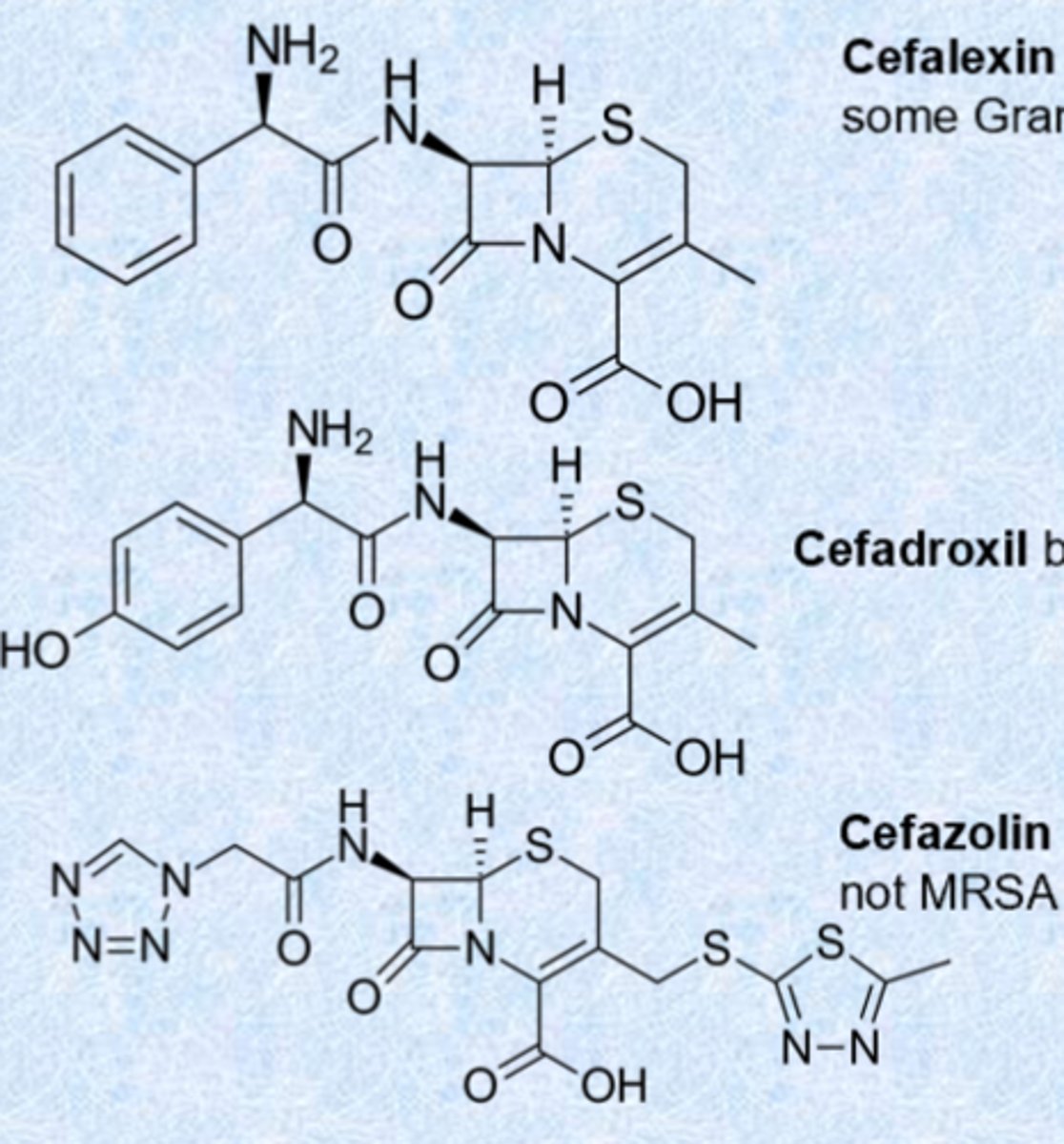
second generation cephalosporins
- cefuroxime has increased resistance to beta-lactamases, retains activity against Streptococci
- cefoxitin and cefotetan have expanded spectrum and are unique in having activity against anaerobic bacteria - given IV/IM
- cefotetan has an N-methylthiotetrazole ring which is a breakdown product and can cause hypoprothrombinemia
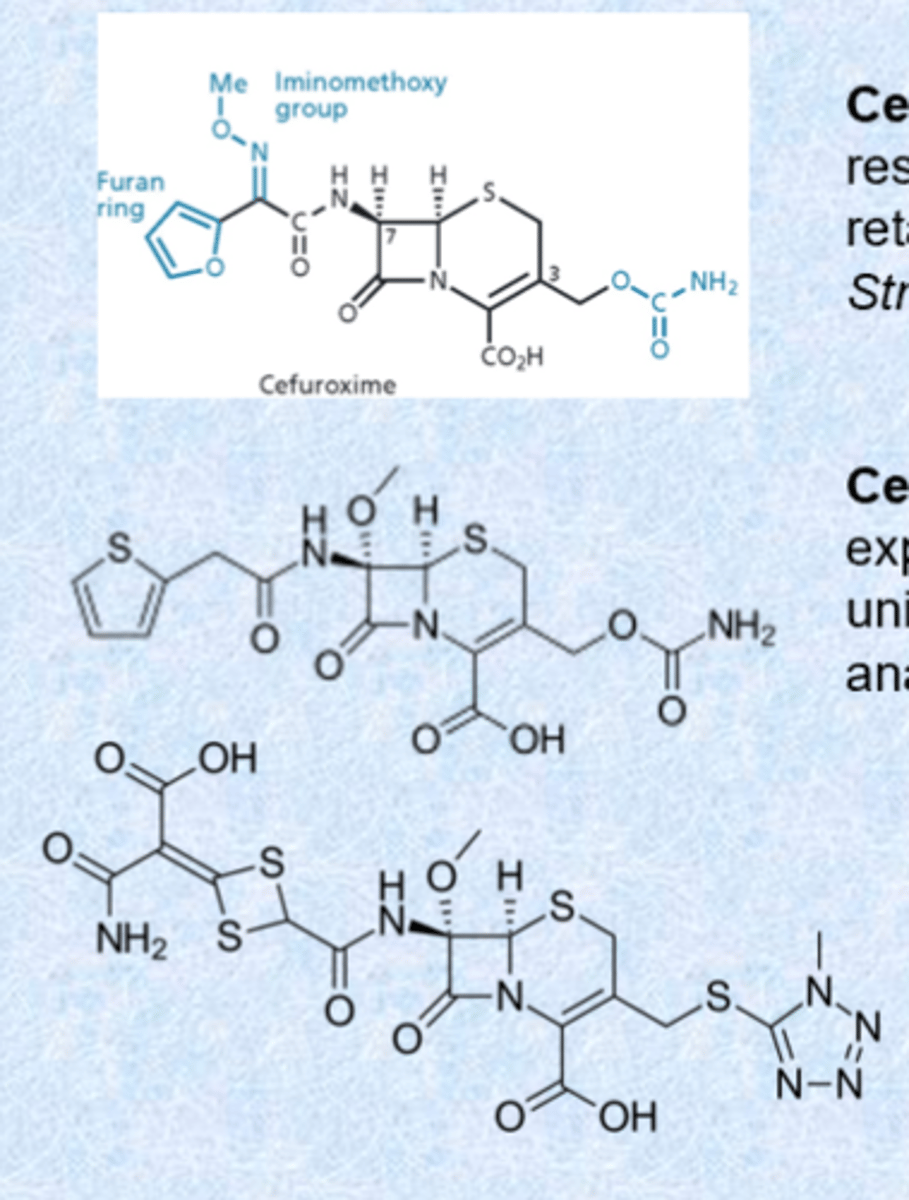
third generation cephalosporins
- inclusion of charge on the right and aminothiazole rings (very basic, positively charged, large amounts of charge, increased permeation into gram-negatives)
- replacing furan ring of cefuroxime with aminothiazole ring enhances penetration through membrane of gram-negative bacteria
- variants at position 3 alter PK
- beta-lactamase work in periplasmic space - targets are also located in periplasmic space so for gram-negative, only want to get into outer ring not inner portion
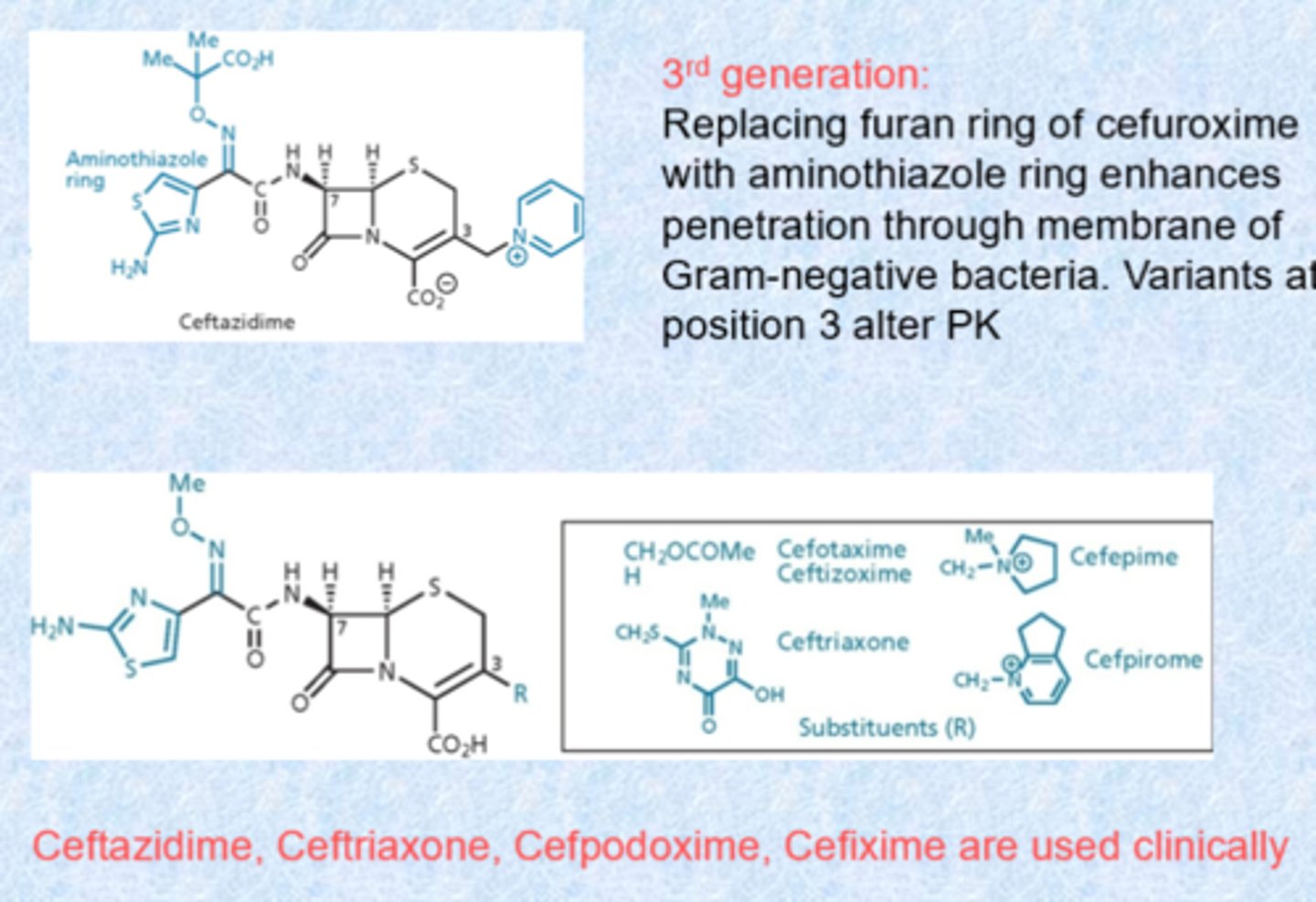
4th generation cephalosporins
- cefepime is the only one!
- offered extended spectrum and higher potency than third generation compounds
- little structural difference with the third generation
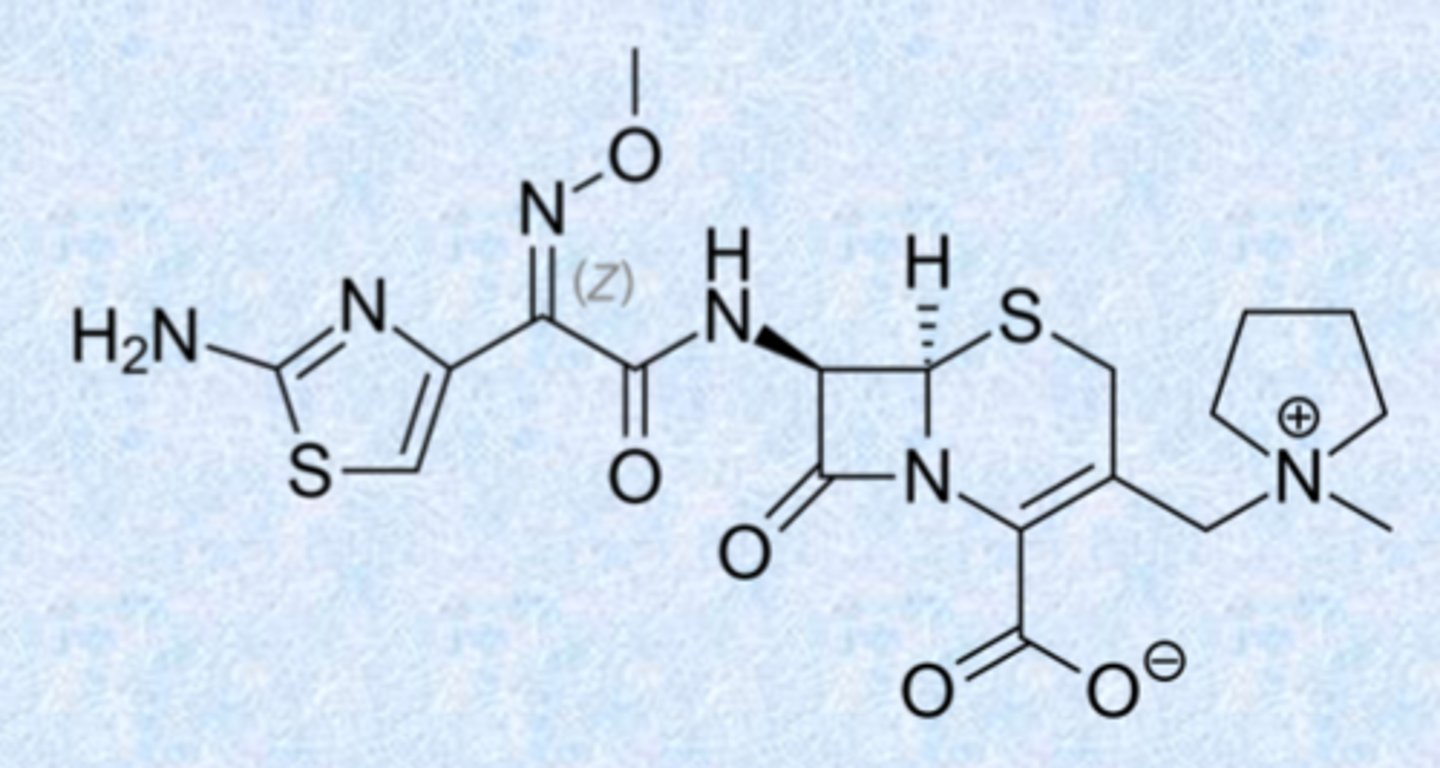
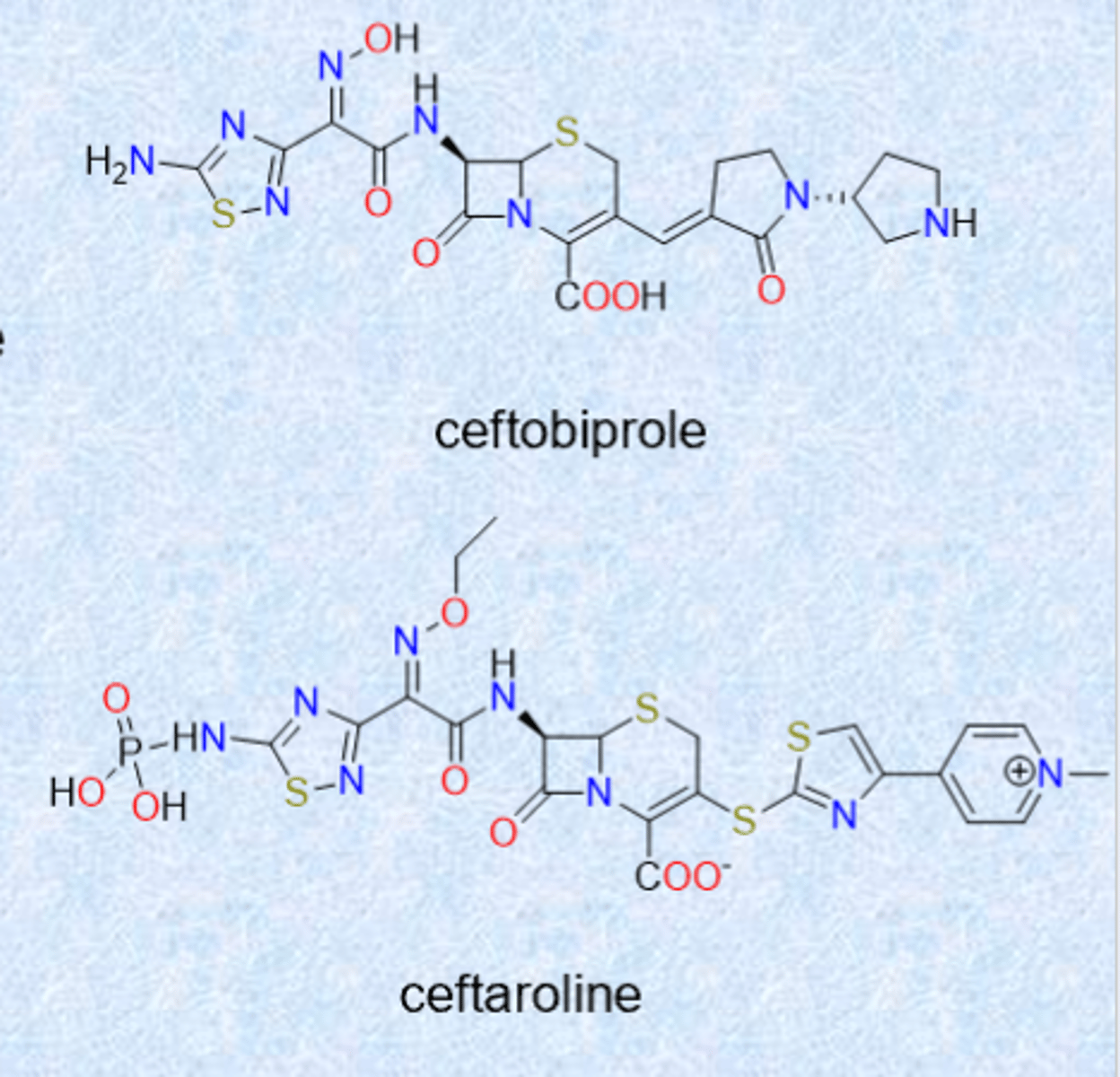
even newer (5th generation) cephalosporins
ceftobiprole (2024 US approval)
– activity against Gram-positive and –negative organisms
– binds and inhibits PBP2a from MRSA
ceftaroline (2010)
– similar spectrum as ceftobiprole
– also inhibits PBP2a
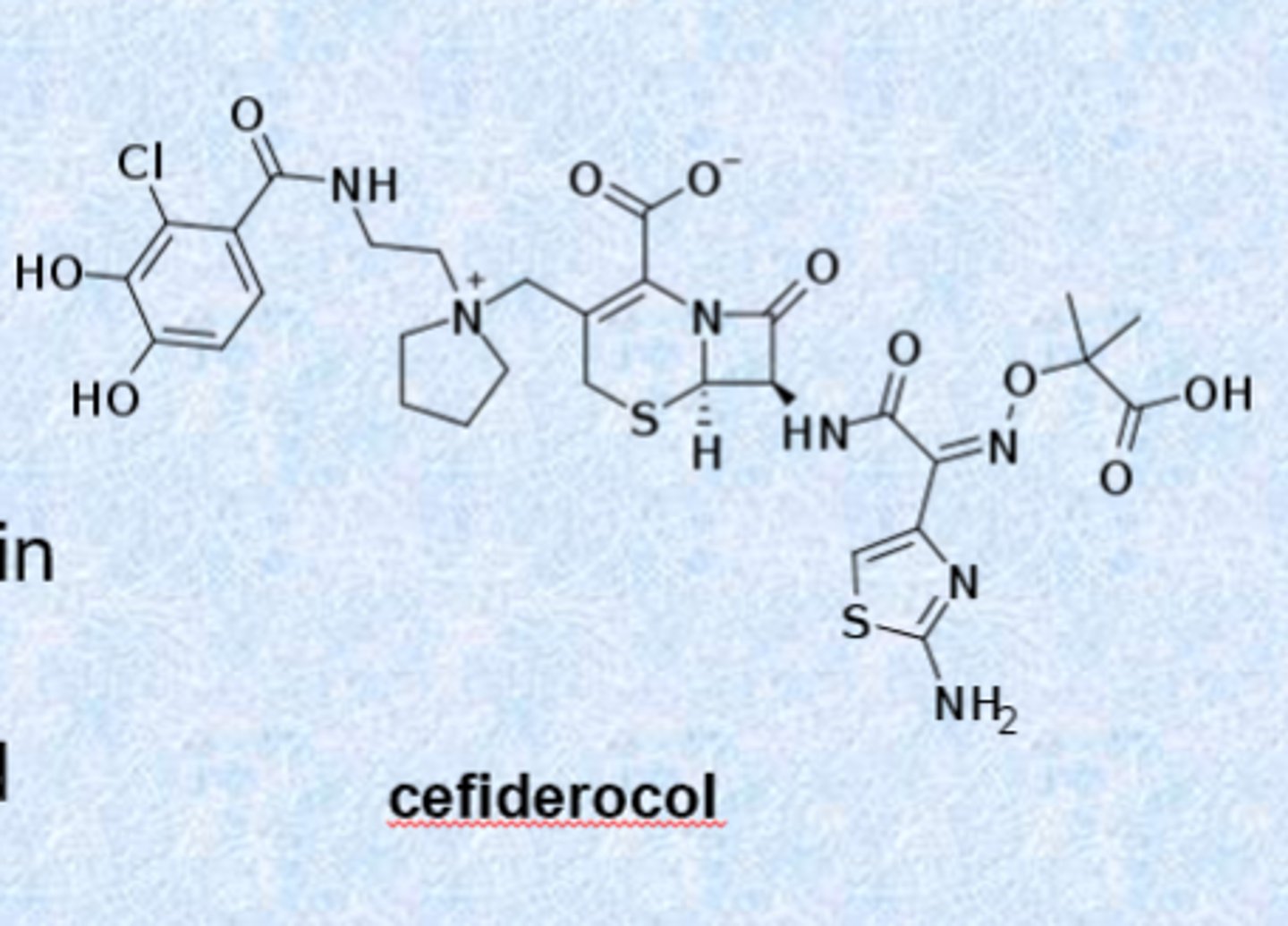
a siderophore antibiotic
cefiderocol (2019)
– activity against gram-negative organisms including P. aeruginosa
– exploits the “war for iron” in the body during infection (bacteria need iron to make new bacteria)
– acts as a siderophore and can chelate iron
- right part acts as iron and the bacteria pulls it in and kills the bacteria
- last resort for cUTI
– iv injection only
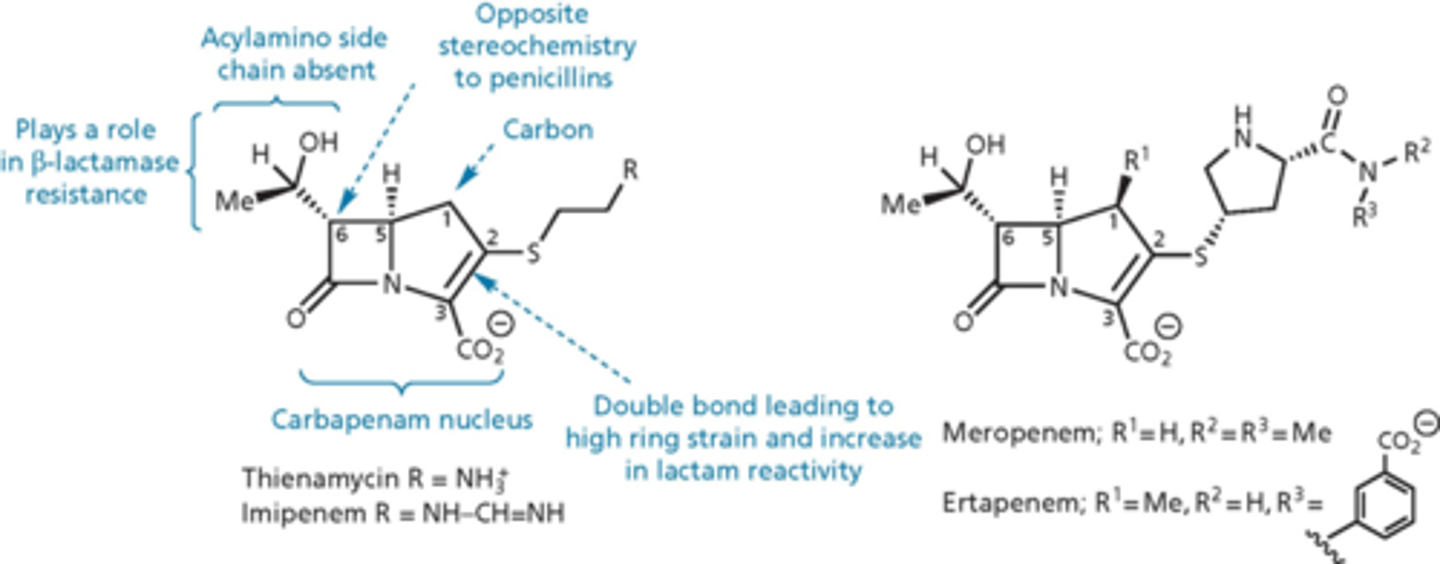
carbapenem
- last major group of b-lactams
- 5 membered ring but instead of sulfur, there is a carbon
- thienamycin isolated from Streptomyces in 1976
- broad range of activity against gram-positive and –negative
- low toxicity, high resistance to beta-lactamases
- poor metabolic and chemical stability
- imipenem, meropenem and ertapenem are clinically useful analogs - changed how reactive the nitrogen is on the right (increase stability by buffering)
- harder to produce
- have to keep in solution because less stable as a solid
3 main classes of b-lactams
- penicillin
- cephalosporin
- carbapenem
