GCC Exam II Lecture VII
1/34
Earn XP
Description and Tags
Lecture 9: Clock, codon usage, protein folding
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
35 Terms
overview paper
read through the purpose

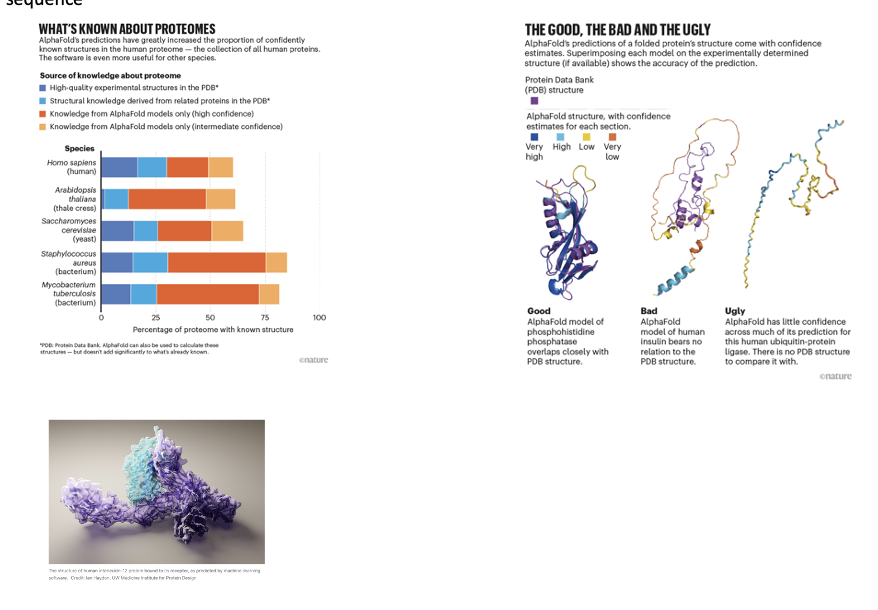
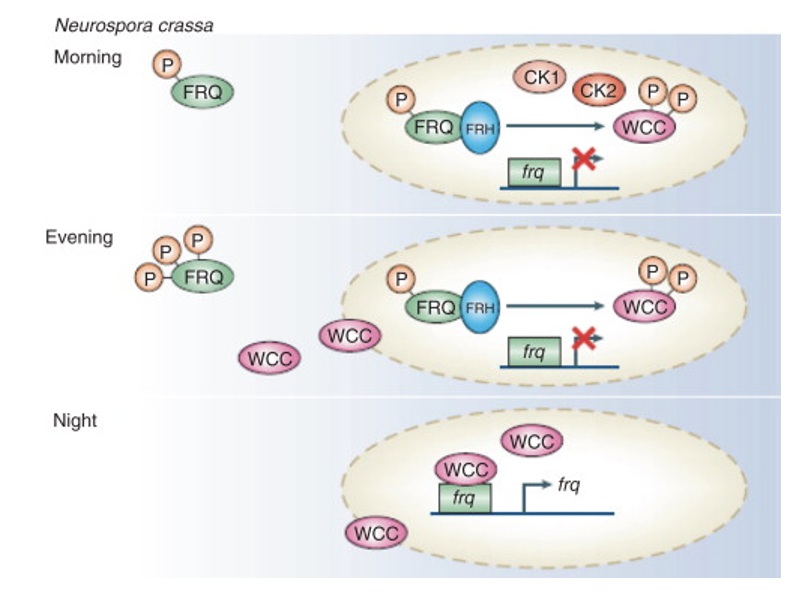
Neurospora crassa as a model system for studying circadian rhythms
Neurospora crassa undergoes an asexual cycle whereby it transitions through different morphological states as it grows.
As the mycel front grows it forms conidia (spores) that can be easily observed.
how N. Crassa models circadian rhythms
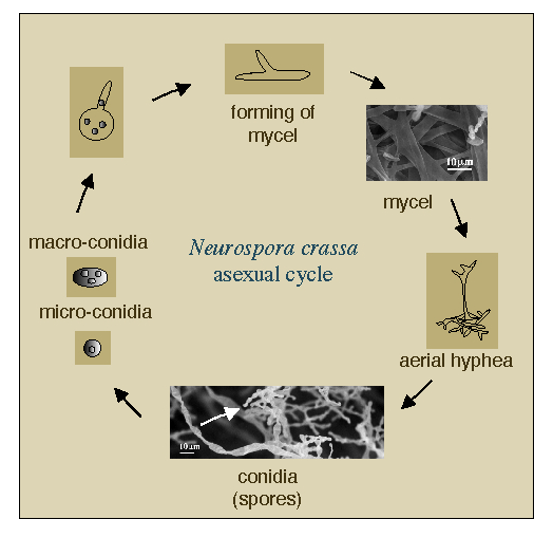
Conidia formation is regulated by the clock and occurs approximately every 22hr as the mycel front continues.
This can be easily observed in a “race-tube”.
