♋ Lecture 17: Cancer Continued
1/13
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
14 Terms
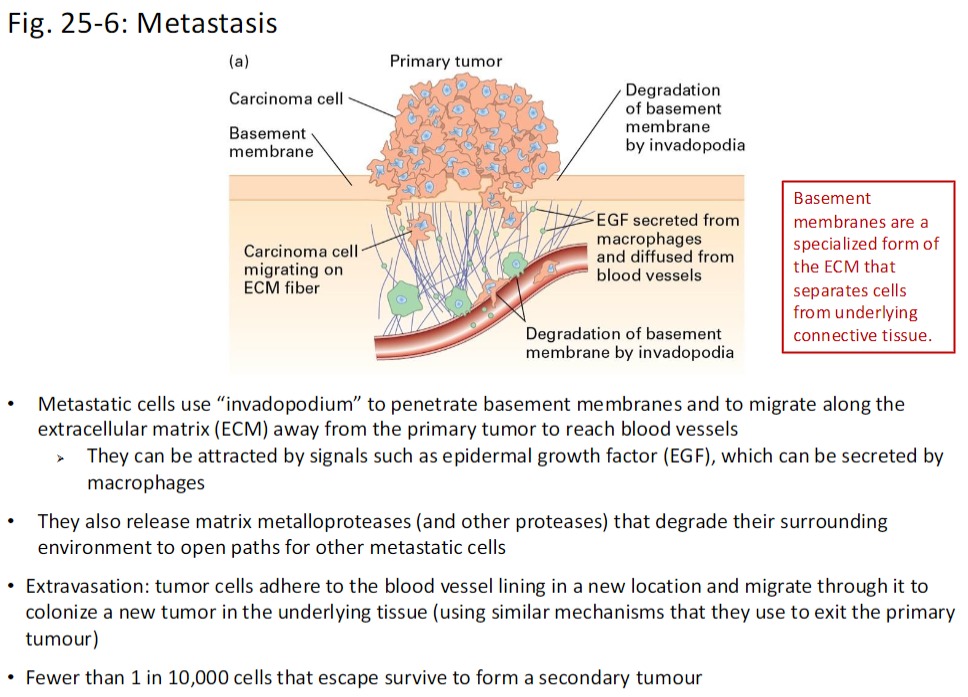
Metastatic Cell Migration
Invadopodia Formation
Metastatic cells form invadopodia to penetrate basement membranes and migrate along the extracellular matrix (ECM) away from the primary tumor
Chemotactic Attraction
Cells can be attracted by signals such as epidermal growth factor (EGF), sometimes secreted by macrophages
Matrix Degradation
Metastatic cells release matrix metalloproteases (MMPs) and other proteases to degrade the ECM, creating paths for themselves and other tumor cells
Extravasation
Tumor cells adhere to blood vessel linings in a new location and migrate through the vessel wall to colonize underlying tissue
Uses similar mechanisms as invasion of the primary tumor
Survival Rate
Fewer than 1 in 10,000 cells that escape survive to form a secondary tumor
Basement Membranes
Specialized ECM structures that separate cells from underlying connective tissue
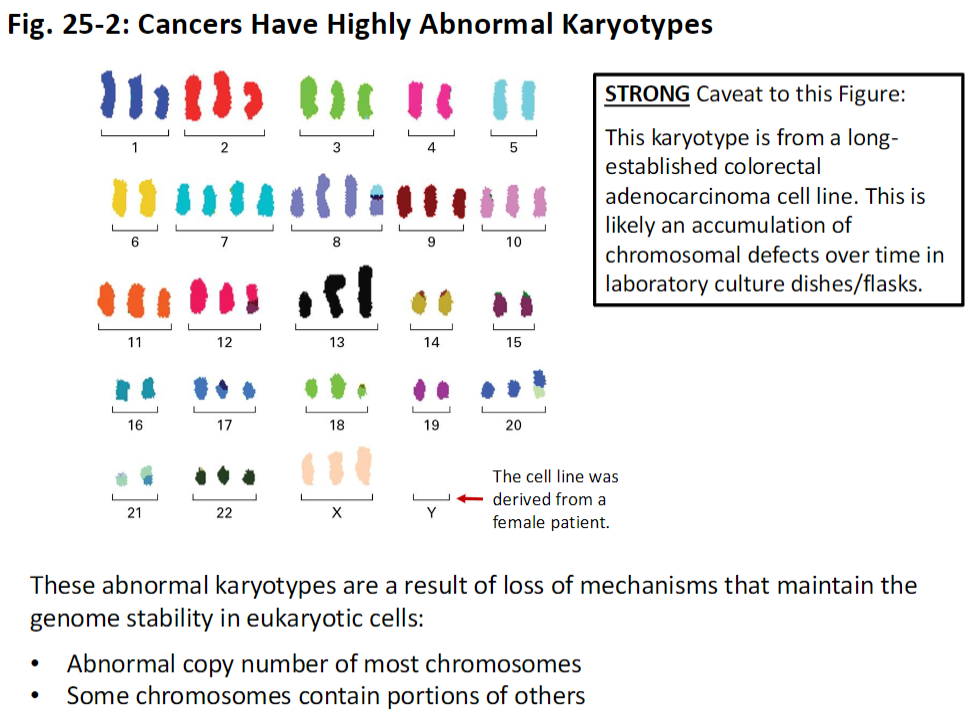
Cancer Cell Karyotypes
Abnormal Karyotypes
Cancer cells often exhibit highly abnormal karyotypes due to loss of genome stability mechanisms
Characteristics
Abnormal copy number of most chromosomes
Some chromosomes contain portions of other chromosomes
Caveat
The figure shown is from a long-established colorectal adenocarcinoma cell line