combination with b-lactamase inhibitors
- blocking b-lactamase can protect the drug from enzymatic degradation
- not useful for MRSA since that resistance is due to the insensitive PBP
- undergo suicide reaction with b-lactamase and slow release of covalent complex (bind and never come off)
- compounds do not inhibit the PBPs
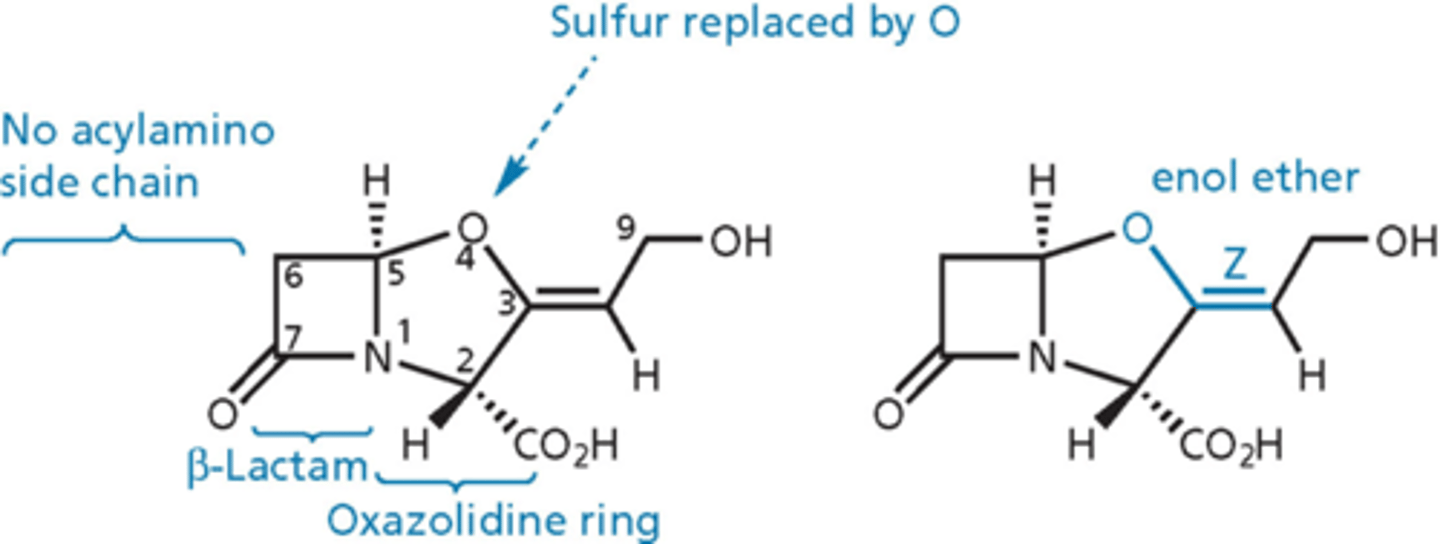
clavulanic acid
- inhibits beta-lactamase enzyme
- has beta-lactam ring but no side chain on the left = not a dipeptide, not fooling PBPs into thinking its D-ala, D-ala
- administered with amoxicllin (Augmentin is amoxicillin + CA)
- mechanism-based irreversible inhibitor
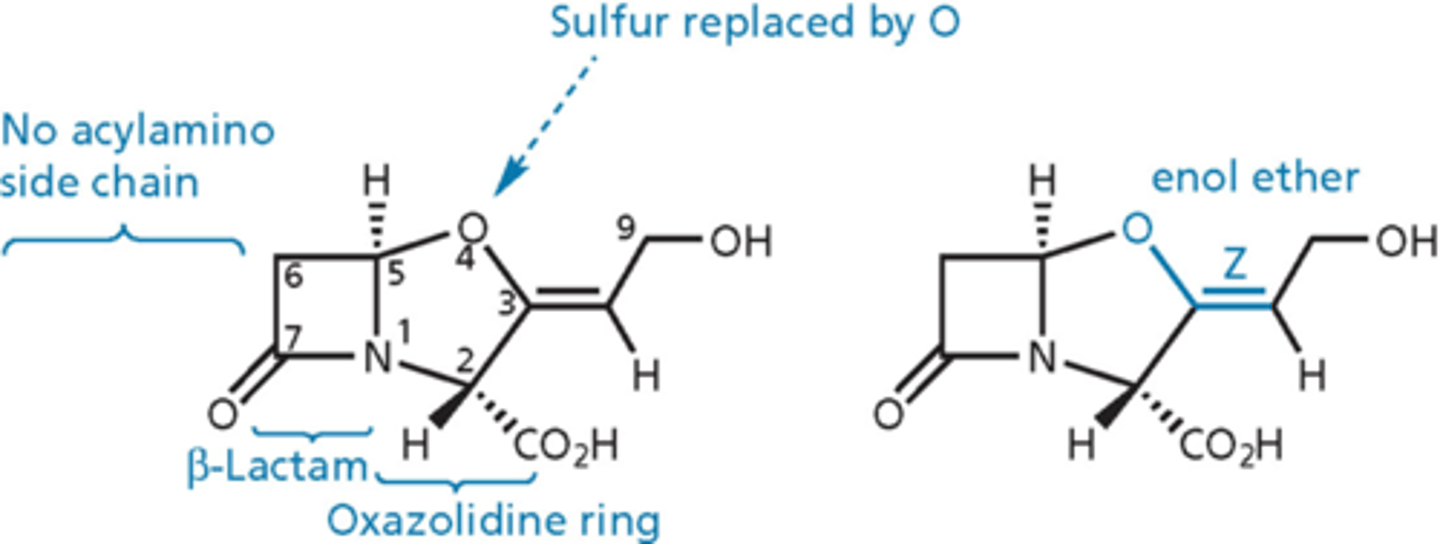
clavulanic acid - essential
- b-lactam ring
- enol ether
- Z configuration for double bond
- no substitution at C6
- R at positions 2 and 5
- carboxylic acid group
b-lactamases and their inhibitors
b-lactamases classified by Ambler (A-D)
- A/C/D similar to serine proteases
- class A inhibitors: no activity on their own
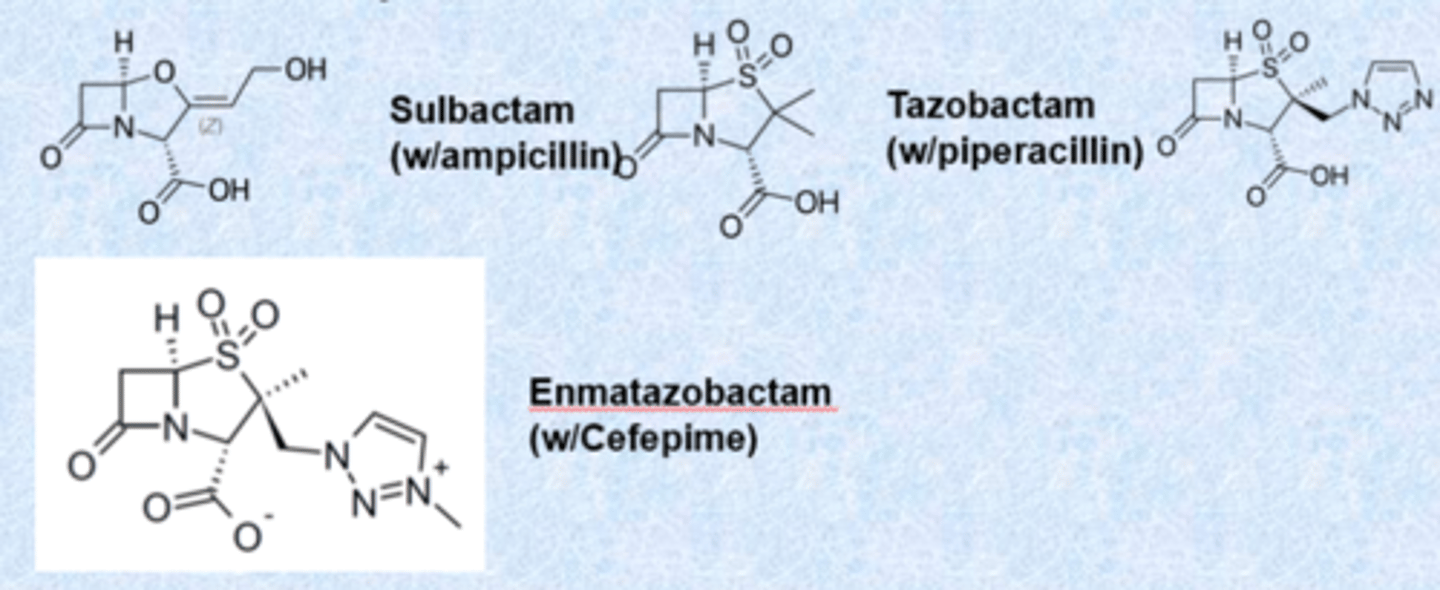
b-lactamase class A/C/D (partial)
- spectrum of coverage is the same just protecting from b-lactamase
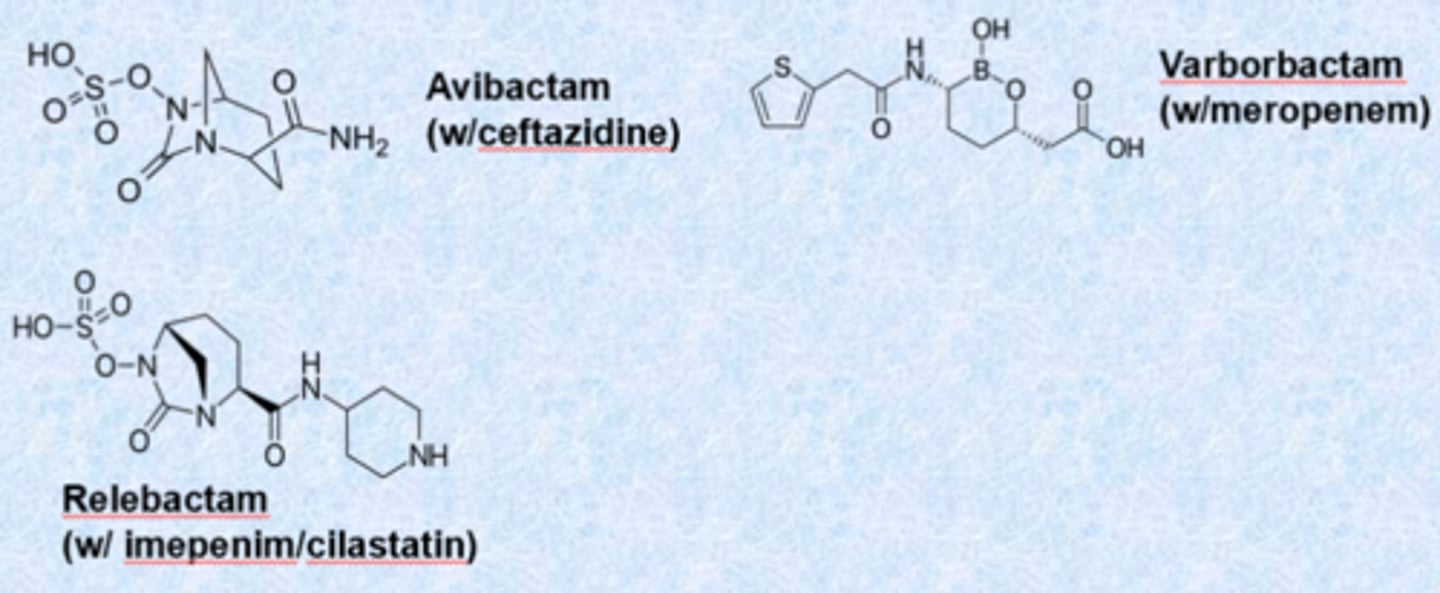
b-lactamase class B
- (NDM-1) are metalloproteases (no covalent adduct) - no inhibitors
- use zinc
- nothing works on these clinically
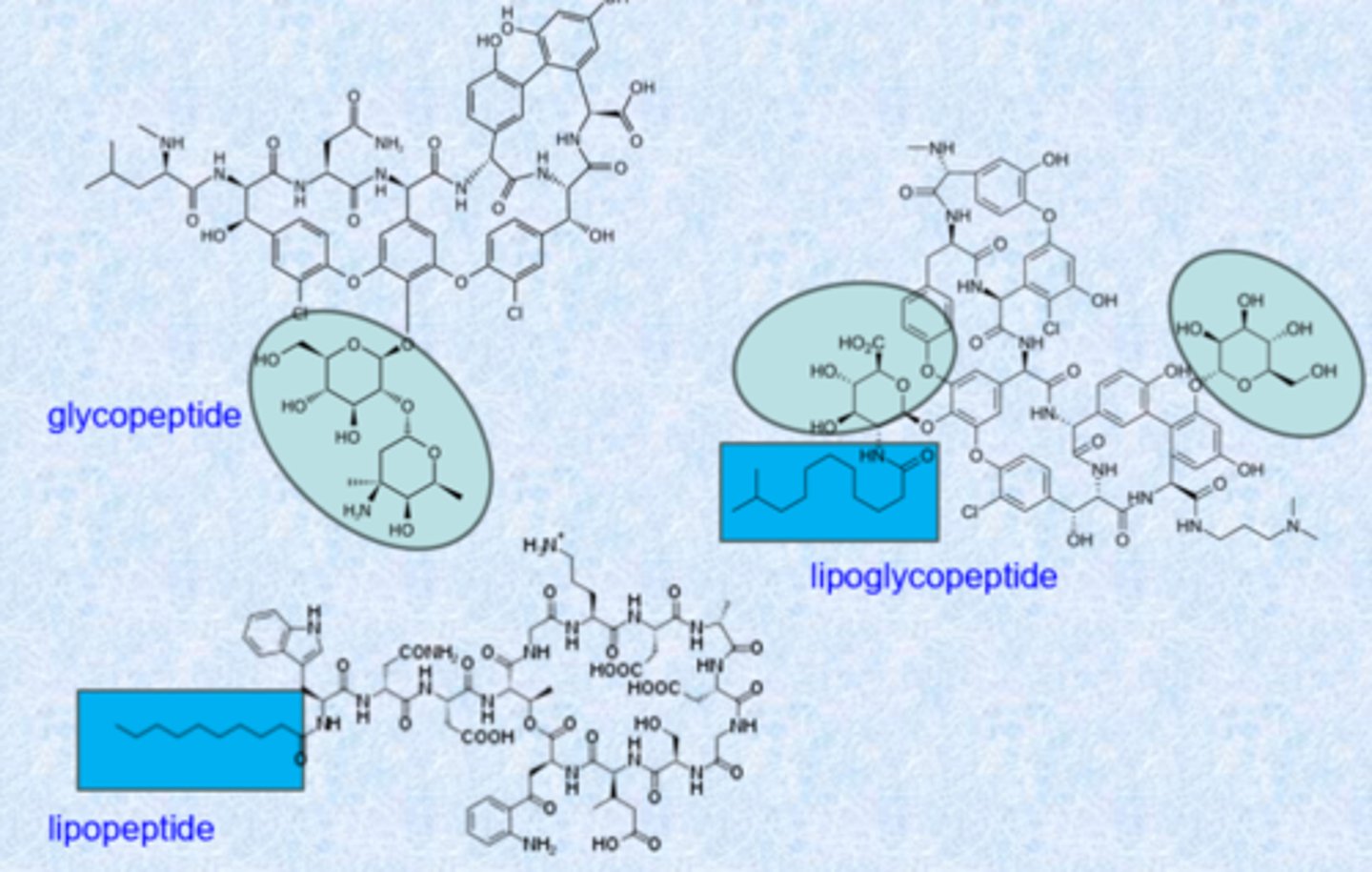
glycopeptides/lipoglycopeptides/lipopeptides
huge molecules
- isolation reveals several species from a single source
- most are cyclic, large
- product of the Non-Ribosomal Peptide Synthesis (NRPS) pathway
- frequently contain D-amino acids and/or unnatural amino acids
- many contain non-amino acid moieties
- work almost exclusively on gram-positives
- important against MRSA
vancomycin
- narrow-spectrum agent from Amycolatopsis orientalis
- introduced in 1956 for treatment of penicillin-resistant S. aureus
- main stand-by drug for MRSA
- size makes it unable to cross outer cell membrane of Gram-negative bacteria or inner cell membrane of Gram-positive bacteria
- since cell wall construction takes place outside cell membrane, effective against Gram-+ bacteria
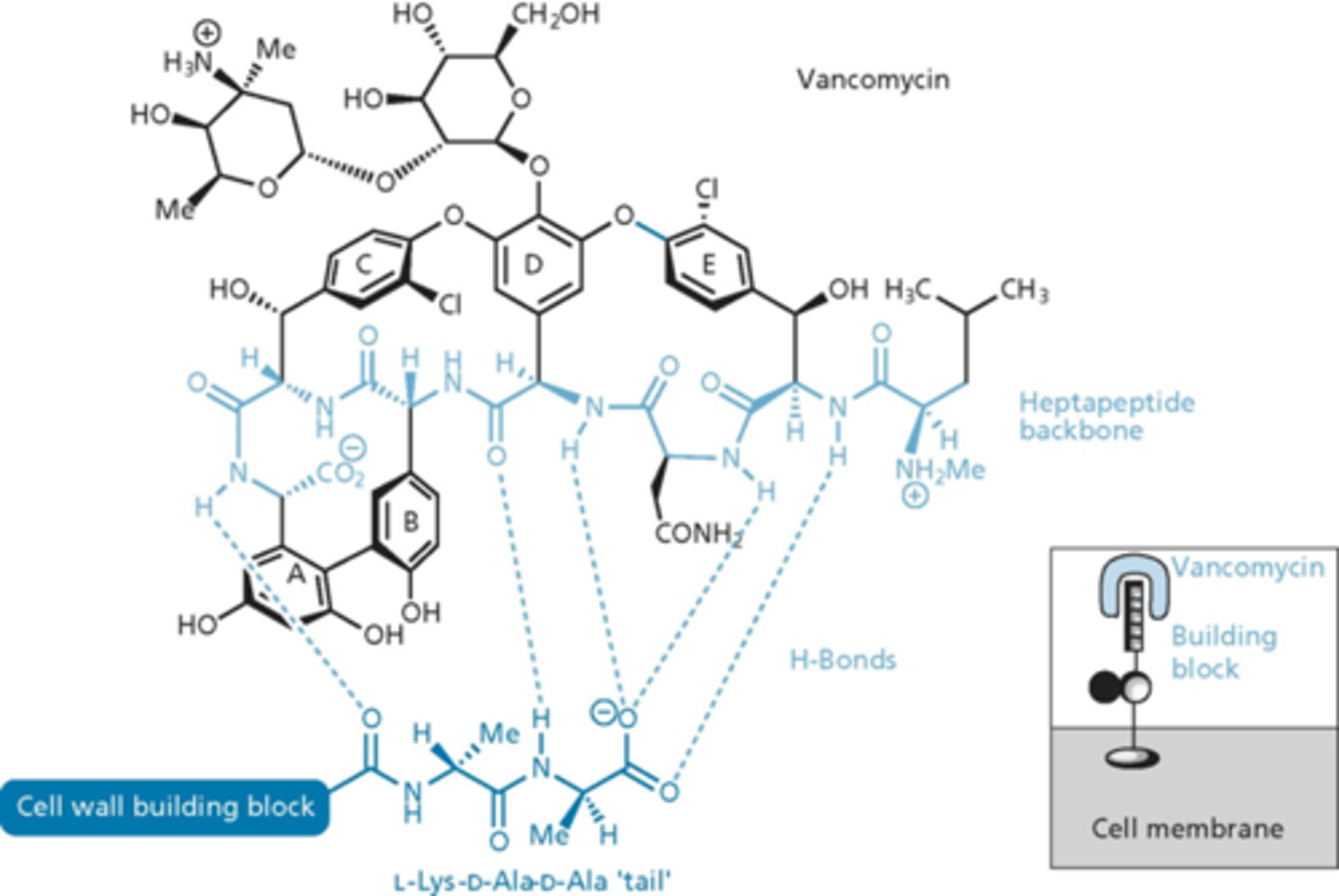
vancomycin structure
- derived from a heptapeptide with 5 aromatic residues
- undergo oxidative coupling to produce 3 cyclic moieties
- chlorination, hydroxylation and addition of 2 sugar units
- cyclizations form rigid unit
- substituents on A,B,C,E important for rigidity (rigid because of cyclic structure)
- hydrogen bonding
- inserts itself into the unit and makes it so the PBPs cannot do anything (hides the substrates) - stops cell wall