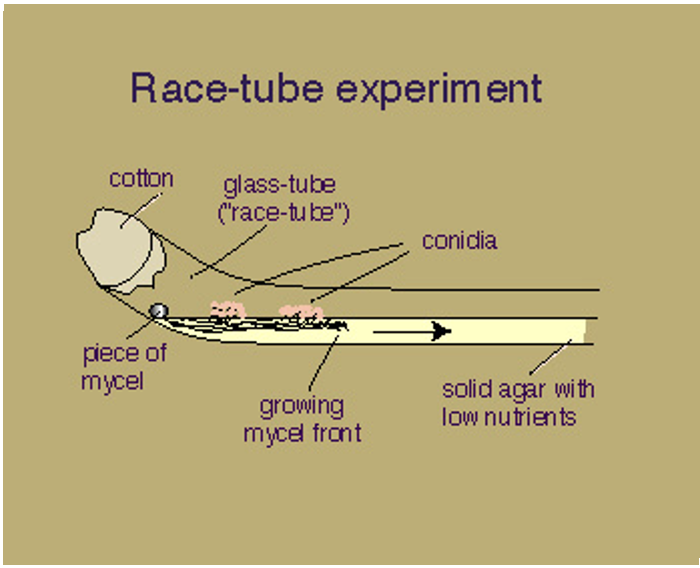
Frequency (frq),
first clock mutant in Neurospora
what does FRQ do in the Neurospora clock
functions in a similar but not identical manner to PER
phosphorylated in morning enters and does not create more
at night WCC attaches to frq allowing for transcription
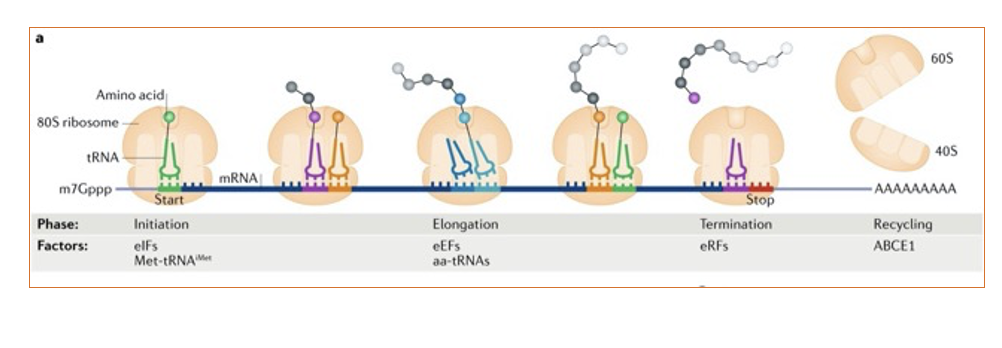
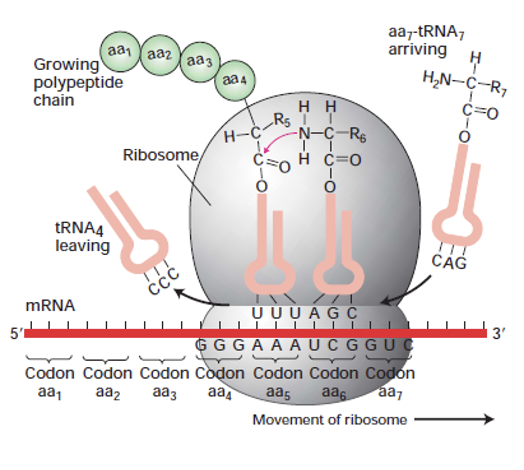
Background for the paper: Simplified view of Eukaryotic protein synthesis
translation simplified
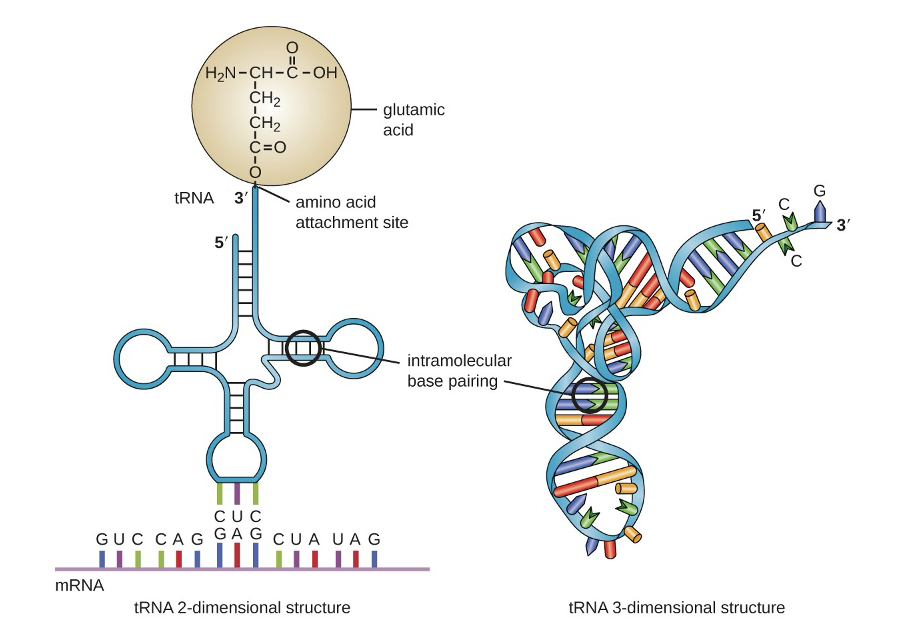
In the paper by Zhou et al they focused on tRNA
anti-codon/codon interactions with the mRNA
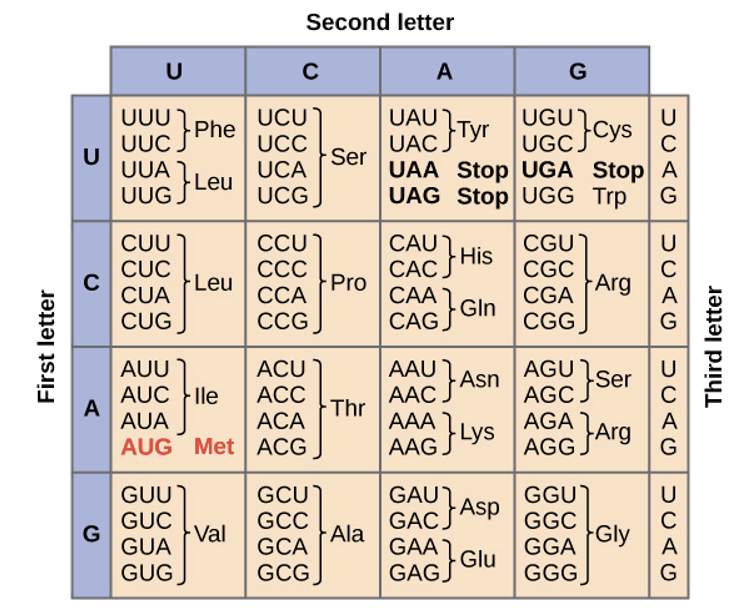
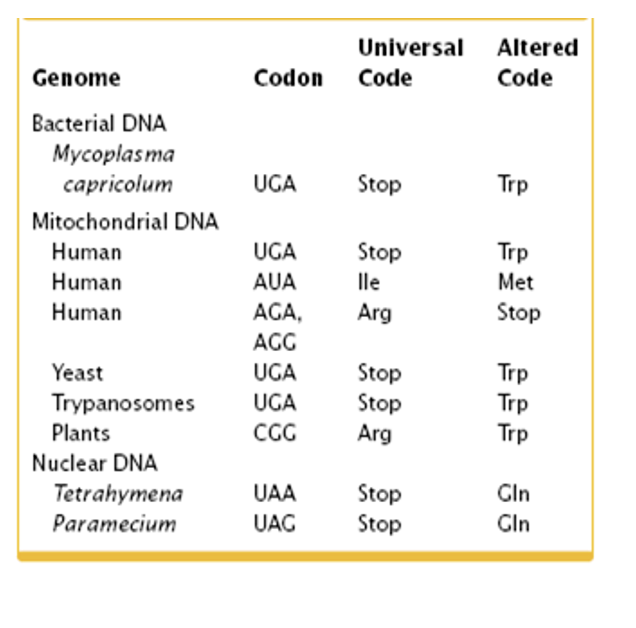
“almost” universal codon code
Note that the first two bases of a codon for a particular amino acid is highly conserved
more on the universal cofon code
differences in stop and other altered codon
The universal genetic code is a nearly uniform system where 64 codons map to specific amino acids or stop signals across almost all life. While largely conserved, altered codons exist, where specific codons (often UGA) are reassigned to different amino acids or function differently in organelles or specific species, primarily due to tRNA mutations or editing.
Degeneracy or redundancy of genetic code –
more than 1 codon for an aa
Quick history:
The idea of codons was first proposed by Crick and colleagues in 1961
Since a codon is three bases, there are 64 possible combinations of codons. Of the 64, 61 code for amino acids and 3 code for translation stop signals.