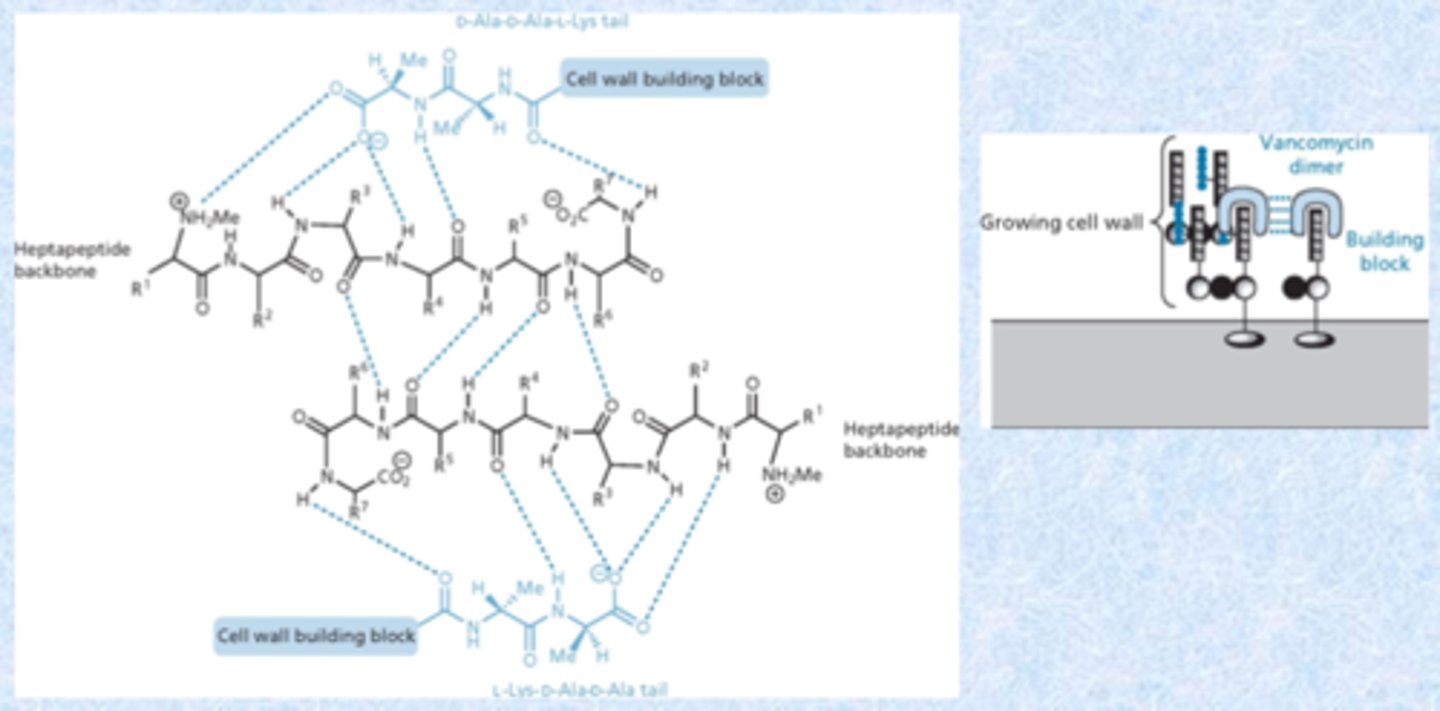
vancomycin dimers
- dimers are an important component in the cell wall
- binds to itself in a dimer and binds to d-ala d-ala units
- shuts down
- sugar and chlorine groups also important in dimerization
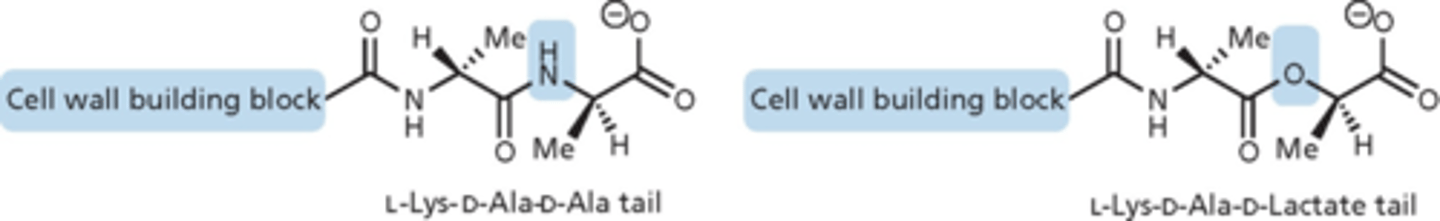
vancomycin resistance
- vancomycin-resistant S. aureus (VRSA, not common) identified in 1996, and vancomycin-resistant Enterococcus (VRE, more prevalent and a problem) in 1989
- modification of cell wall precursors where terminal D-alanine group replaced by D-lactic acid
- removes an NH group in H-bond, destabilizes the complex and H-bond cannot stick to the D-ala
telavancin
lipoglycopeptide
- synthetic derivative of vancomycin
- approved 2009 for cSSSI
- inhibits bacterial cell wall synthesis
- disruption of bacterial membrane - makes it more leaky
- large lipid tails that associate into gram-positive walls (more gram-positive agents)
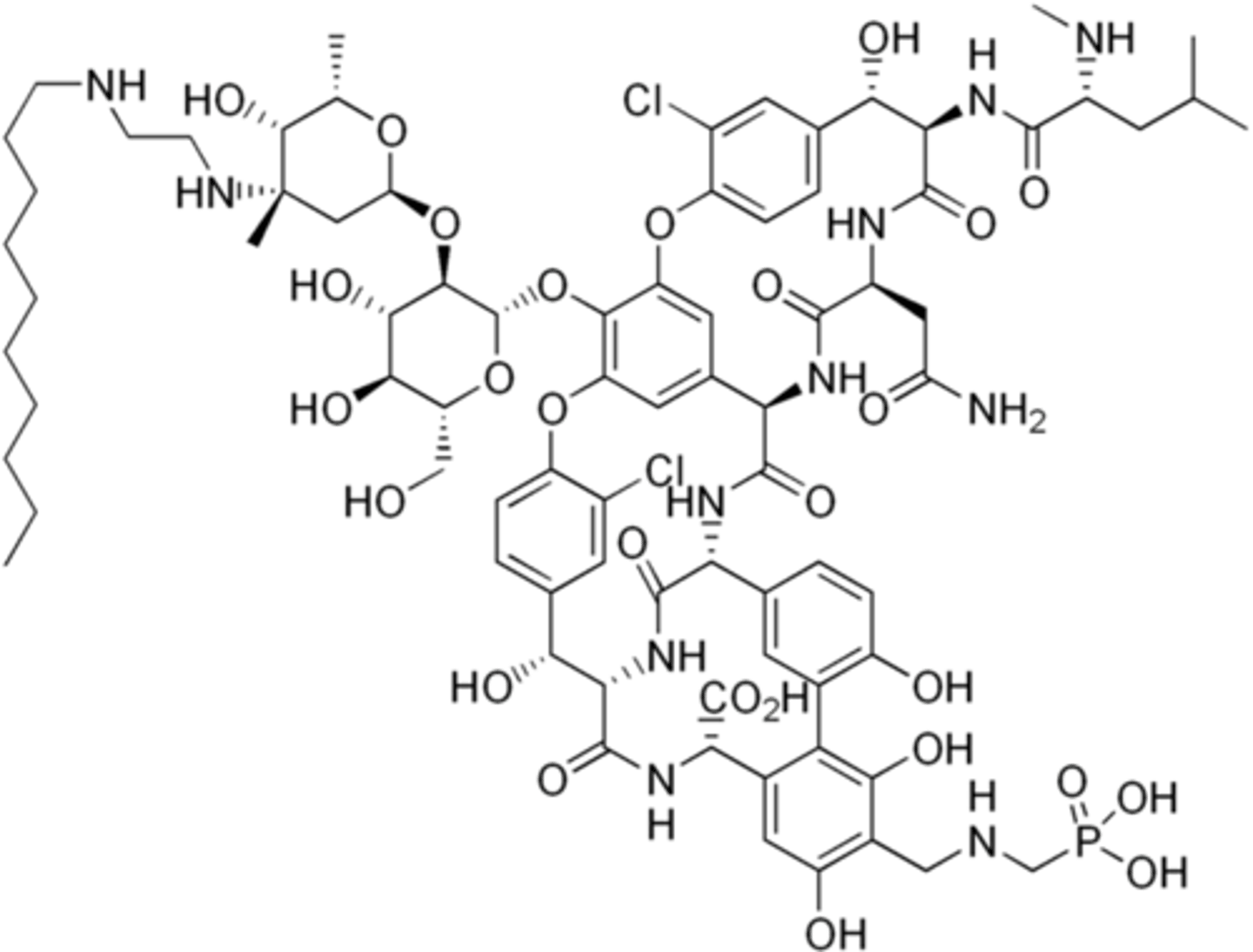
long-acting lipoglycopeptides
- very long half-lives (14-15 d), bind strongly to membrane via lipid tail
- uses in skin infections (single dose)
- because they are associated with the membranes, they are not excreted from the body
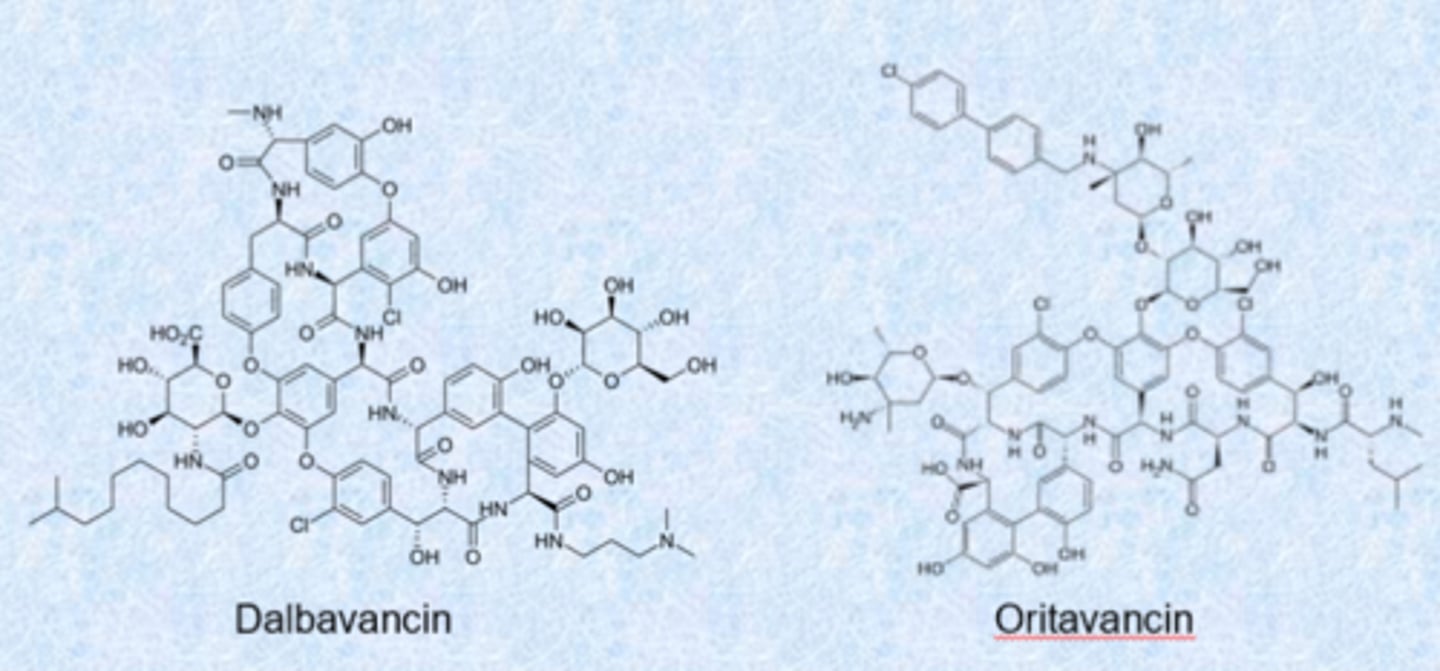
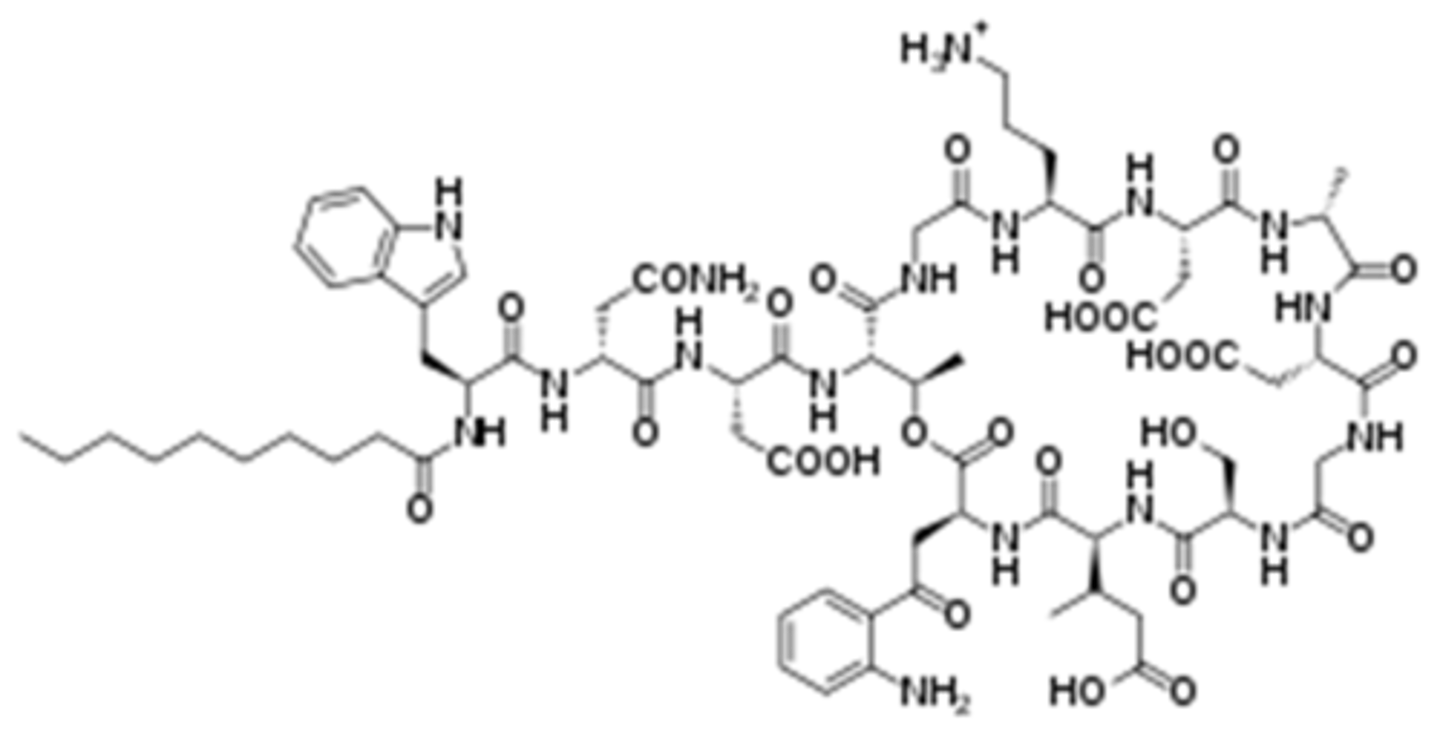
daptomycin
- lipopeptide antibiotic
- active against gram-positive bacteria
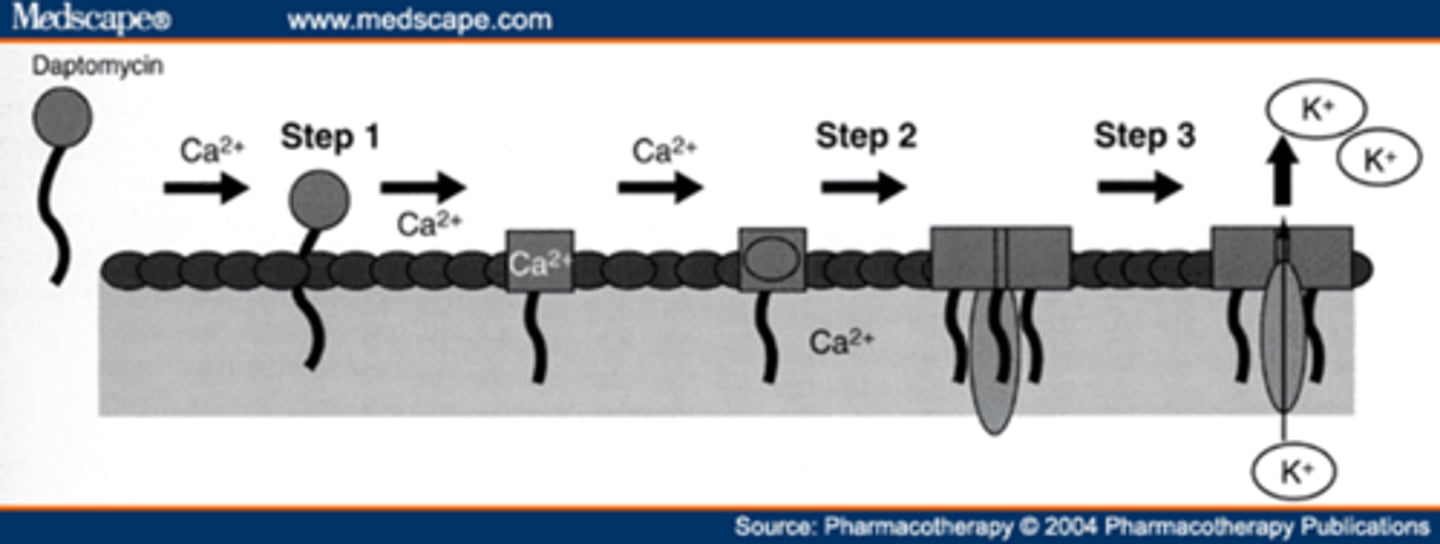
daptomycin MOA
- binds to and causes depolarization of the bacterial cell membrane
- efflux of K+ and other ions, inhibiting macromolecular synthesis
- leaves “ghost cell”
how to tell these apart
- all have peptide
- sugars = glyco
- long fatty tail = lipo
- if it has both = lipoglycopeptide
- name tells you a lot about the structure but probably not on the exam
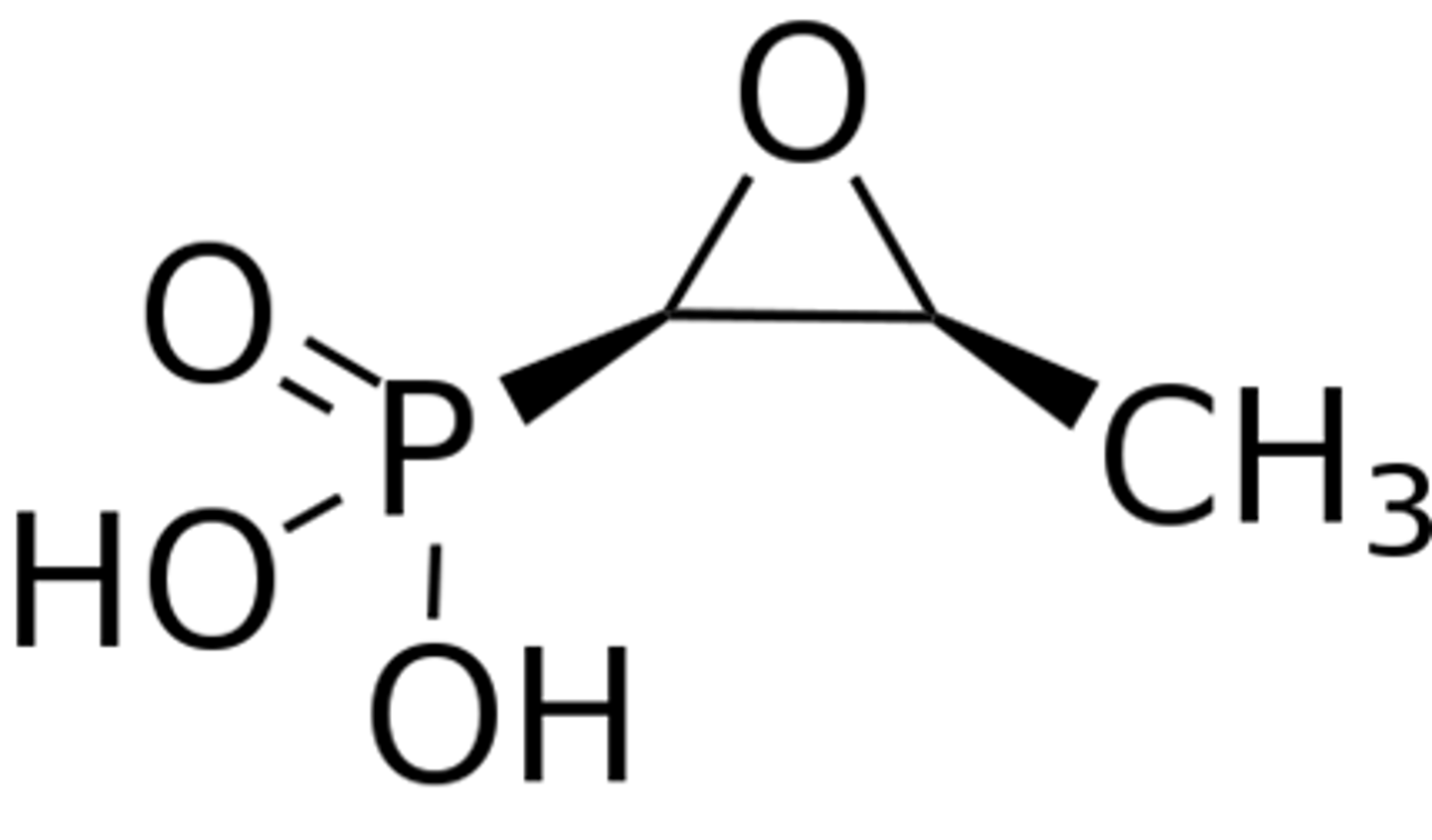
fosfomycin
- epoxide 3 membered strained ring and super negatively charged
- inhibits the bacterial enzyme MurA needed for synthesis of peptidoglycan
- mimics phosphoenolpyruvate
- irreversible Inhibitor, needs active transport
- used for UTI, concentrates in the urine
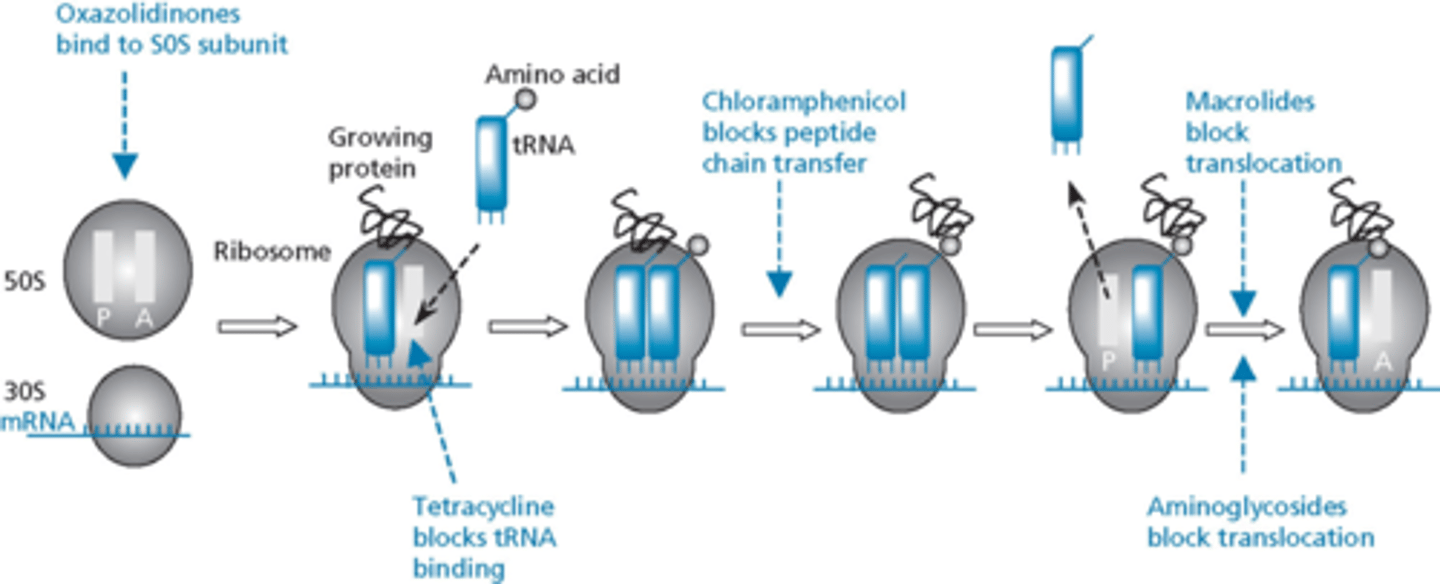
antibiotics that inhibit peptide synthesis
- tetracyclines (doxycycline) - most important/most used
- macrolides (erythromycin)
- aminoglycosides (gentomycin)
- oxazolidinones (linezolid) - the only synthetic ones
- lincomycins (clindamycin)
- most are natural product derived
- the way bacteria makes peptides and proteins are the same exact way we make peptides and proteins
- very safe compounds, most drugs have no action against human protein synthesis
- some shut off mitochondrial synthesis
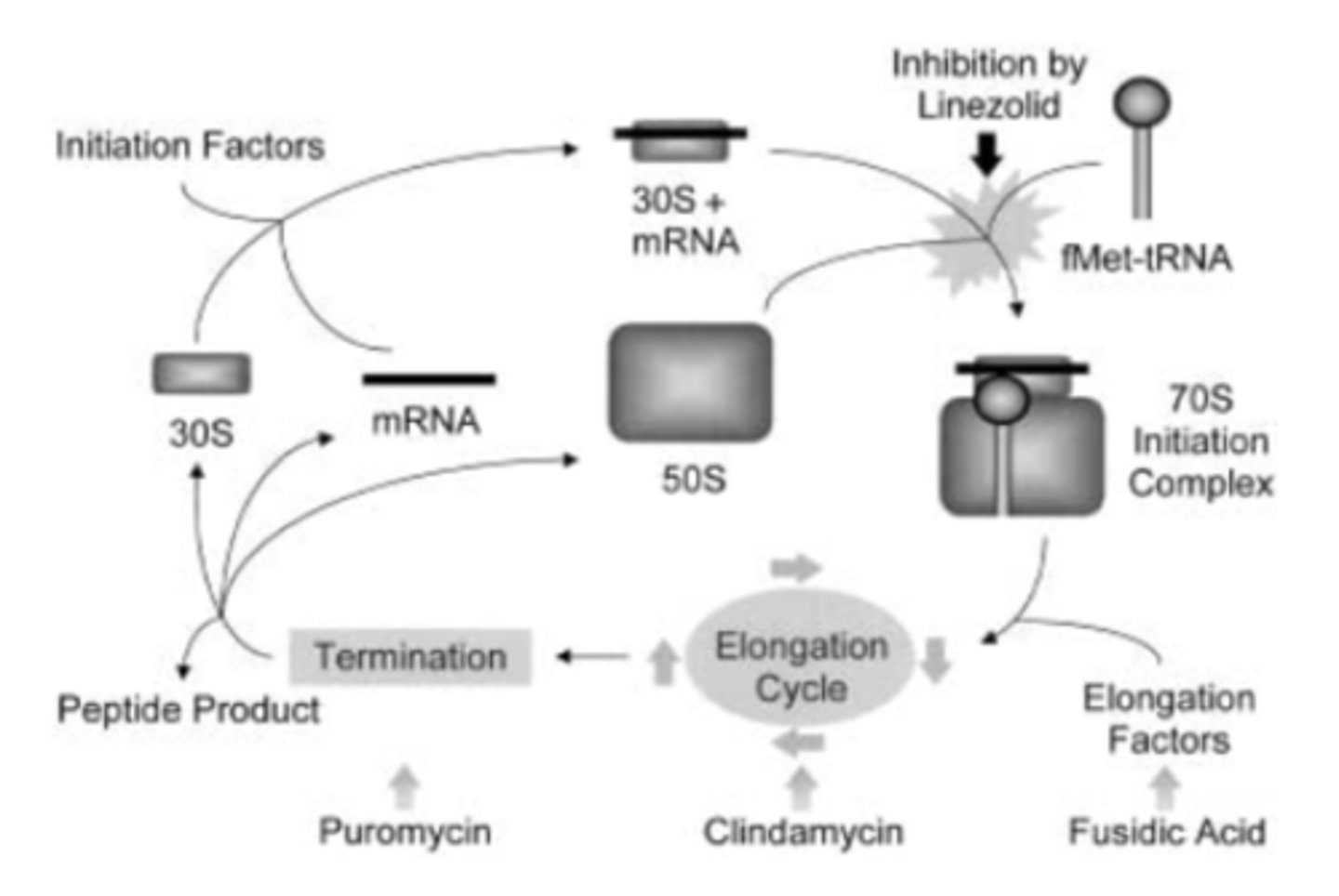
ribosomal assembly
- bacteria have 2 subunits of ribosomes
- 30S is mRNA binding site
- more than one binding site in ribosome for drugs (know where they work) - different in how they inhibit protein synthesis
- not a lot of cross resistance
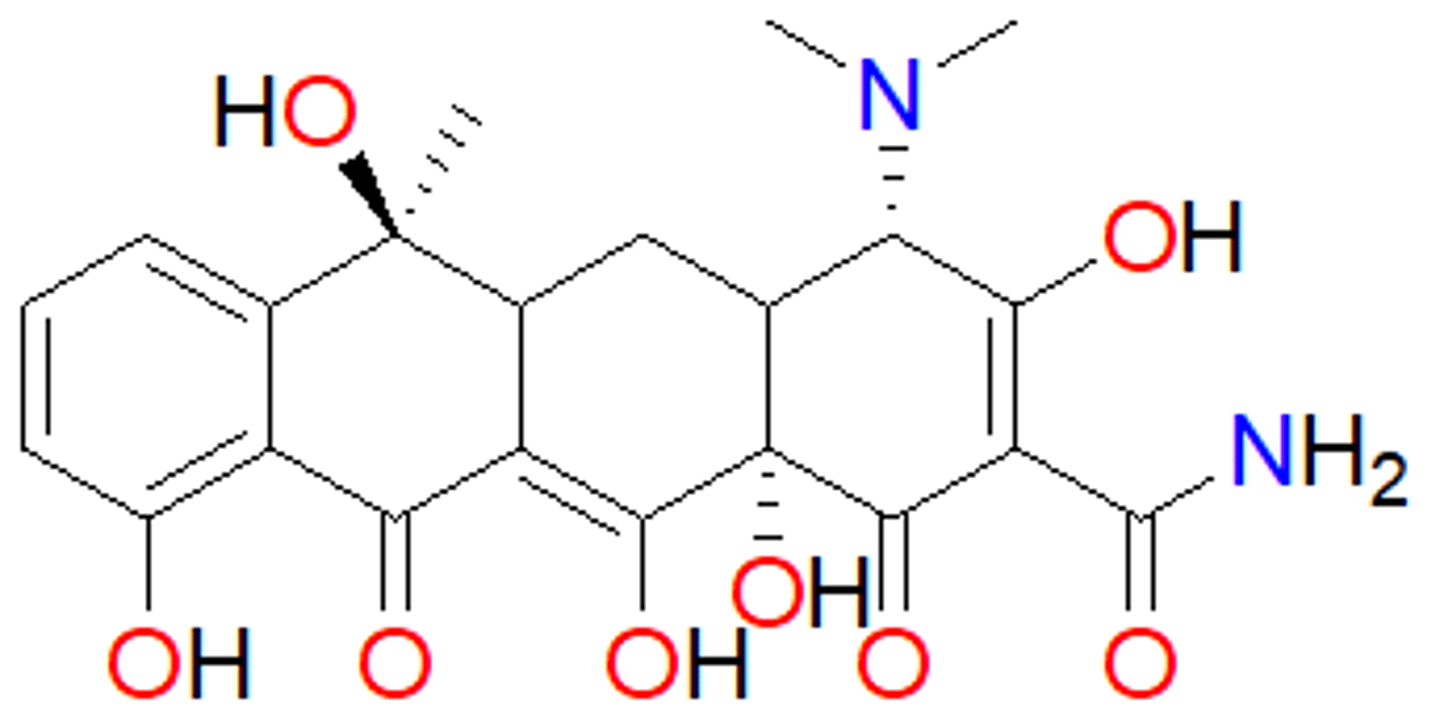
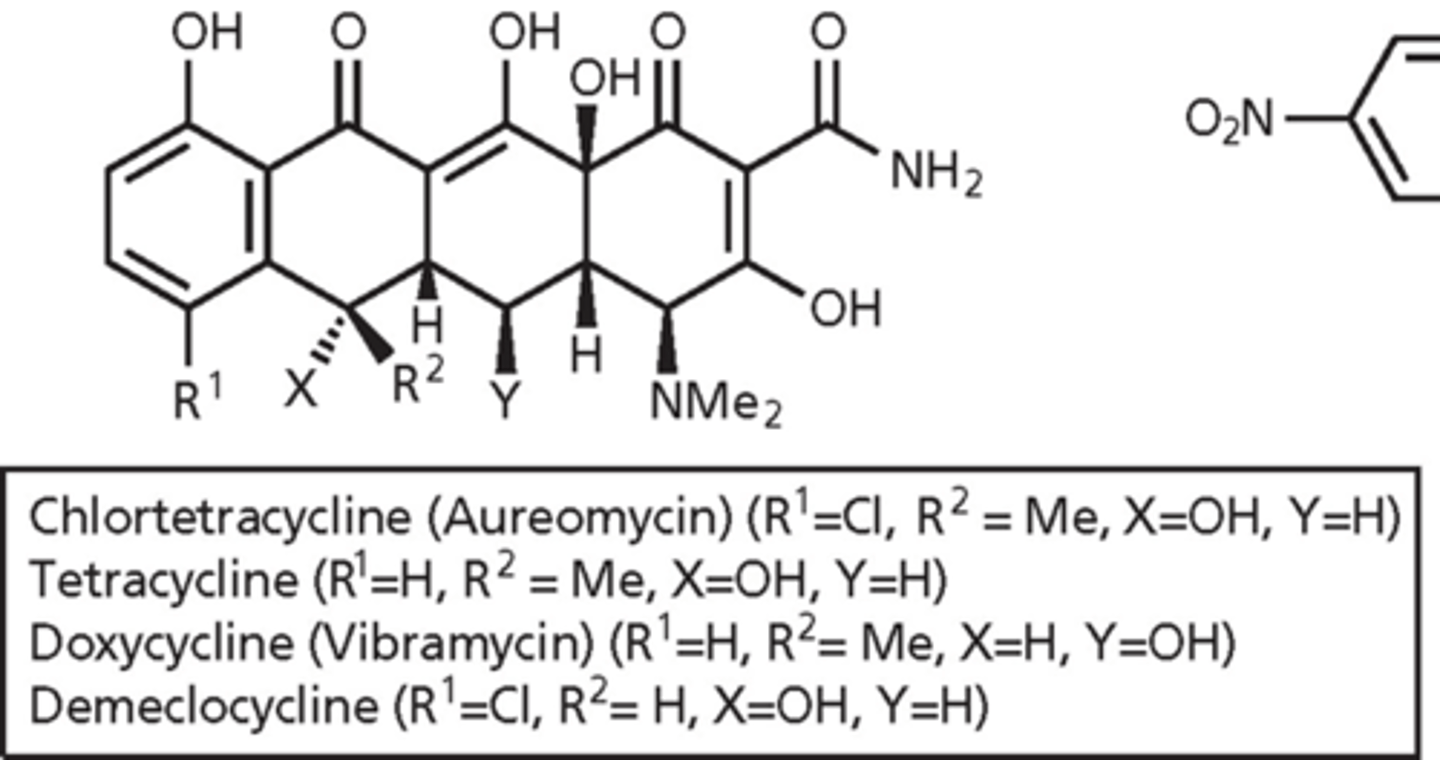
tetracycline (1953)
- produced by Streptomyces
- tetracycline means 4 rings
- oxygens on the bottom are 1,3 or 1,2 from each other - get rid of oxygens and they tend not to work
- protein synthesis inhibitor – binds to 30S subunit of bacterial ribosome - bacteria cannot replicate
- used to treat infections caused by Gram-positive and –negative bacteria
- also used for acne
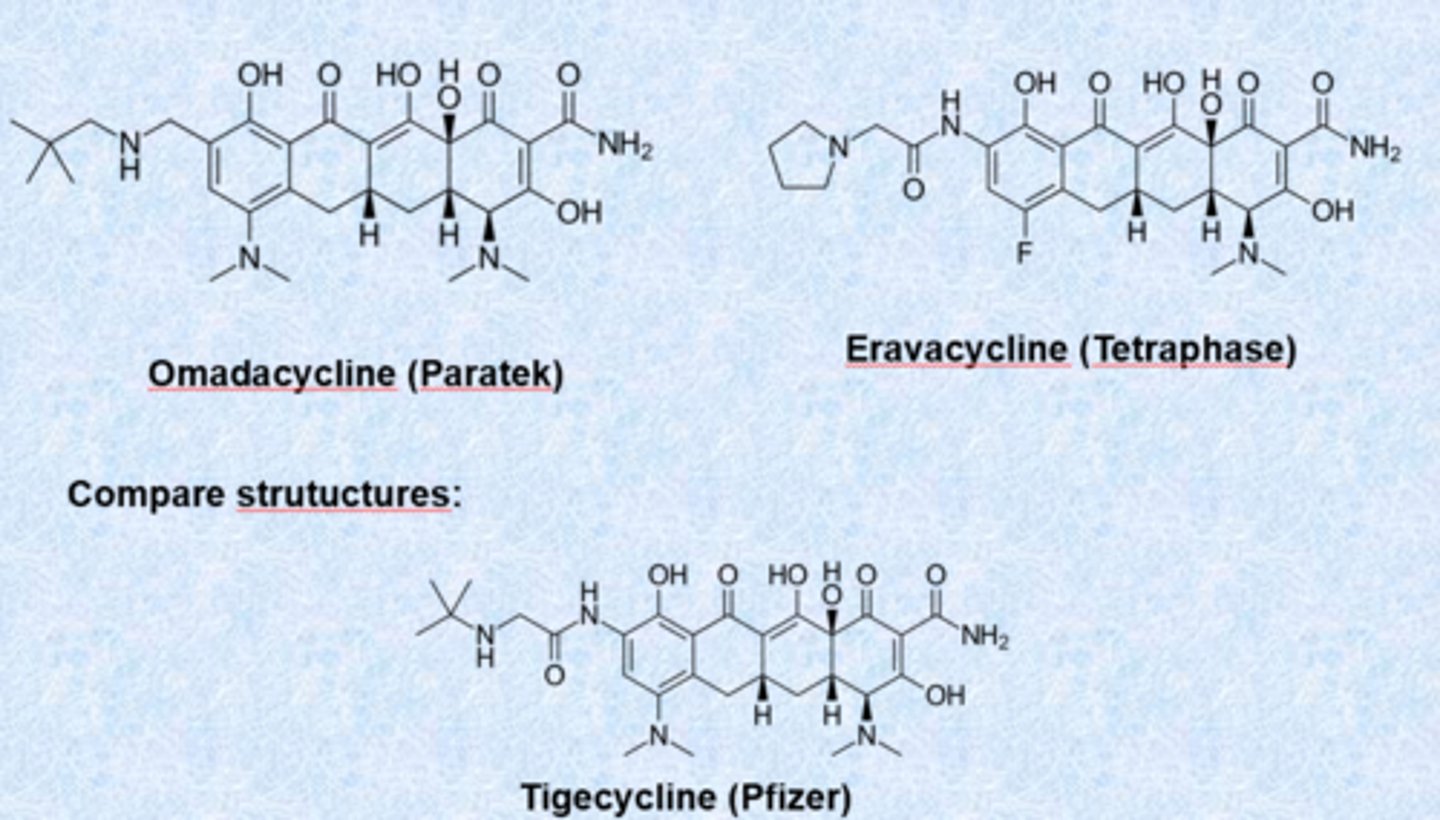
- clinically used embers of the family: tetracycline, doxycycline, minocycline, tigecycline, eravacycline and omadacyline
tetracyclines - clinically
- amphoteric (hydrophilic and hydrophobic surface) - hydrophilic surface is extensive array of oxygen atoms and hydrophobic is ring system
- oxygens can bind metal ions - form chelate as part of MOA with phosphates neutralized by magnesium
- impaired absorption in presence of milk, Ca2+, Mg2+, Al3+ - containing antacids (antagonize action via metal chelation)
- side effects: GI problems
- incorporation into bones and teeth (because of Ca2+ binding) - discoloration of the teeth
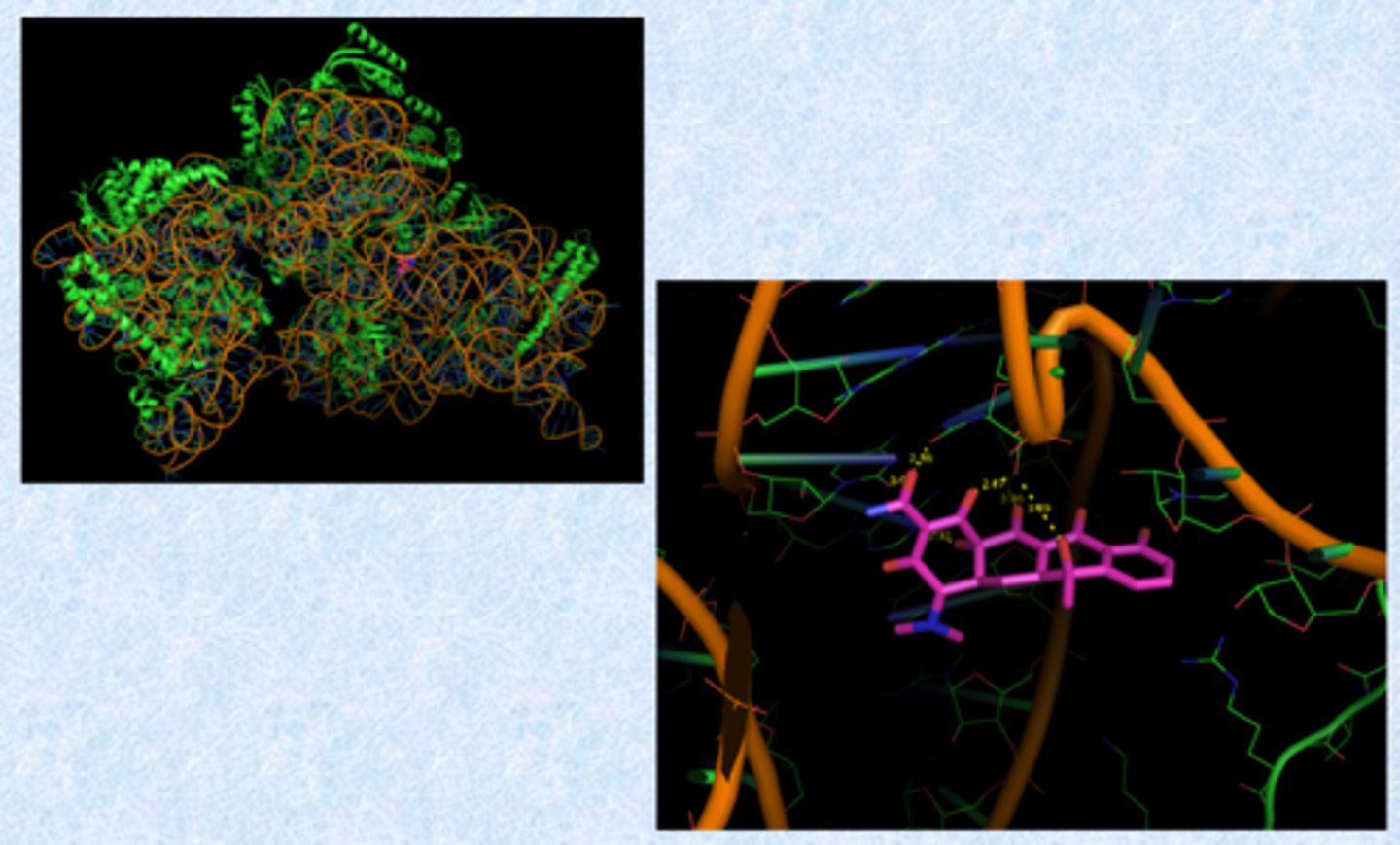
tetracyclines - mechanism of action
- bind to 30S ribosome
- prevent binding of aa-tRNA to the mRNA-ribosome complex (block adding the next amino acid to the peptide chain)
- binding requires Mg2+ ions (ribosome is Mg2+ rich because of phosphates)
Where/how does tet bind?
- oxygen atoms coordinate Mg2+
- Mg2+ embedded in phosphates on ribosome and coordinate the drugs
- protonated nitrogen also binds negative phosphate
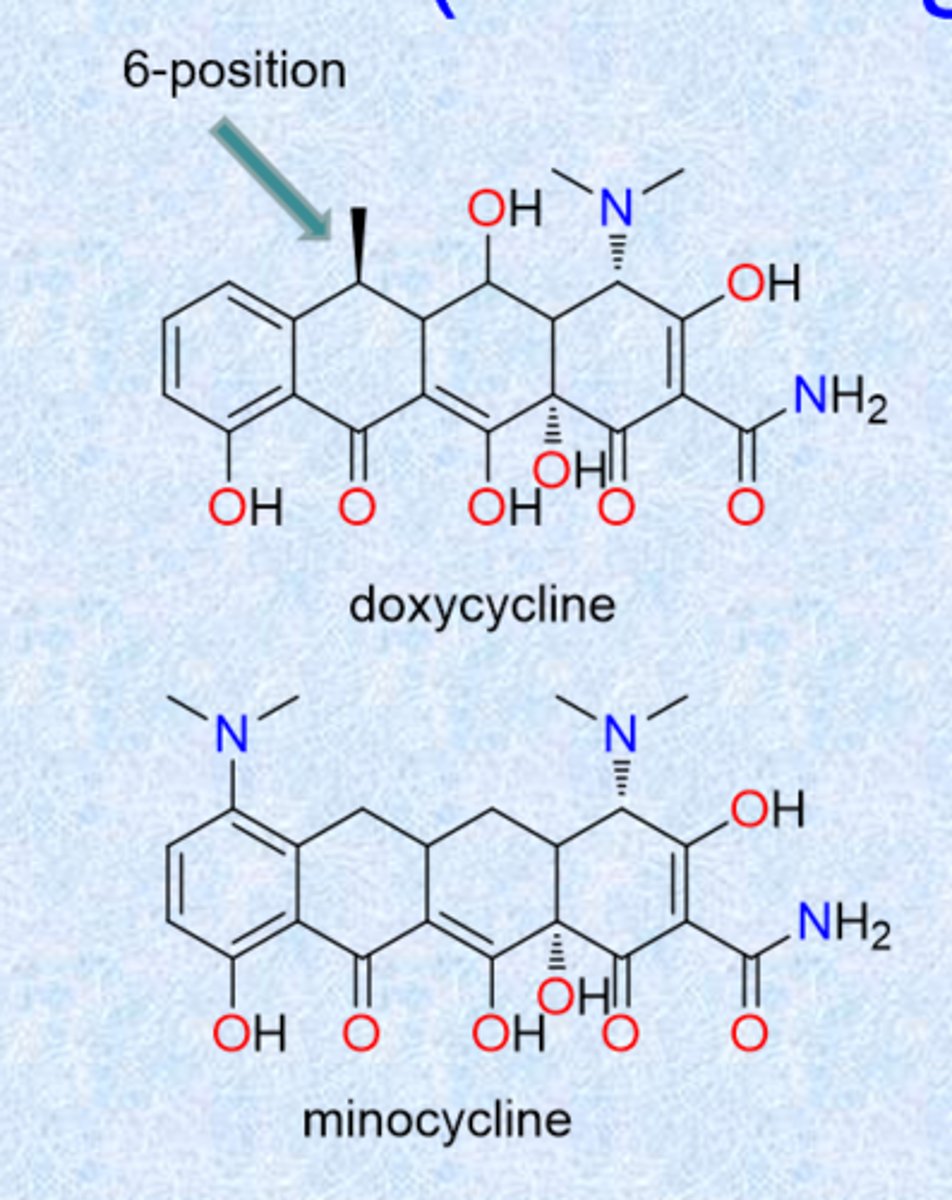
doxycycline and minocycline (second-generation)
modification of c 6-position
- hydroxyl in tetracycline gave some instability - these 2 derivatives get rid of that
- absence of the 6-OH group increases stability in acids and bases, yields long biological half-life
- absorbed well from GI tract
- doxy = methyl
- mino = minus (nothing)
- minocycline most potent tetracycline (1971)
- these show least Ca2+ binding - advantage
tetracycline - resistance
efflux: tet-A, -E, -G, -H, -K, -L (not important which one each is)
- reduce intracellular concentration of the drug to below critical threshold amount that allow bacteria to survive
- large, massive upregulation