Since there are only 20 (actually 21) amino acids that means that many amino acids can be coded by several different codons, hence degeneracy or redundancy in the code (which helps to preserve the integrity of the code by being more resistant to single mutations.
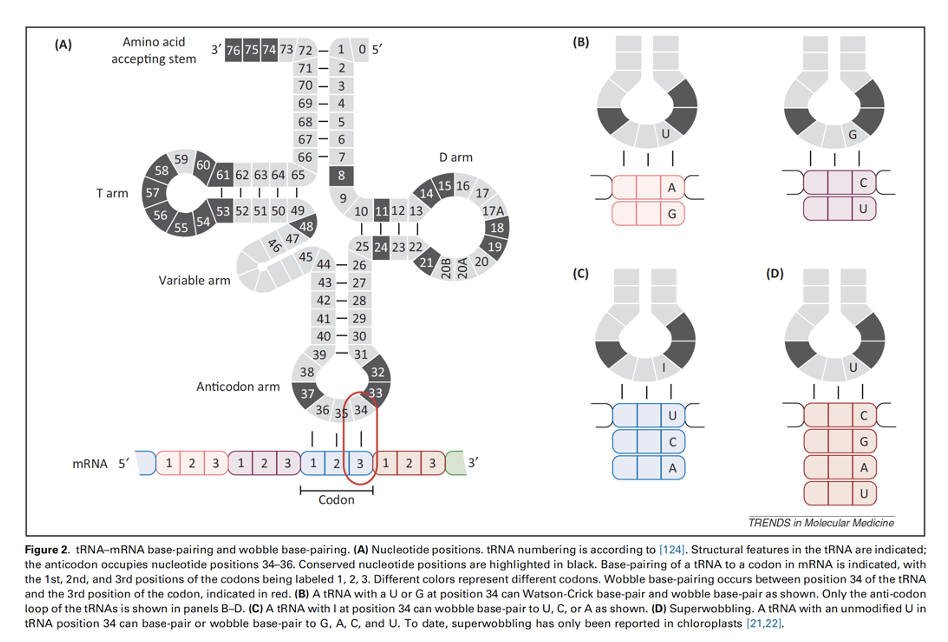
“wobble” hypothesis—total number of tRNAs is less than 61 (most organisms have ~45)
Wobble codon-anticodon interactions are up to 3-fold less efficient compared to those using only Watson-Crick interactions
The third base on the codon is less important than the first two and allows for non-Watson-Crick base-pairing (rules for wobble interactions are shown to right and sometimes involve an inosine (I) in the 1st position of the tRNA anticodon)
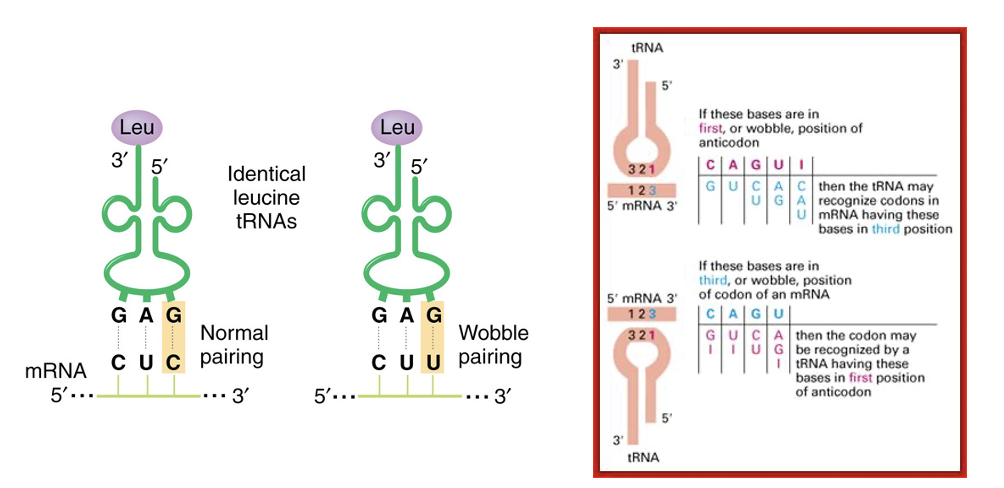
wobble hypothesis
better idea of why it doesn’t matter too much
explains how one tRNA anticodon can recognize multiple mRNA codons. While the first two codon bases pair strictly with the anticodon, the third base ("wobble position") pairs flexibly, allowing fewer tRNAs to translate all 61 amino-acid-coding codons, reducing the total tRNA required
The number of genes for each tRNA differs between species
If the expression patterns of different tRNAs reflect their gene copy number, then you can imagine that mRNAs containing many codons with low codon/tRNA ratios could have slower translation elongation rates compared to those with high.