- efflux pumps = pumps the drug out
ribosomal protection: tet-M, -O, -S
- bacteria modify the ribosome to disrupt binding (ex. methylation)
- bacterial protein synthesis apparatus rendered resistant by an inducible cytoplasmic protein
enzymatic oxidation
- bug destroys the drug
- oxidases
- less important than the previous 2 methods
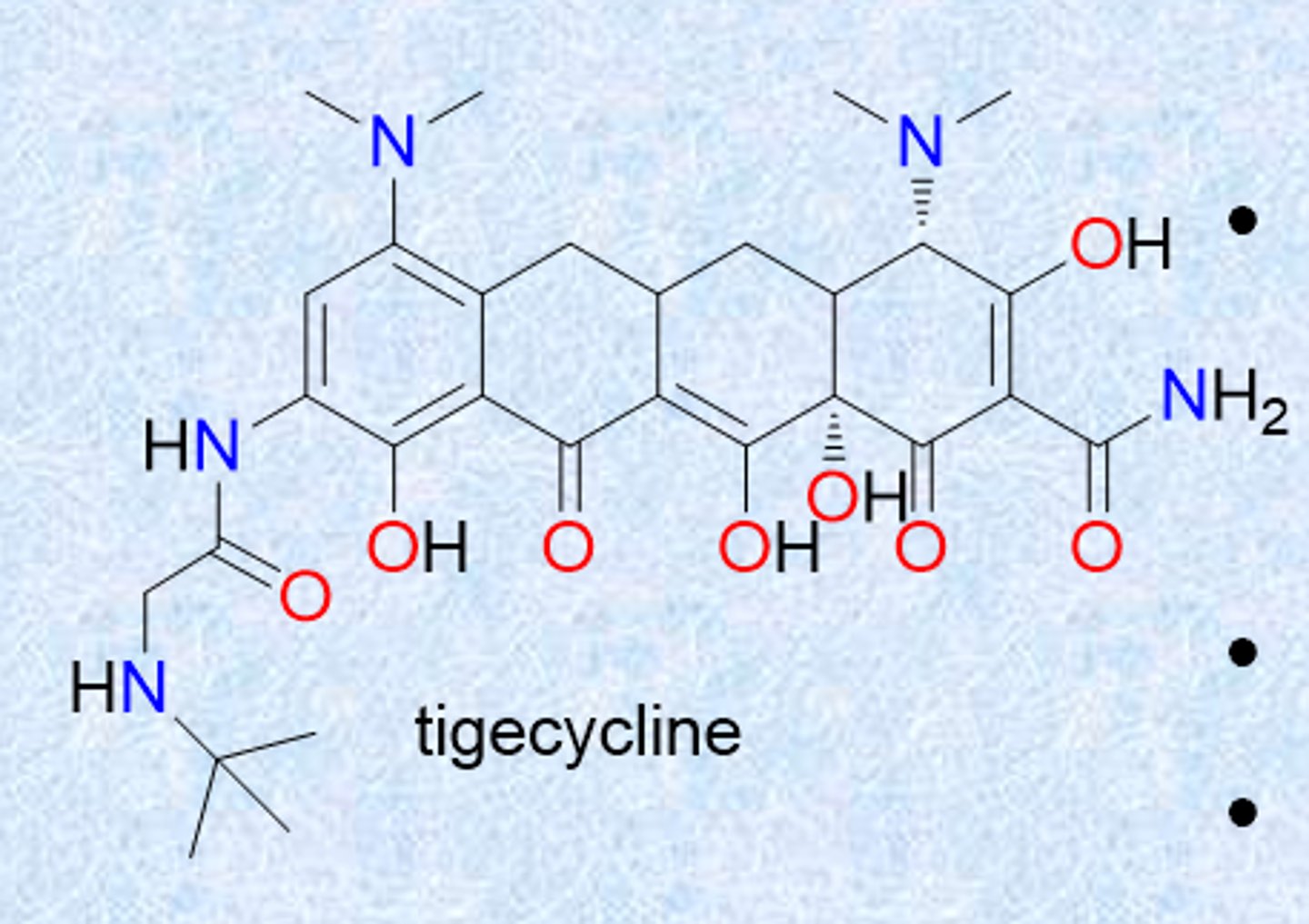
tigecycline (third generation)
- got rid of c 6-hydroxyl
- dimethylglycylamino substitution (group called glycylcycline)
- designed to be active against resistant strains (both Gram-+ and Gram-neg)
- charge helps with gram-neg permeability
- retains broad spectrum
- approved 2005
- 2010 Black Box warning-increase risk of death (cause unknown)
recently approved third generation tetracyclines
- modifications of tigecycline
- more positive charge
- omadacycline, eravacycline
macrolides
- metabolites isolated from soil microorganism, Streptomyces
- early work: isolation, later work: semi-synthetic derivatives to improve PK
- macrolide = big ester (ester linkage in large ring)
- large lactone ring, ketone group, glycosidically linked amino sugar
- lactone ring has 12, 14 or 16 atoms and is often unsaturated (large rings)
- floppy, amphipathic characteristic (all methyls on one side and all oxygens on other side - one binds to target and others face outside)
- dimethylamino group on sugar makes the macrolides bases that form salts (needed, does not work when it is taken away because weakens binding to ribosome)
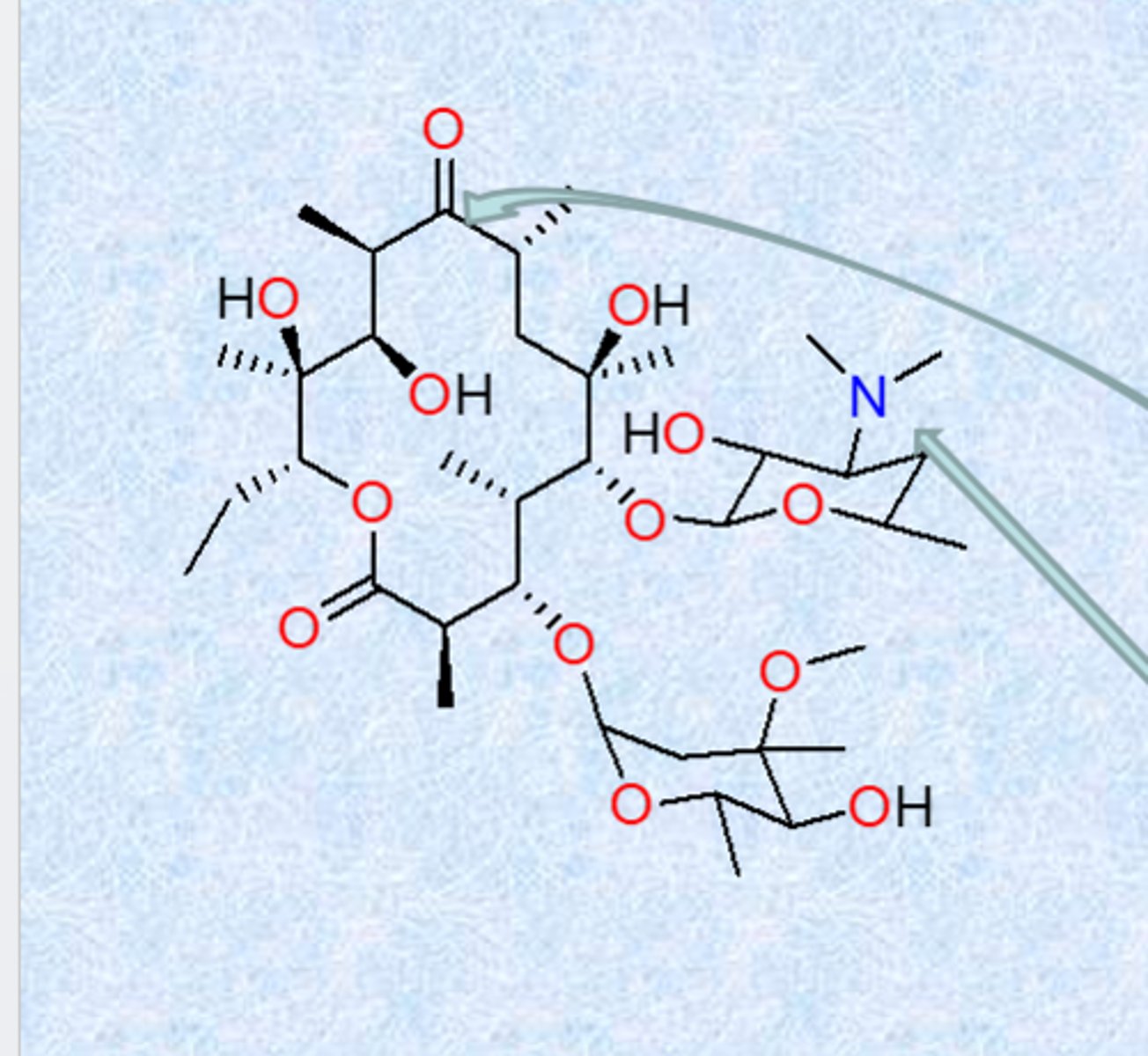
erythromycin
natural compound from bacteria
- 14-member macrocyclic lactone ring with a sugar and an aminosugar attached
- sugar residues critical for binding and activity
- erythromycin binds the 50S ribosome subunit, inhibits translocation
- unstable to stomach acids – because of ketone and alcohol groups (GI problems, poor pk, massive doses) - internal ring forming reaction (ketal formation and completely inactive but impacts GI motility)
- next generation protected the alcohol
- nitrogen is important, hydroxyl groups form strong contacts
- largely used for gram positive (no permeation, not good for gram negatives)
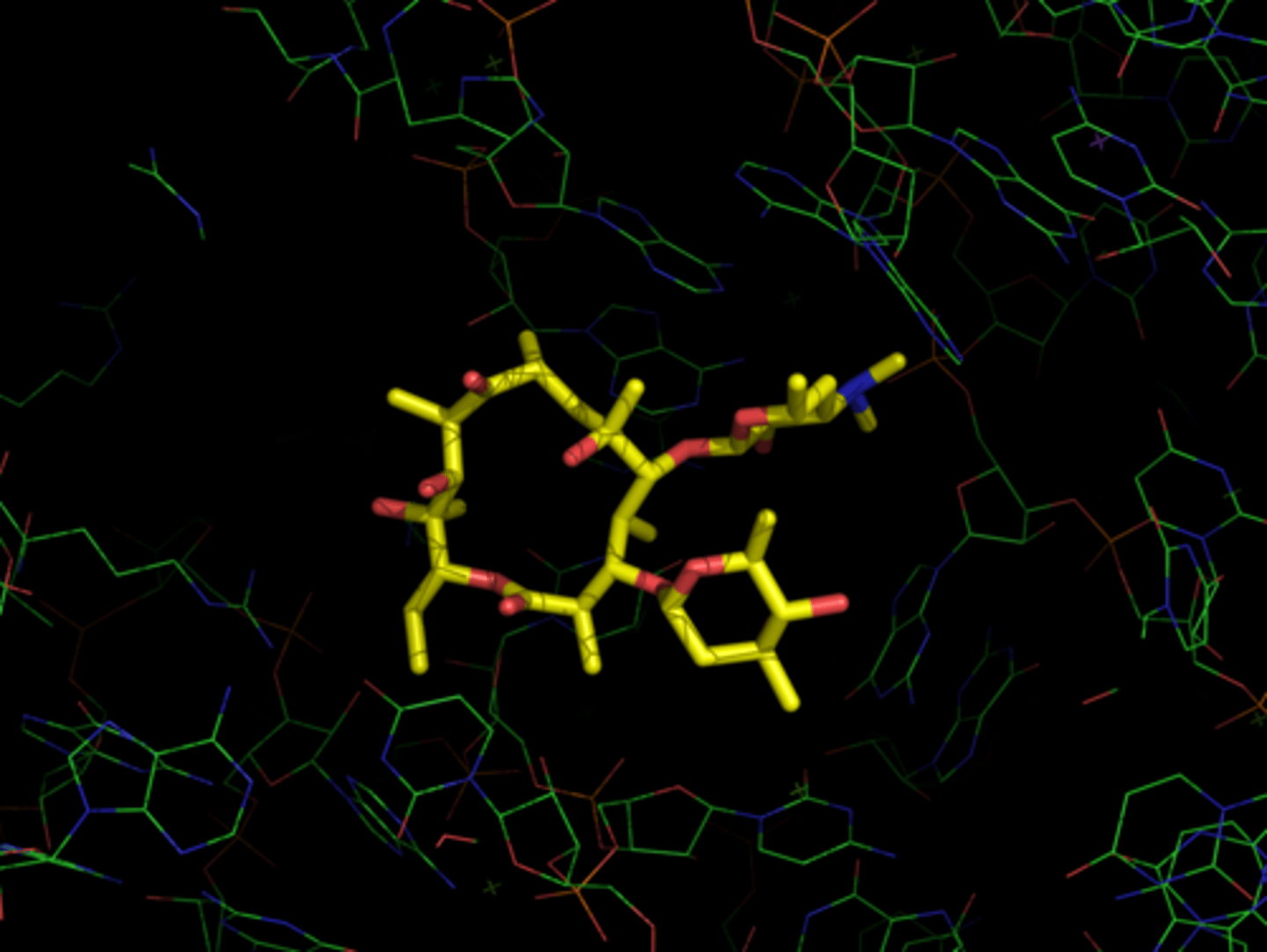
erythromycin/ribosome complex
- all oxygens point to one face and all methyls face back
- hydroxyls face cytosol, methyls into ribosome - stabilize
clarithromycin
- methylation of alcohol was first trick to block acetal formation
- commonly used (ex. strep throat)
- instead of OH becomes OCH3
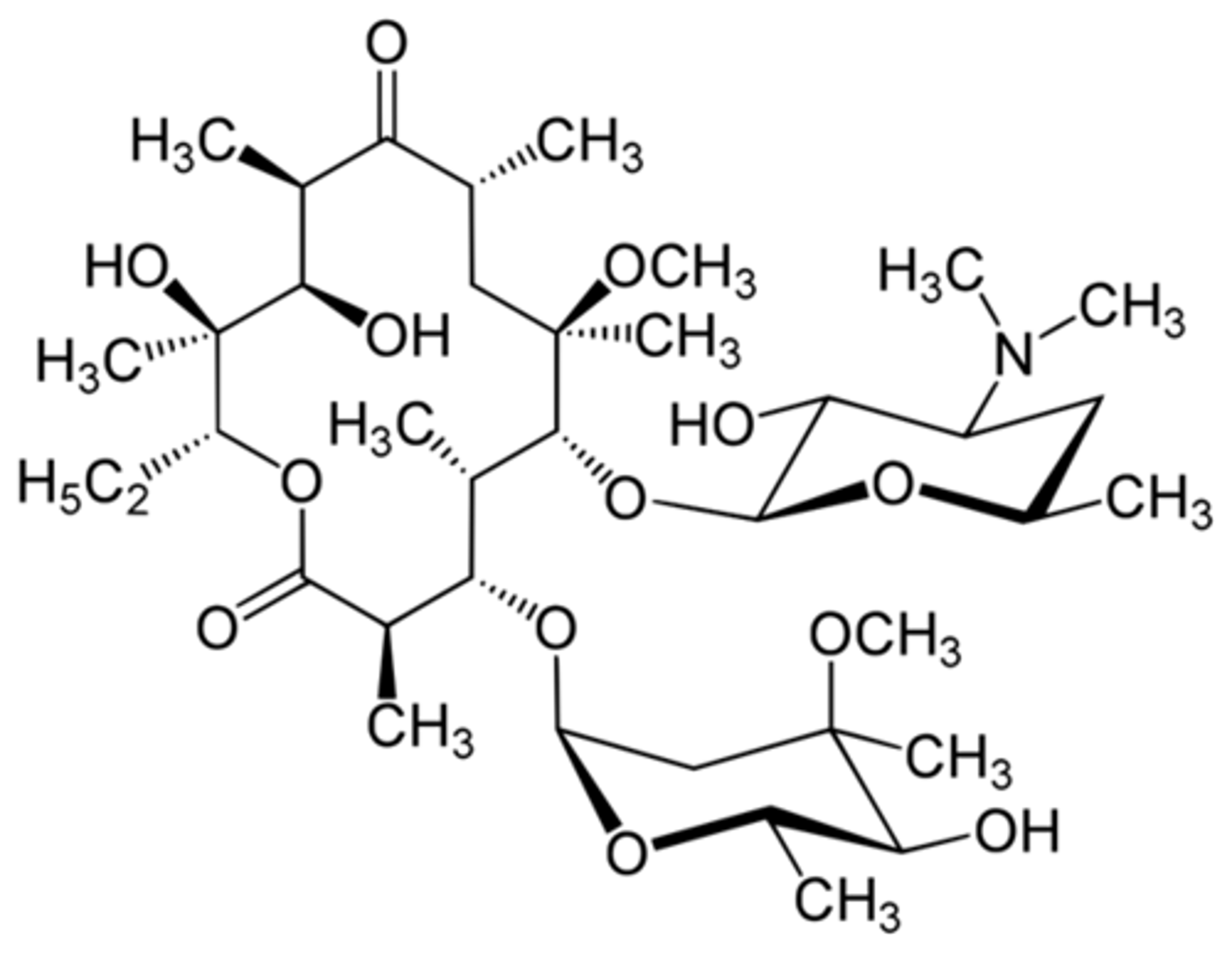
azithromycin
- one of the best-selling drugs
- 15-membered ring, N-methyl group incorporated (larger ring)
- carbonyl from erythromycin was not essential - removed carbonyl and inserts a nitrogen
- blocks reaction that destroys drug (smaller dose, better pk)
- more active against Gram-negative infections (incorporation of basic nitrogen, can become protonated and will help with permeation)
- PK: slow release from tissues
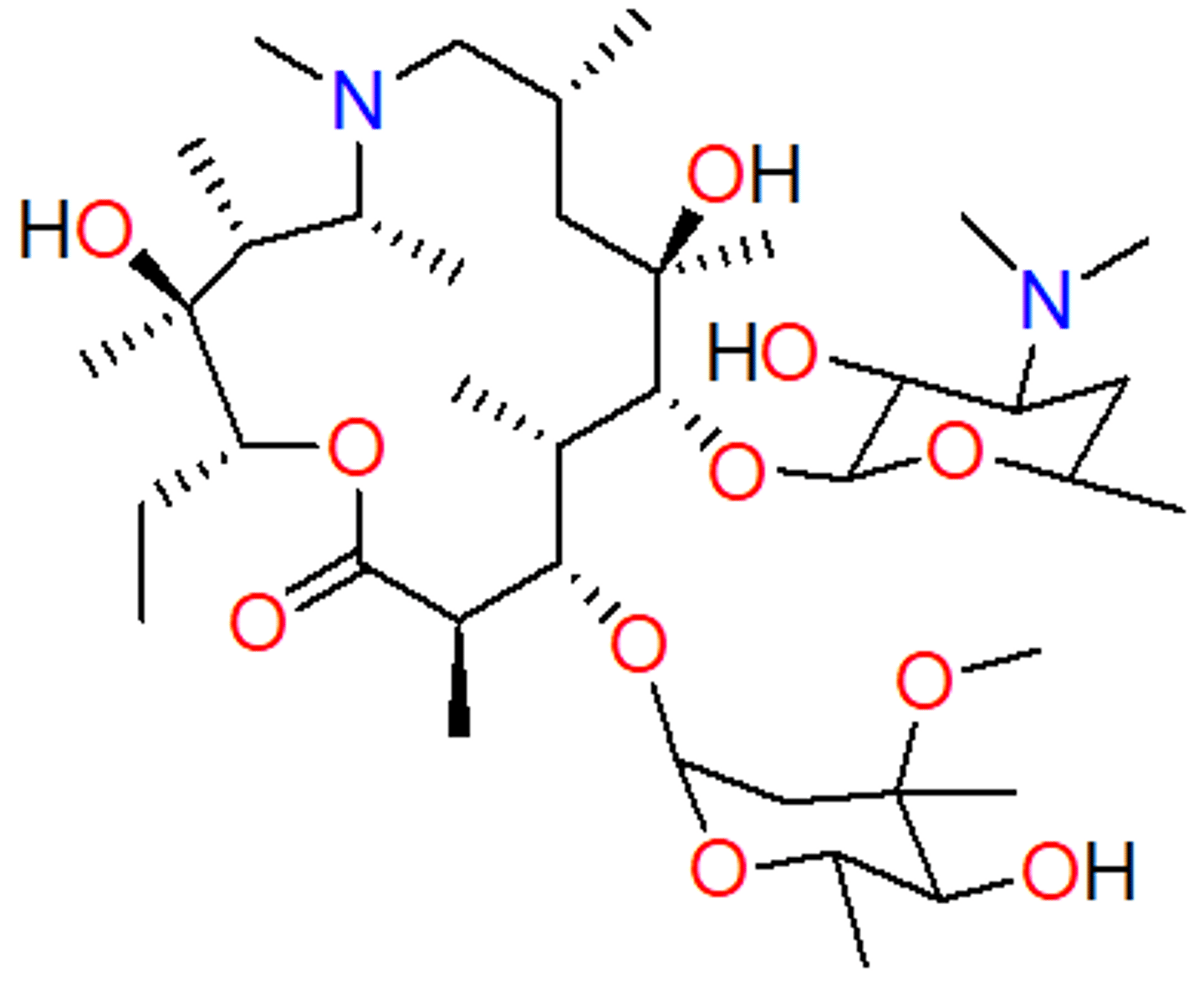
macrolide - resistance
MLSB – resistance to macrolides, lincosamides and streptogramin B (acronym) - modifying the target
– methylation of single adenine in 50S subunit - confers large resistance
- destabilizes binding to ribosome
– can be inducible or constitutive
– erm gene (erythromycin ribosomal methylase)
M – resistance to macrolides
– efflux pump
- mef gene (macrolide efflux pump)
- lowers concentration
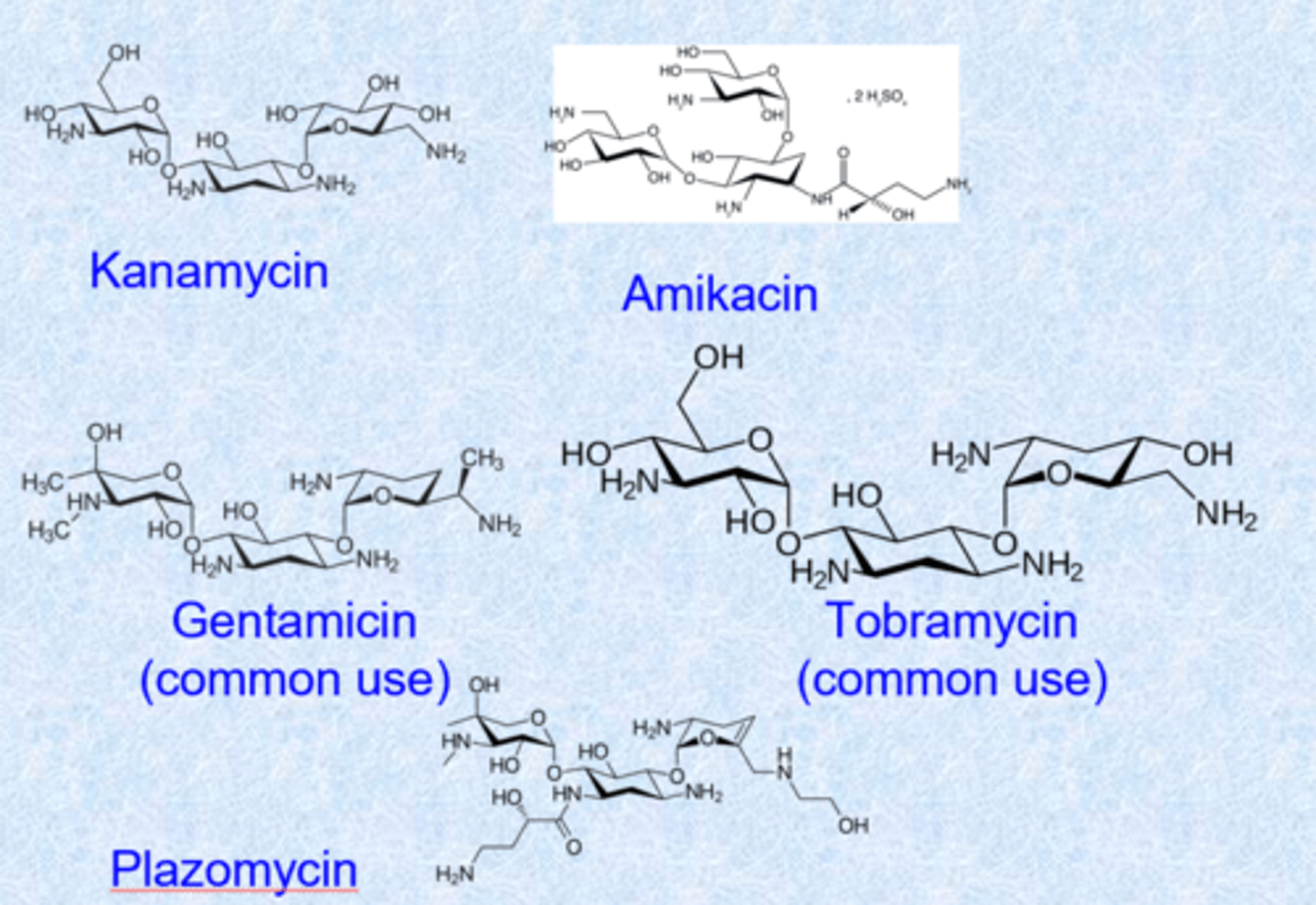
aminoglycosides
- natural products containing amino sugars (nitrogens on sugars)
- activity against Gram-negative pathogens - a lot of their use
- blocks peptide elongation at the 30S subunit of the ribosome (similar to macrolides)
- not absorbed well, given IV, IM or topical or drops (hard to give oral - too polar) - makes more highly concentrated solutions
- major side effect is hearing loss (ototoxicity - impact of mitochondrial protein synthesis) and renal toxicity
aminoglycosides - drugs
- complex shape
- 5 or 6 membered rings with oxygens = glycosides (sugars)
- glycosidic linkage but many basic nitrogens (NH2 becomes super highly positive charged, penetrate into gram negative bacteria)
- all basic nitrogens make this work (bind tightly to phosphates)
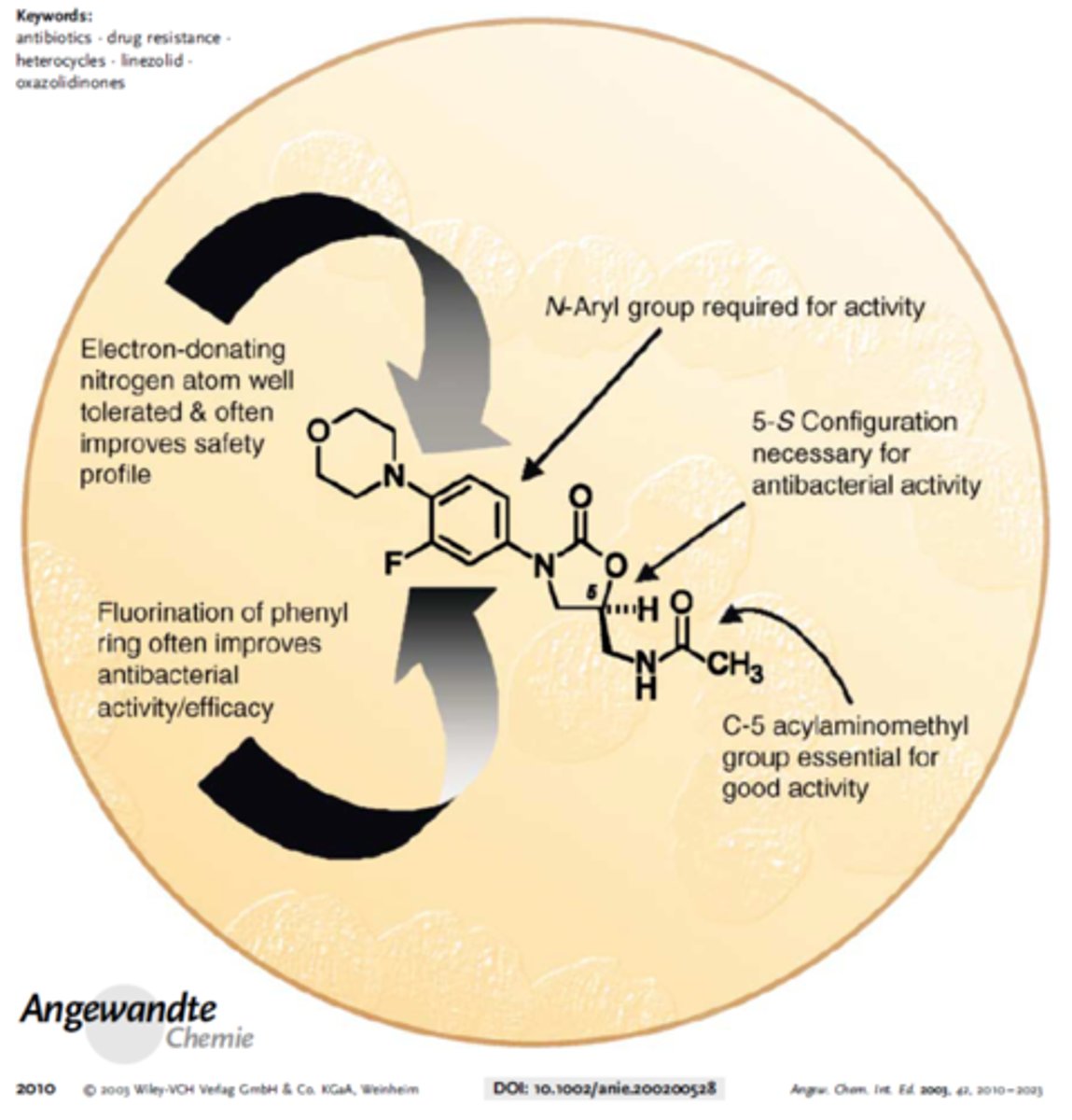
oxazolidinones
- fully synthetic
- oxazolidinones 5 membered ring with nitrogen and oxygen defines the structure (carbonyl = -one)
- linezolid
oxazolidinones - mechanism of action
- linezolid inhibited phage-specific in vitro translation, peptide chain termination and polypeptide chain elongation in a cell-free E. coli protein synthesis assay
- determined that it most likely binds 50S ribosomal subunit and prevents formation of a functional initiation complex
- other classes impact elongation
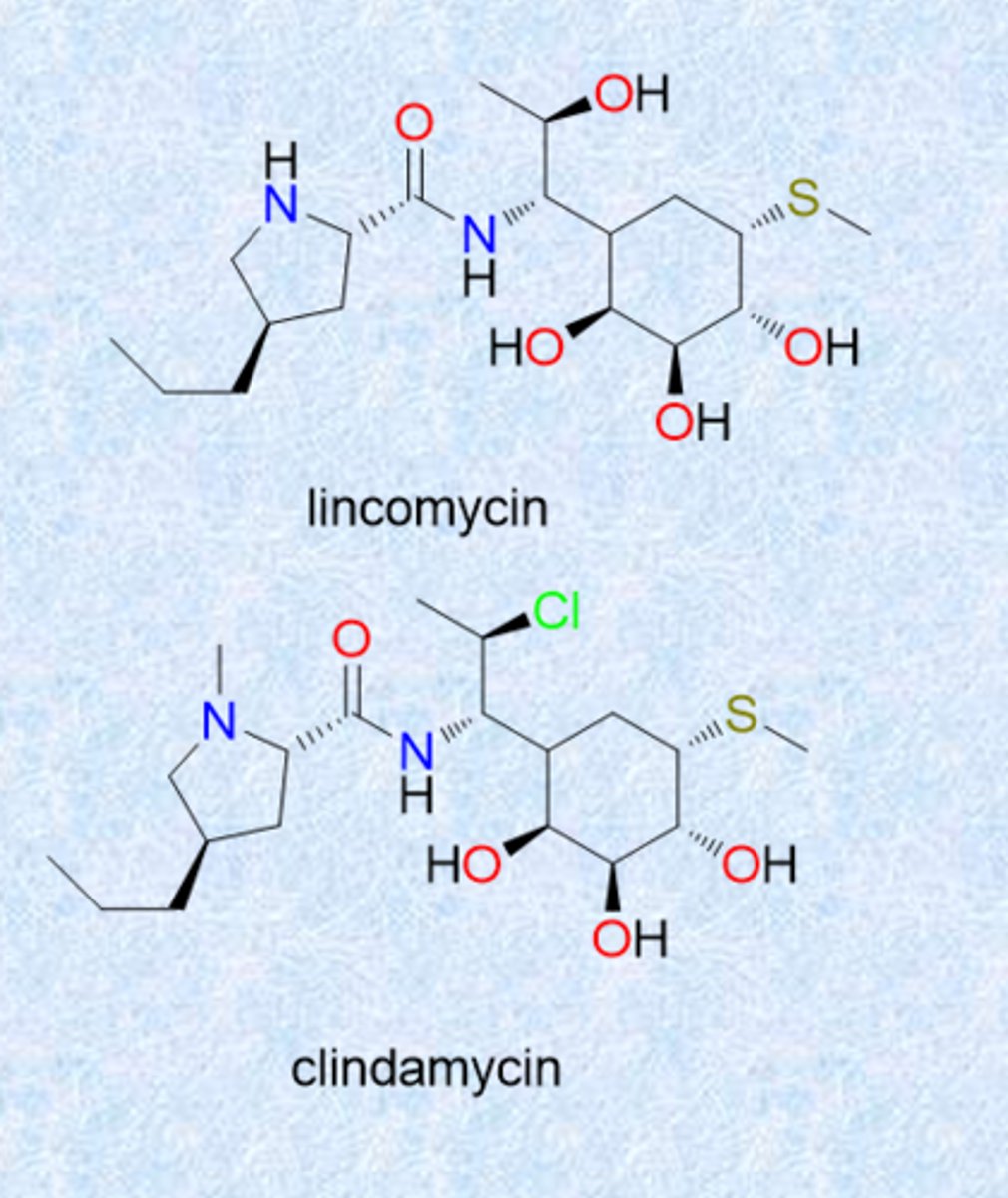
lincomycins
- isolated from Streptomyces
- lincomycin and clindamycin
- resemble macrolides in spectrum and MOA
- Cl in clinda is chloride (improved stability and activity)
- MOA similar to macrolides (bind ribosome and block elongation)
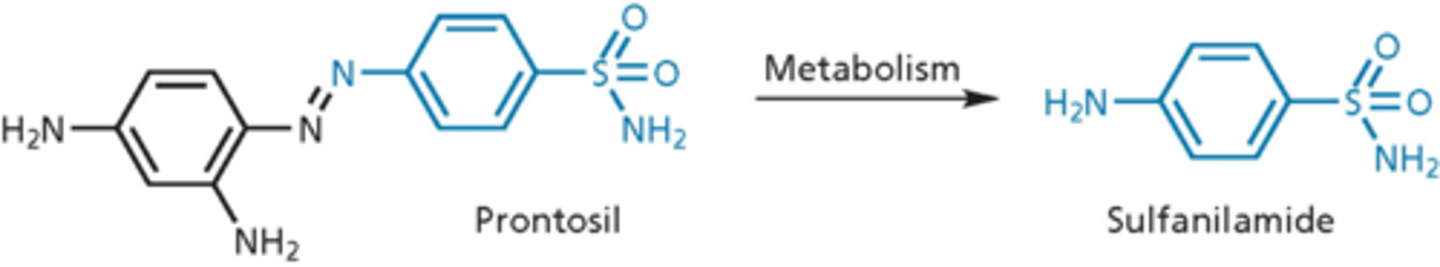
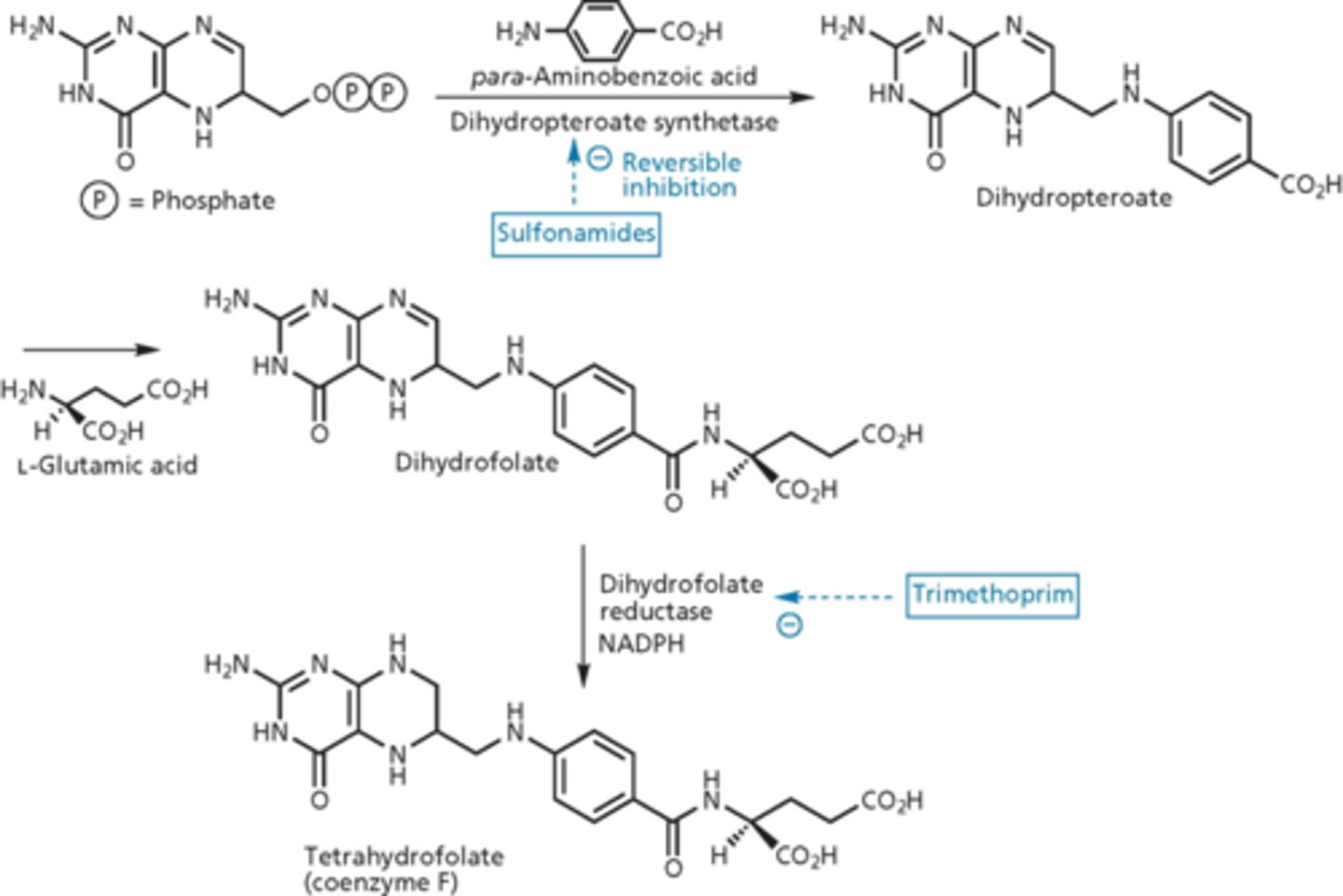
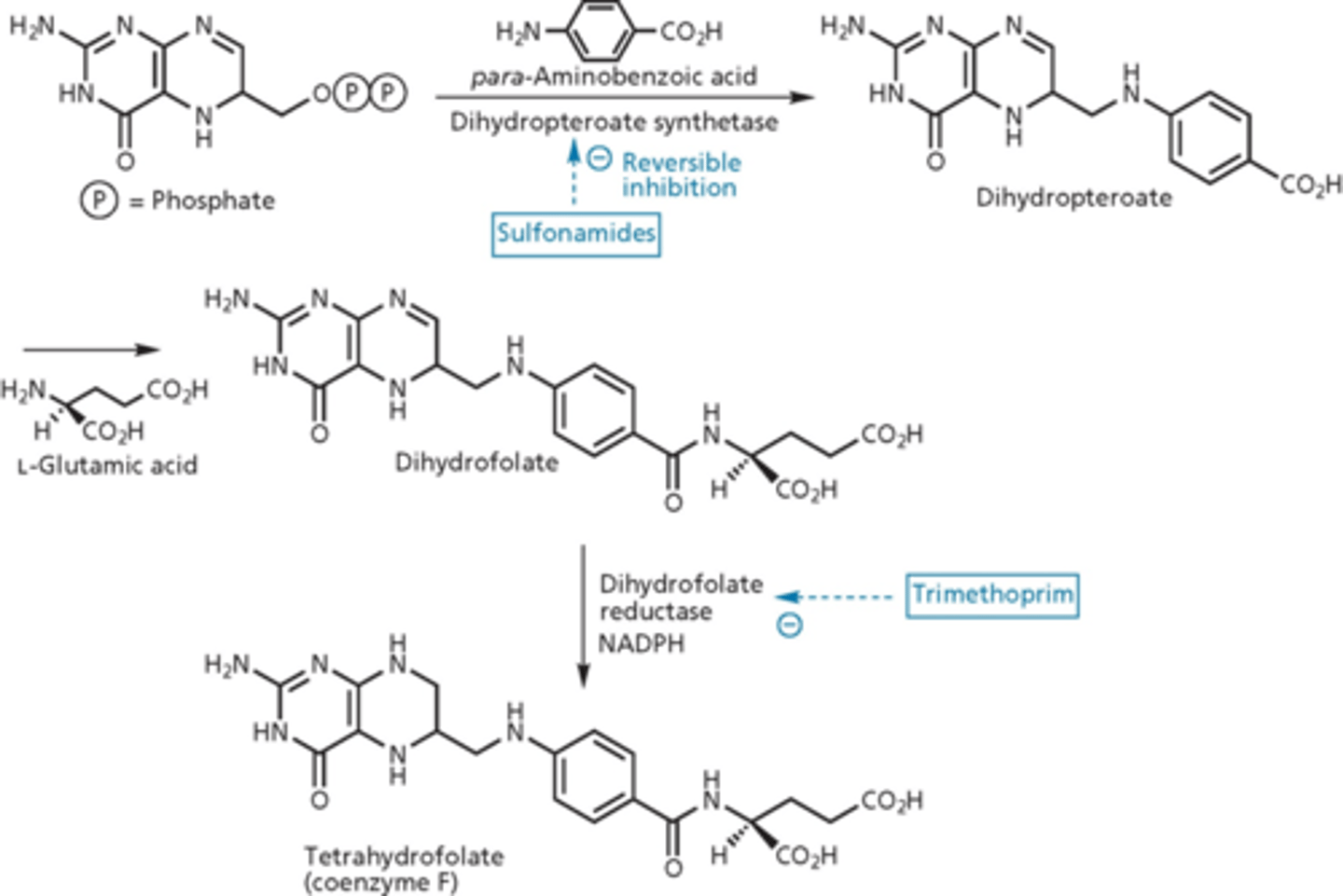
metabolic inhibitors: sulfonamides