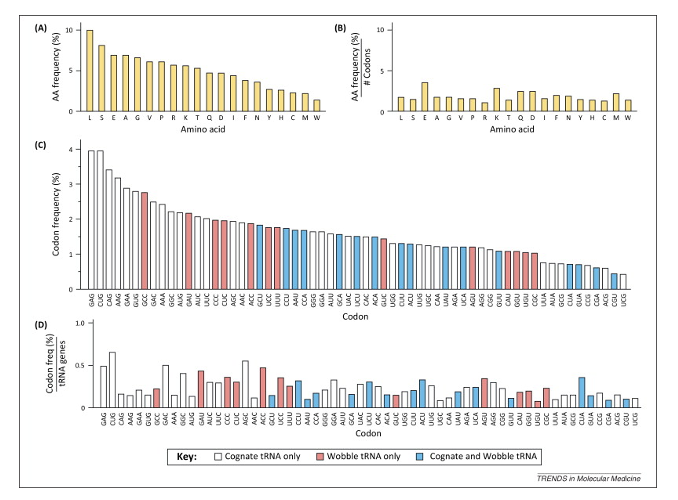
The predicted ratios for the human genome are shown below
Use for that AA means the relative frequency that a particular codon is used for an amino acid in the human genome
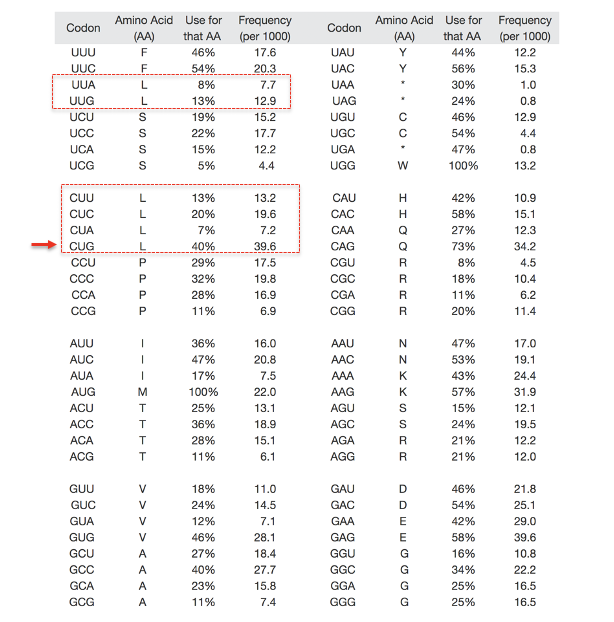
The abundance of tRNA differ depending on tissue, developmental stage, etc
An example for nuclear and mitochondrial encoded tRNAs is shown below for ovary and spleen
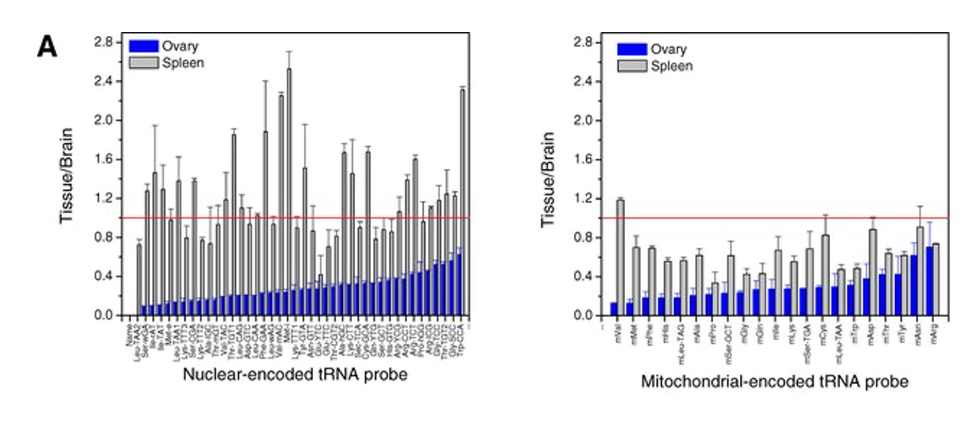
IN GENERAL: mRNAs with non-optimal codons are associated with
less protein production and higher mRNA degradation
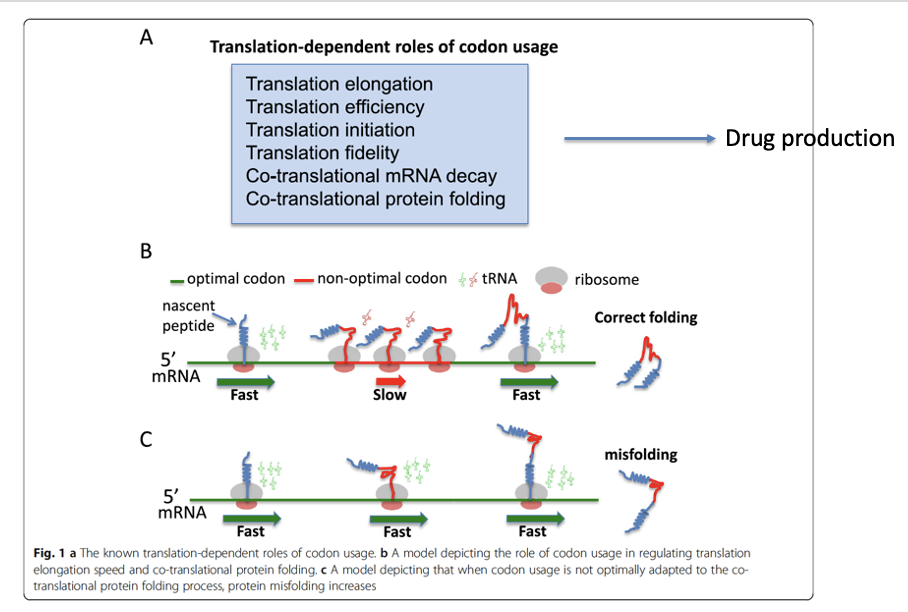
Proteins begin folding as they are being translated, which can affect protein folding
The average rate of protein synthesis is ~20 amino acids/s in E. coli and ~6 amino acids/s in eukaryotic cells.
In comparison, experimentally measured rates of spontaneous folding of single-domain globular proteins range from microseconds to hours.
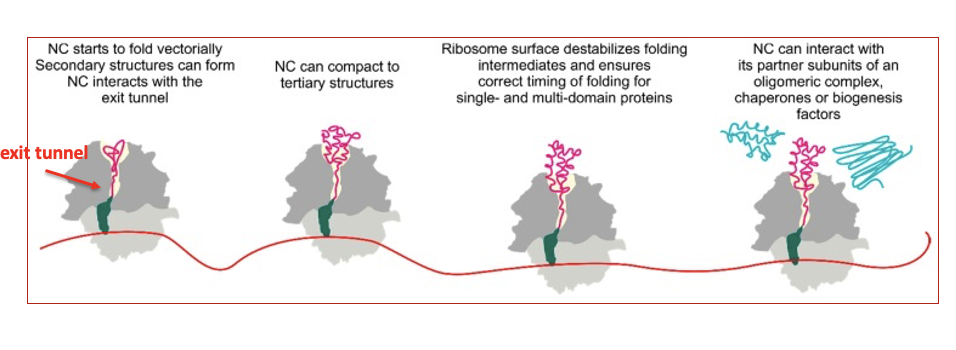
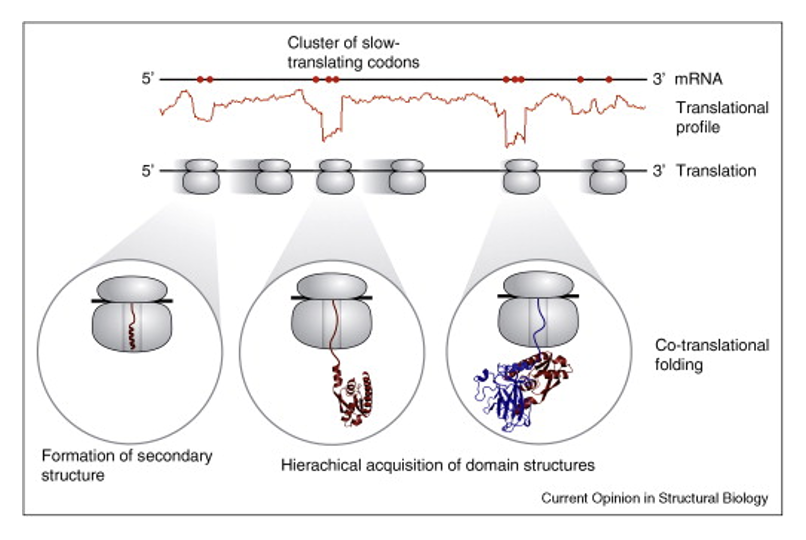
Slow translating codons might give enough time to stabilize a certain structure, whereas fast translating codons
might diminish the chances of a “bad” fold!
75% of proteins in eukaryotes are multi-domain. Folding domains sequentially as they protrude from the ribosome can ensure a correct overall structure
impetus for study - paper discussed
Codon usage optimization for luciferase in Neurospora
Noticed frq open reading frame predominately has non-optimal codons (see below; Supplemental figure 1 from paper).
This led them to wonder what would happen if they optimized the codon usage for FRQ and tested it for clock function in Neurospora.