blocked metabolic pathway needed for replication of nucleic acid of genome of bacteria
- Prontosil discovered to have antibiotic activity (1935) in vivo but not in vitro (2 nitrogens, from a dye)
- metabolized to sulfanilamide in the body
- Prontosil was a prodrug
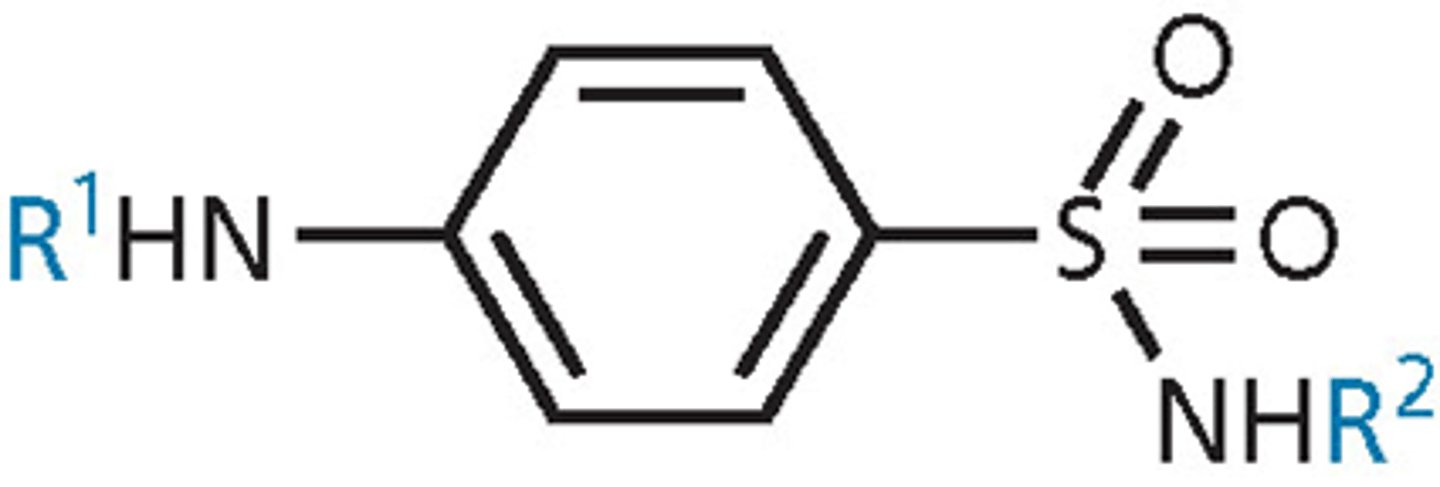
sulfonamide SAR
- para-amino group essential – must be unsubstituted (R1=H or acyl tolerated) - para orientation
- aromatic ring and sulfonamide functional group essential and must be directly attached
- aromatic ring must be para-substituted only
- R2 is the only possible site for variations - other than that, limited SAR
- varying R2 affects plasma protein binding and solubility
mechanism of action of sulfa drugs
blocked folic acid pathway
- tetrahydrofolate = vitamin
- prokaryotes need to make folate (de novo folate synthesis)
- para-aminobenzoic acid looks similar to sulfonamide
- dihydropteroate synthetase makes dihydropteroate (we do not do this, we get it from our diet)
- if you increase dihydropteroate synthetase (target), it increases effectiveness of the drug
- suicide reaction, sulfonamide acts as a fake substrate (dead end product)
- over expression of DHPS - exhausts substrates of folate synthesis
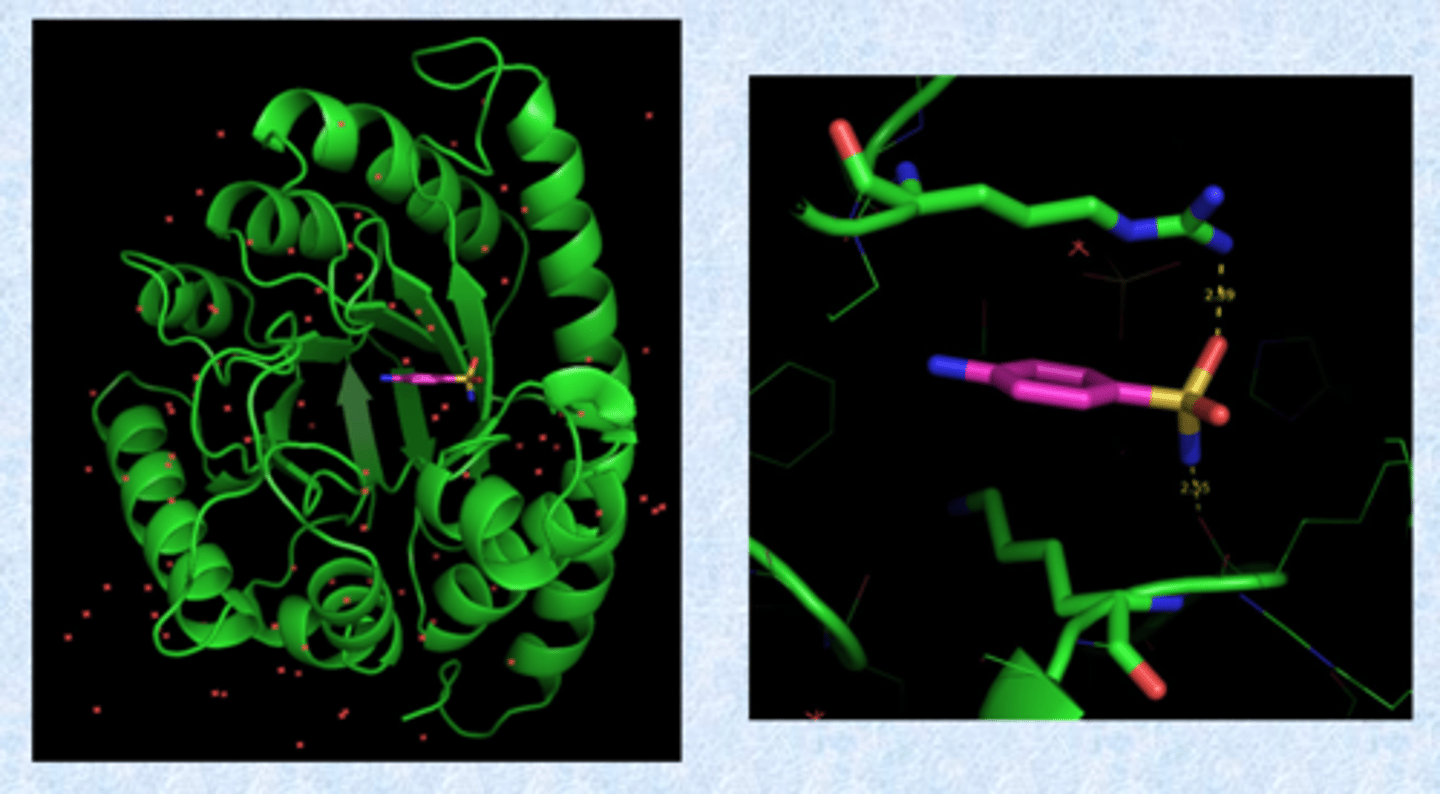
crystal structure of DHPS bound to sulfanilamide
- pseudo substrate for that site
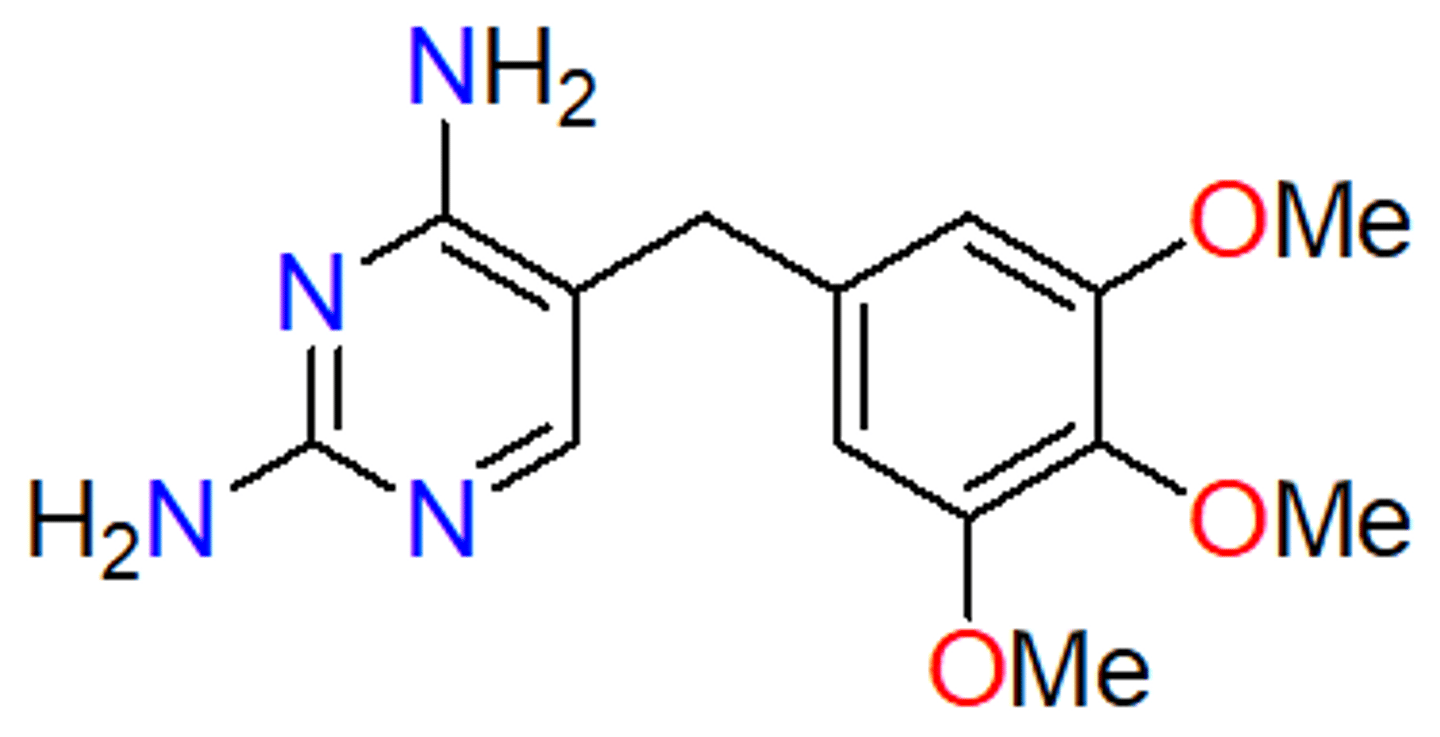
trimethoprim
- inhibits dihydrofolate reductase (folate biosynthesis) - no bonds, traditional competitive inhibitor
- antifolate (similar to sulfonamides)
- selective for bacterial DHFR
- bacteriostatic to bactericidal (kills bacteria, more powerful)
- sulfa and trimethoprim static agents used alone - cidal when used together (synergistic combination) - resistance pops up more slowly but still present
- specific for prokaryotic enzymes
- reflect small metabolites in the cell
- binds more strongly than folate, but cannot be reduced
- no activity against our enzyme
- good against E. coli and Staph aureus - UTIs --> always used as a combination (Bactrim)
dihydrofolate vs. tetrahydrofolate
- extra hydrogen on nitrogen
- reducing agent with NADPH = dihydrofolate reductase (adds hydrogen)
- target of trimethoprim
- accumulate more dihydrofolate and cannot reduce it = shuts the cell down
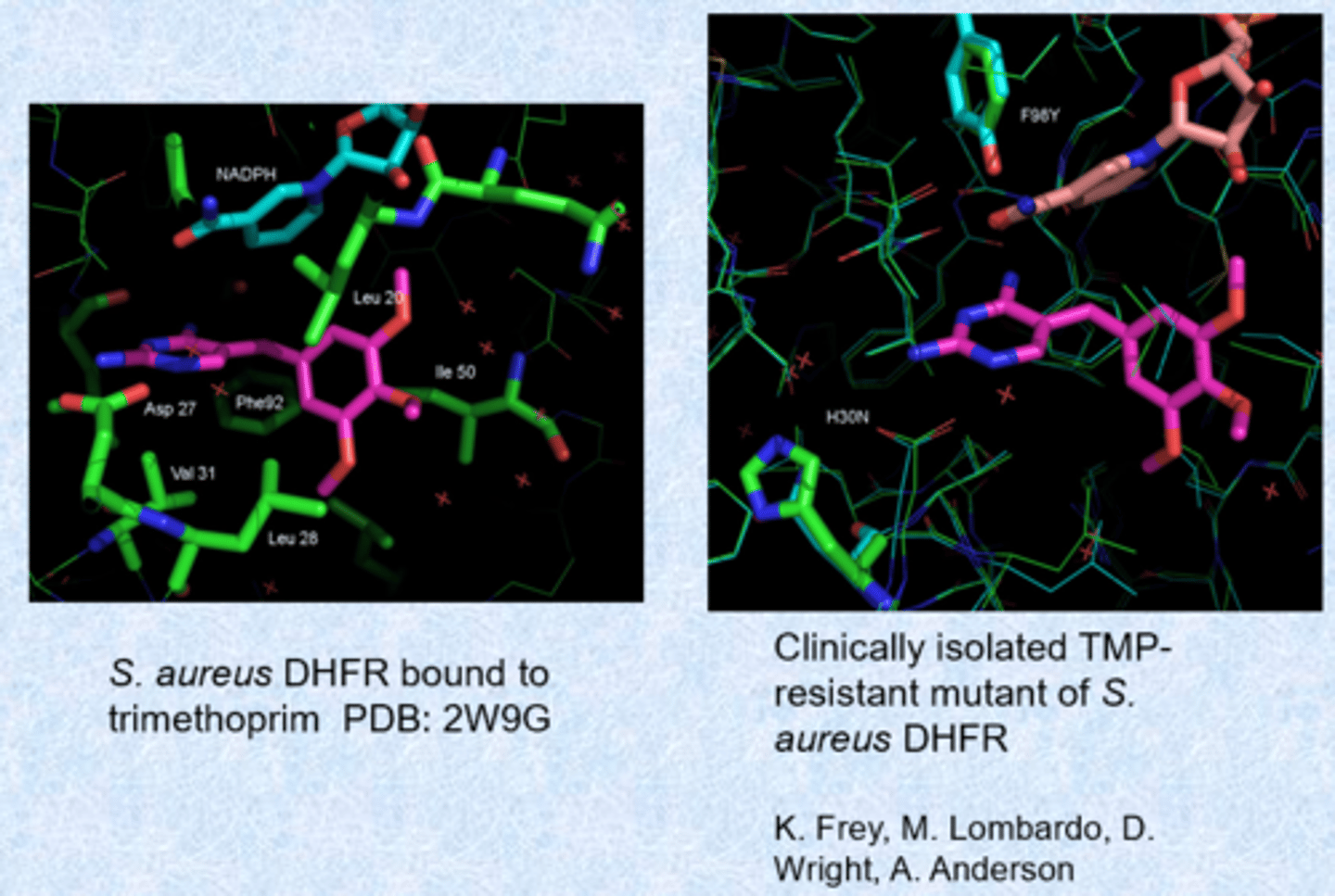
wild-type binding and a mutant
- changes in the enzyme active site for resistance - chromosomal mutations
- other dihydrofolate reductases and DHPS - bypass mechanisms that are resistant to the drug
- extensive resistance - may not longer be used
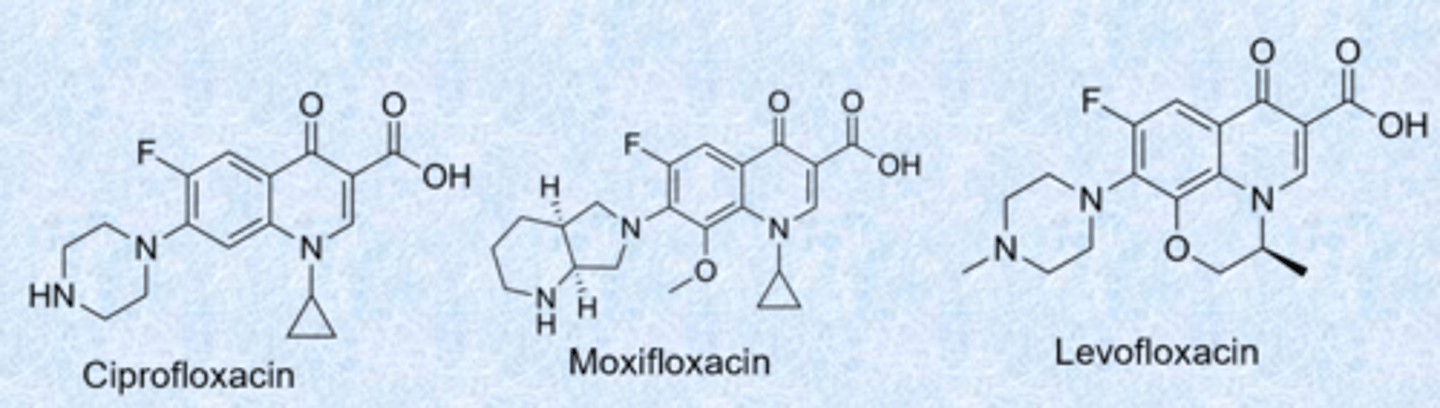