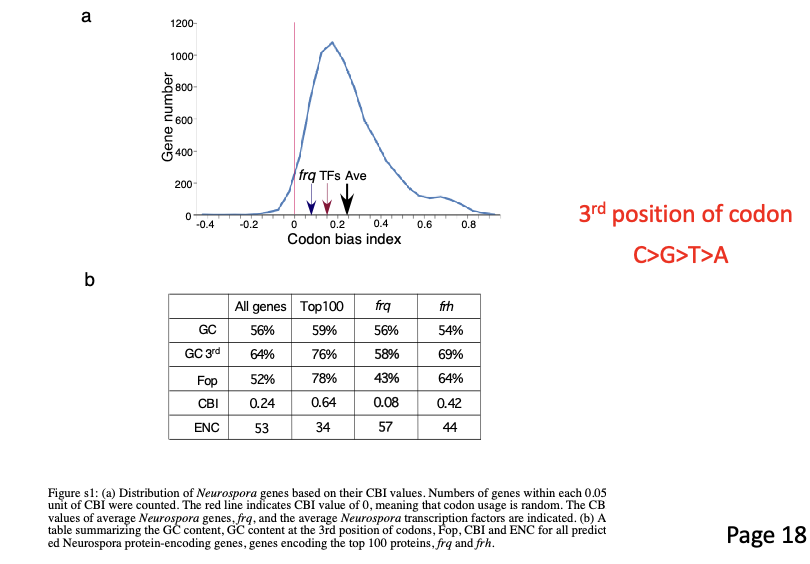
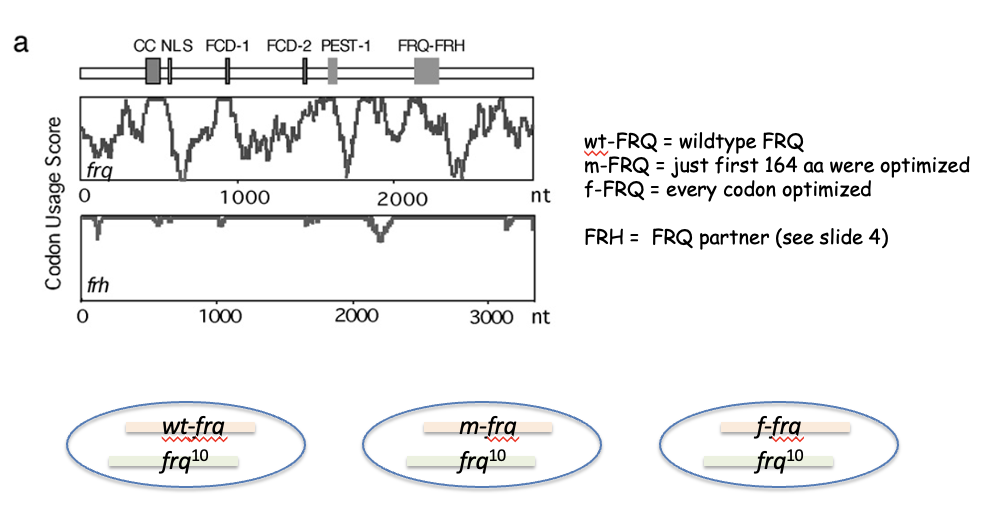
Figure 1A Testing codon optimization of frq
wt-FRQ = wildtype FRQ
m-FRQ = just first 164 aa were optimized
f-FRQ = every codon optimized
FRH = FRQ partner (see slide 4)
higher levels of FRQ protein from
reading frames with optimized codons
Overall frq mRNA levels not affected but circadian feedback is abolished/reduced
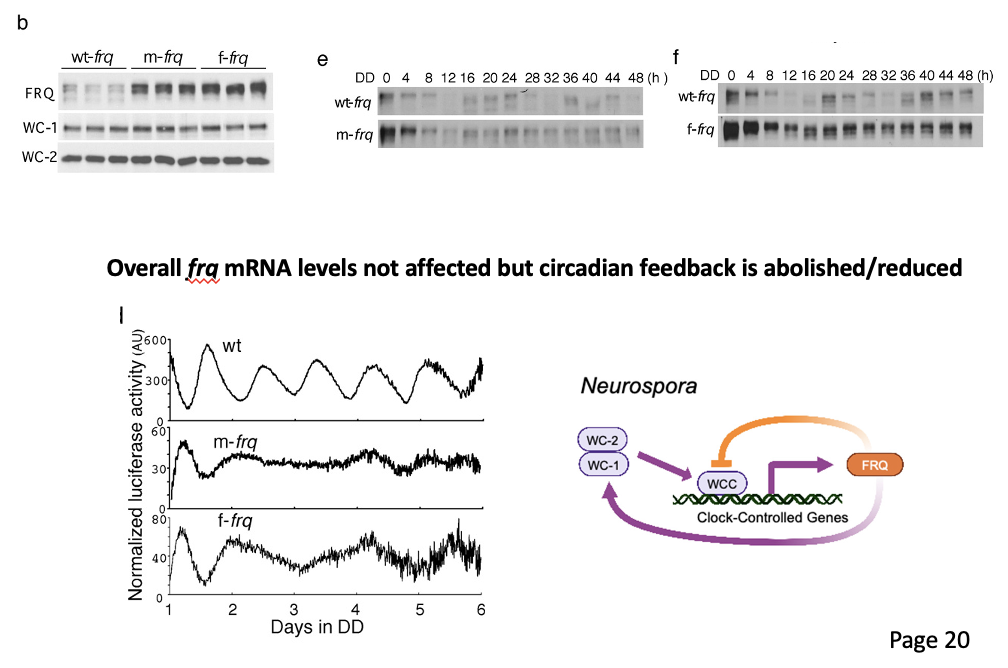
Altered rhythms in neurospora
with codon optimized frq

Fig 2: Codon optimized FRQ has impaired binding to WC complex
f-frq has much less bonding with WC2 with codon optimization
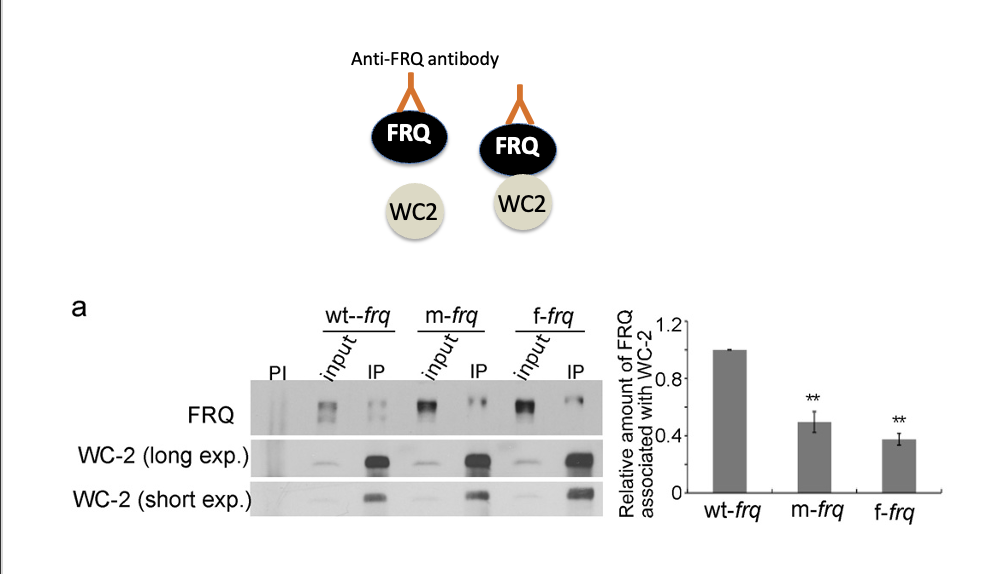
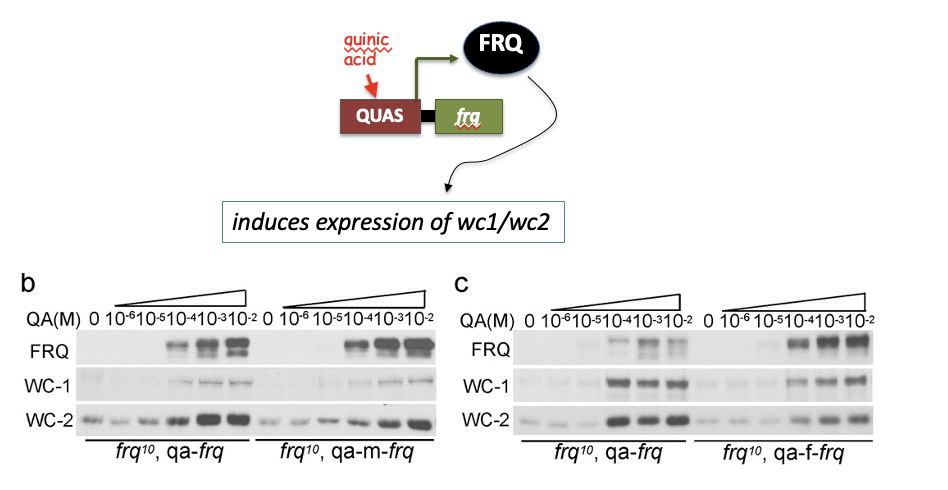
Fig 2: Codon optimized FRQ has impaired ability
to stimulate wc1/wc2 expression