fluoroquinolones
- nalidixic acid was the first quinolone (synthesized 1962)
- quinolone ring (benzene, 6 membered ring, carbonyl) - fluorine always at that position
- Ciprofloxacin, Levofloxacin and Moxifloxacin most commonly used today - introduced a lot of basic nitrogen for gram-neg activity
- 1,3 dioxygen - similar to the tetracyclines, really good at binding metals (bind to target through Mg2+)
- cyclopropyl group further increased spectrum (also active against Gram-neg) - a lot of gram-pos activity
- used less because of side effects
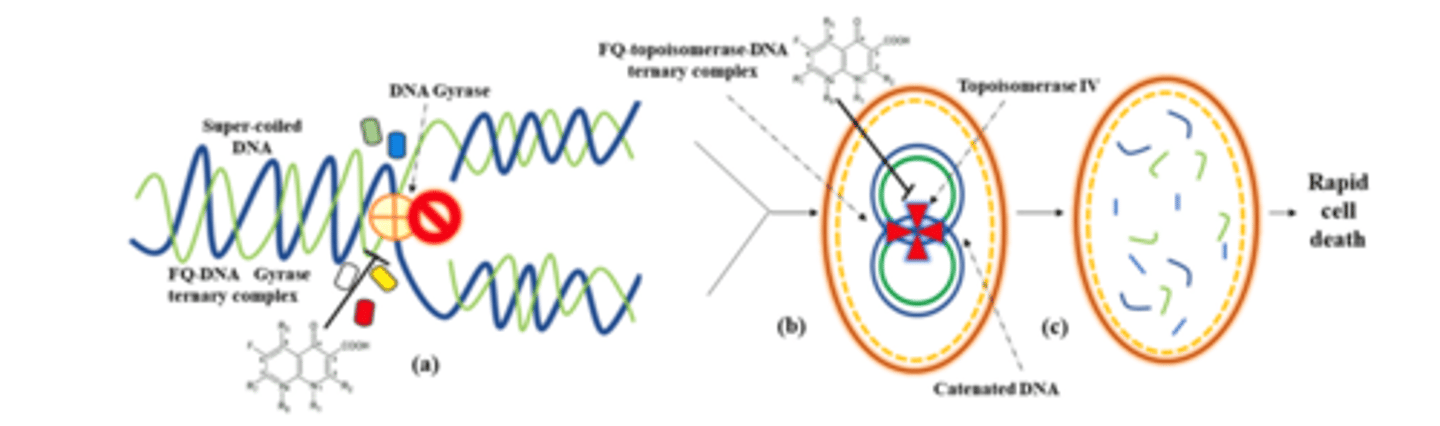
fluoroquinolones MOA - revisited
- inhibit replication and transcription of bacterial DNA by stabilizing complex with DNA and topoisomerases (topoisomerases and gyrases relieve strand strain) - same pathway in bacteria and us but enough differences that we do not get cross reaction
- gram-pos bacteria: DNA and topoisomerase IV
- gram-neg bacteria: DNA and gyrase (required when helix is supercoiled after replication and transcription)
- cleave DNA, allow to unwind, the reattach it back together
- incompatible with antacids and any foods or supplements with Ca2+, Mg2+, Zn2+, Fe2+, Fe3+, Bi3+ - similar to tetracyclines
- resistance = mutations to gyrases, changes to drug uptake and efflux
fluoroquinolone (FQ) MOA
- (a) A ternary FQ/DNA/DNA gyrase complex leads to slow death of the bacteria
- (b) Formation of an FQ/topo IV/DNA ternary complex leads to more rapid bacterial cell death
- fragmented DNA
fluoroquinolone - resistance
- altered porins (gram-neg) that decrease uptake
- energy-dependent efflux (usually always)
- amino acid substitutions in gyrase and topoisomerase - reduce overall drug affinity (highest level of resistance)
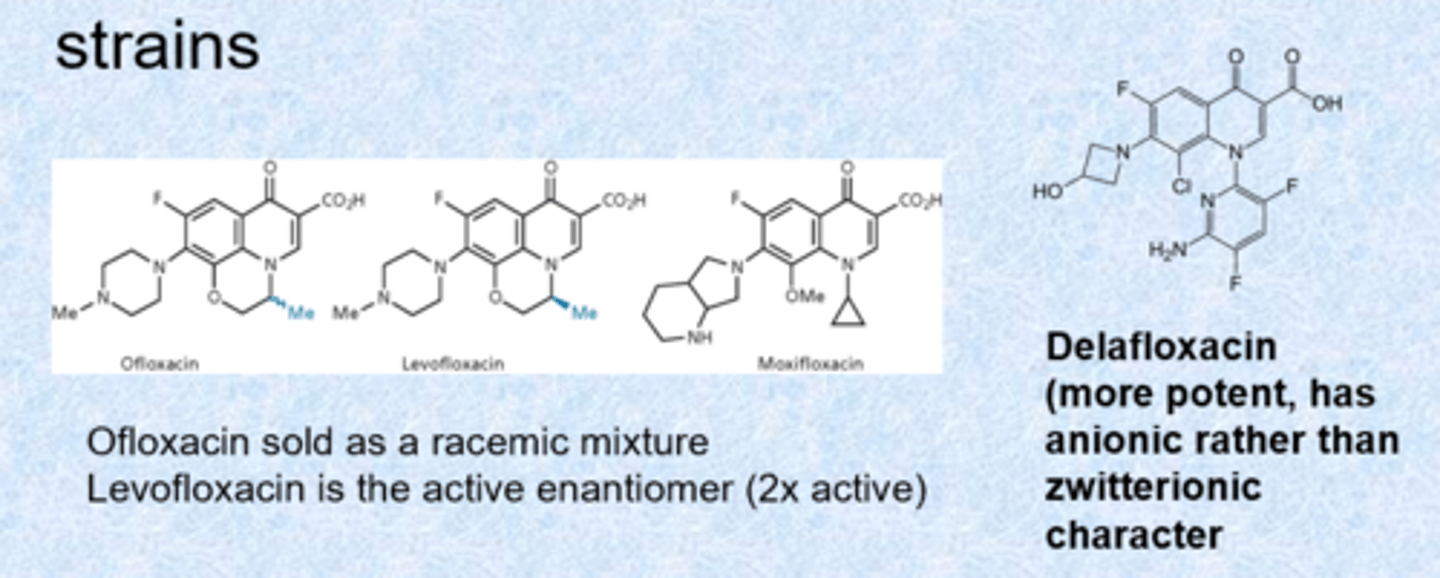
next generation fluoroquinolones
- always include a bicyclic pyridinone system and carboxylic acid at C3
- delafloxacin more potent for gram positive but loses a lot of gram negative activity (super activity, retained more in bacteria than outside)
- developed to show activity against S. aureus, S. pneumoniae and resistant strains
- aniline cannot be protonated - only aniline nitrogen on delafloxacin (negative charge)
- zwitterionic = overall neutral but very charged on either side (helps with transport through porins)
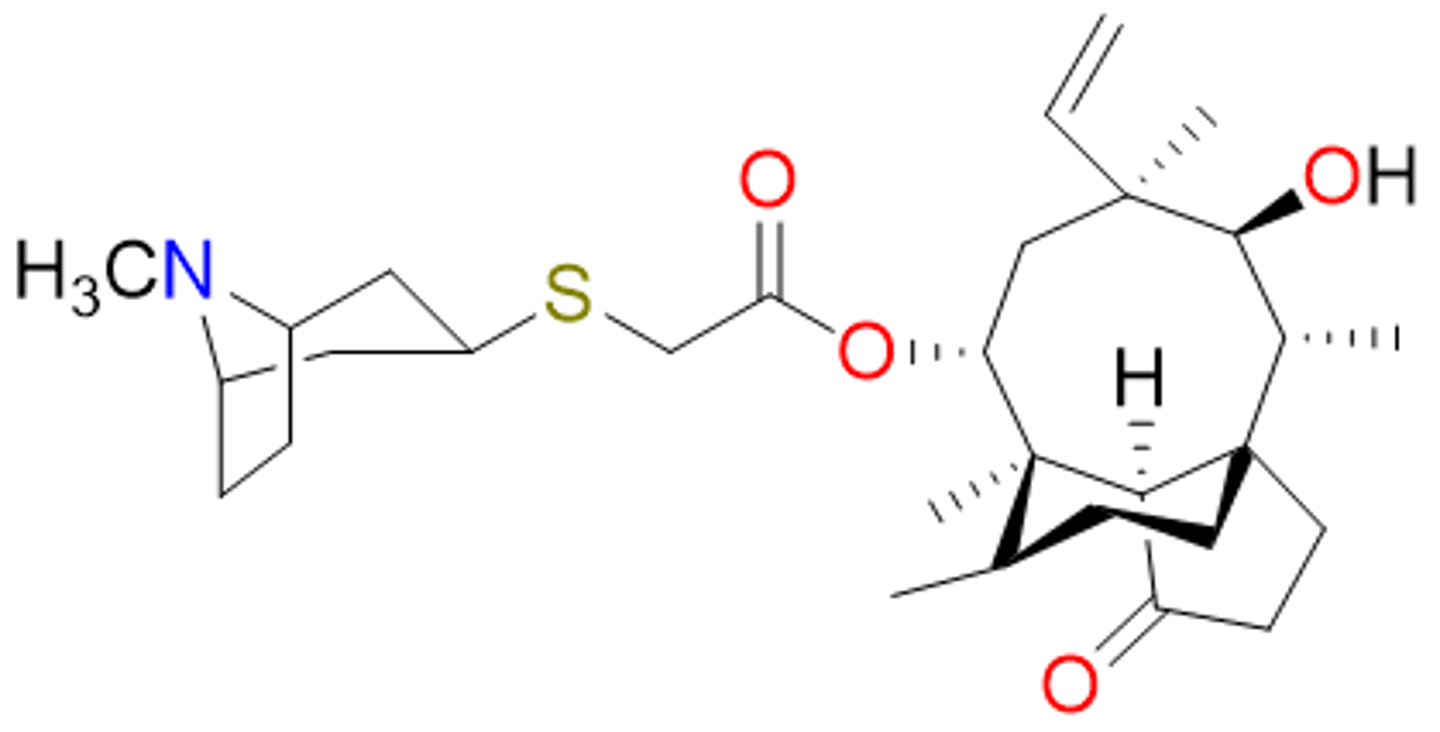
retapamulin (2007)
belongs with protein synthesis inhibitors
- first antibiotic from the pleuromutilin class
- semi-synthetic derivative from a mushroom
- topical; effective against Gram-pos
- binds 50S subunit, inhibits protein synthesis
- low resistance profile