The impaired FRQ functions despite higher FRQ levels in codon-optimized strains suggest that FRQ’s structural conformation is altered.
Codon optimized FRQ protein is less
stable compared to wildtype
CHX = cycloheximide
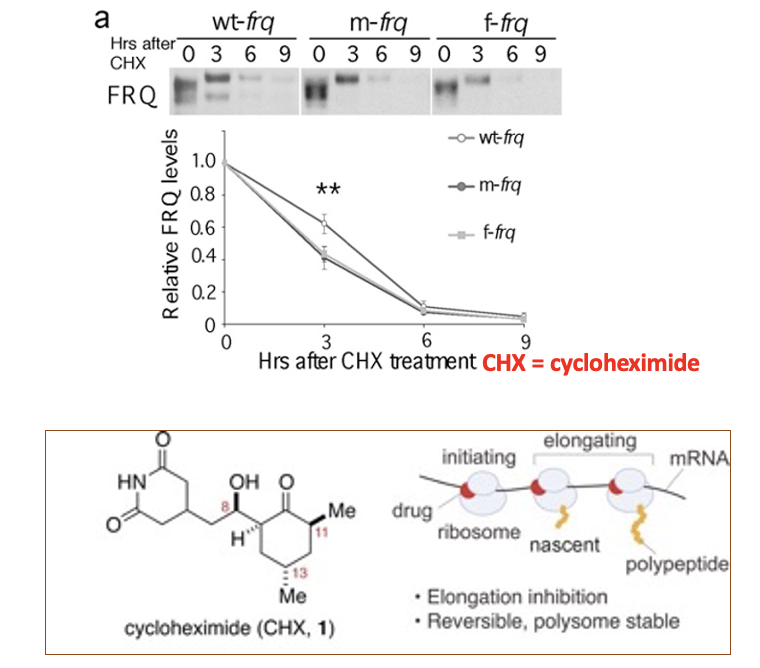
FRQ with optimized codons was more susceptible
to loss after multiple freeze-thaws
Fig 3b. FRQ protein in the codon-optimized strains is less
stable and more sensitive to trypsin digestion.
(b) Western blots showing sensitivity of FRQ from codon-optimized strains to freeze-thaw cycles.
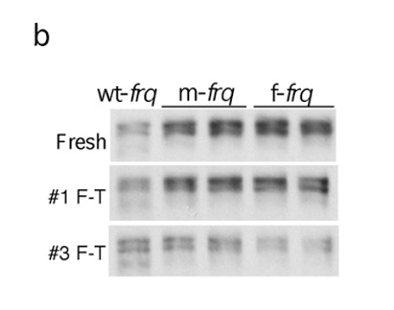
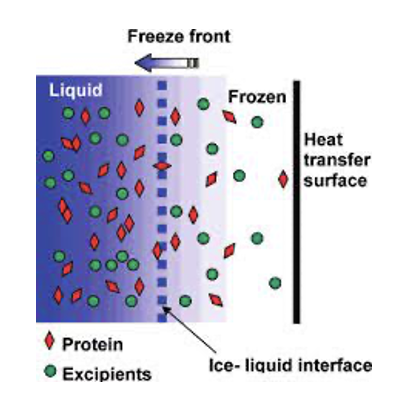
During freezing the formation of ice crystals from the water component of your sample can leave high salt or protein concentrations in the aqueous phase.
This is known as freeze concentration and causes severe stresses to protein stability.
It has even been shown to cause protein unfolding at the ice: aqueous interface and the aggregation of unfolded proteins,
Figures c-e
Sensitivity to limited trypsin digest (panel c)
Is codon optimized FRQ less sensitive to freeze-thaw and limited trypsin digestion when produced at lower temperature where translation rates are decreased in Neurospora? (panel d and e)
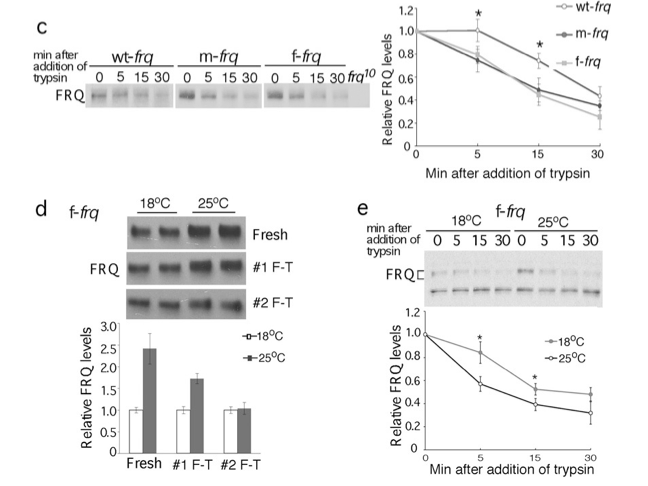
Fig 3c-e. Western blots showing sensitivity of FRQ from codon-optimized strains to trypsin (1 µg/ml) digestion
(c). A longer exposure for the wt-frq strain was used in (c). Densitometric analyses of FRQ levels of three independent experiments in are shown.
(d & e) Western blots showing that FRQ from the f-frq strain grown at 18°C is more resistant to freeze and thaw cycles (d, n=2) and to trypsin digestion (e, n=4) than that from 25°C. Two asterisks indicate p value <0.01, and one asterisk indicates p value <0.05.
Fig 4. Codon optimization of the middle region of FRQ
impairs FRQ phosphorylation and stabilizes FRQ
In Fig 4, a mid-frq strain, in which the middle region (aa 185–530) of frq ORF is optimized. This region contains two CK1 interaction domains and most of the phosphorylation sites that are important for FRQ stability and period determination
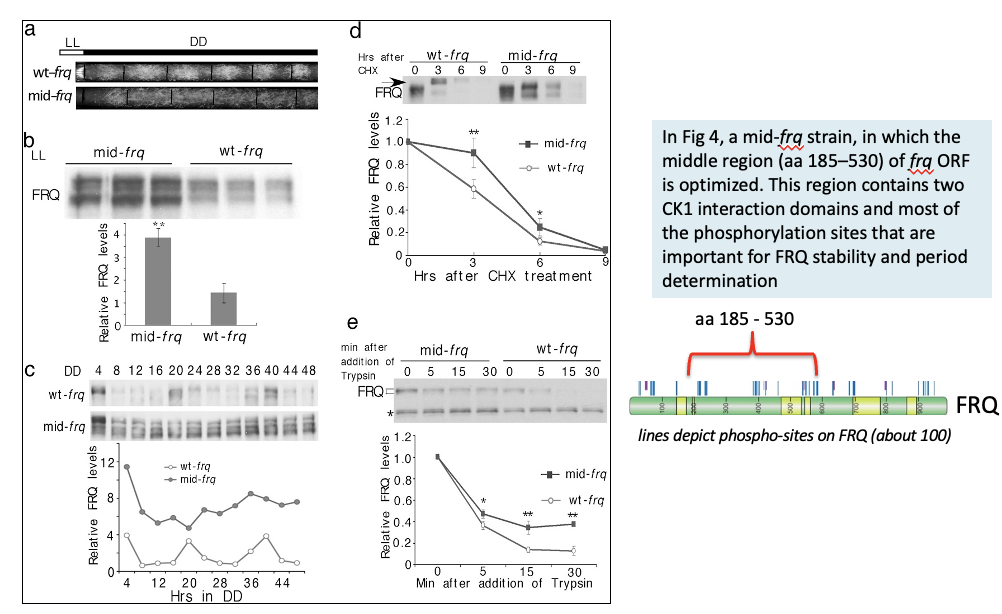
SUMMARY: The opposite molecular phenotypes of mid-frq and the N-terminal optimized strains indicate that changes in FRQ structural conformation in these strains are due to location-specific codon optimization.
Therefore, frq non-optimal codon usage should be a mechanism that allows the proper folding of FRQ by reducing translation rate in these predicted disordered regions. Synonymous aa differences in genome might still have effects!
similar findings shown for Drosophilia PERIOD protein
codon usage affects the structure and function of the Drosophilia circadian clock protein PERIOD

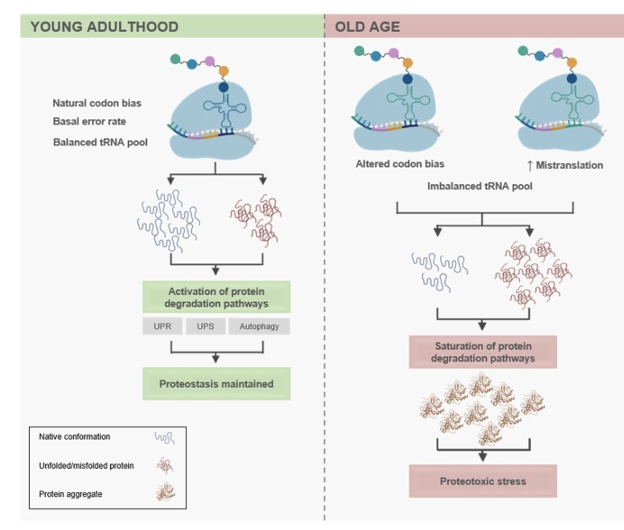
Translation rates change with
age and disease states, leading to more misfolded proteins
Age-related alterations in protein synthesis and proteostasis.
During aging (right panel), this equilibrium is disrupted by increased mistranslation.
The misfolded/unfolded proteins then accumulate in the cell as protein aggregates (depicted in dark orange), triggering proteotoxic stress
Recent breakthroughs in predicting protein structures
DeepMind a London based company linked to Google uses machine learning AI to predict structures from primary sequence