Many chromosomal defects may have accumulated over time in culture
The original cell line was derived from a female patient
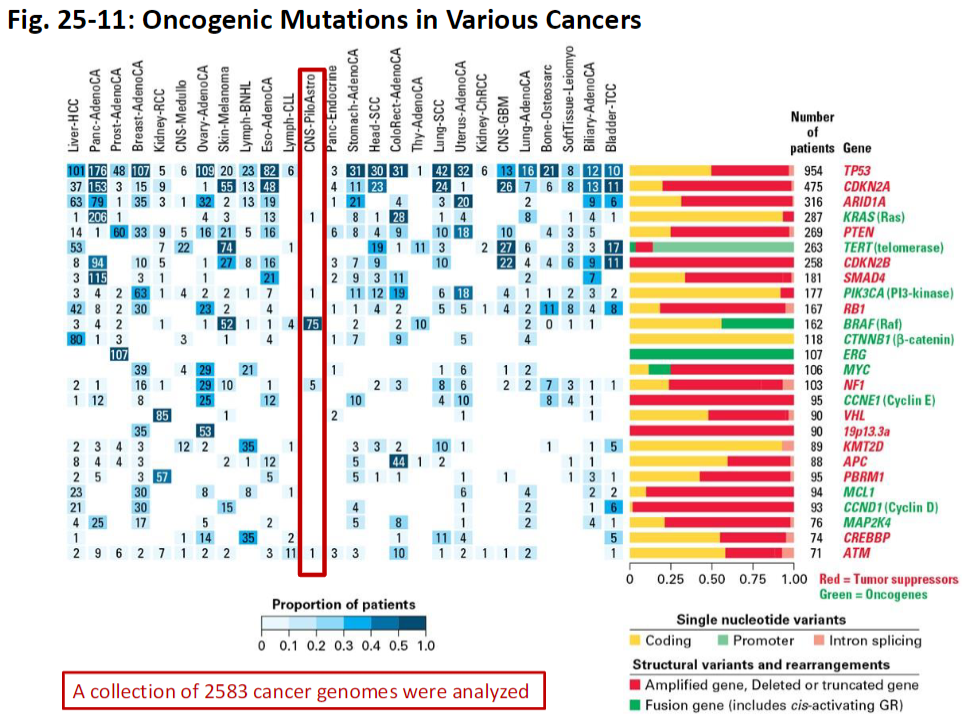
Oncogenic Driver Mutations in Cancer
Data Overview
Analysis based on 2,583 cancer genomes
Key Concept
Identifies oncogenic driver mutations that contribute to cancer development and progression
Helps distinguish driver mutations from passenger mutations that do not confer growth advantage
Cancer Incidence and Age – Multi-Hit Model
Concept
Cancer rates increase with age due to the Multi-Hit Model, where tumors arise through a recurring clonal selection process
Mutation Progression
First Mutation
Gives a cell a slight growth advantage
Second Mutation
Allows cells to grow more uncontrollably and form a small benign tumor
Third Mutation
Enables cells to overcome constraints imposed by the tumor microenvironment
Fourth Mutation
Allows cells to enter the bloodstream and establish daughter colonies at other sites, a hallmark of metastatic cancer
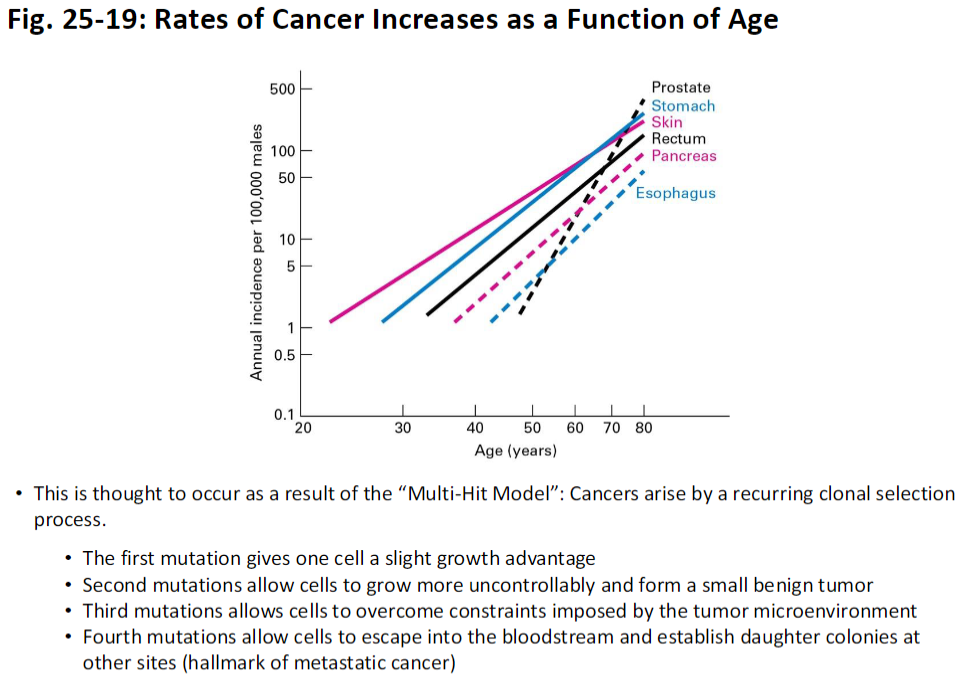
Development and Metastasis of Human Colorectal Cancer
Concept
Example of the Multi-Hit Hypothesis explaining tumor development and progression
Genetic Basis
APC = adenomatous polyposis coli protein
(Not to be confused with Anaphase Promoting Complex)
Key Idea
Mutations accumulate in specific genes like APC, driving the formation of colorectal tumors and potentially metastasis
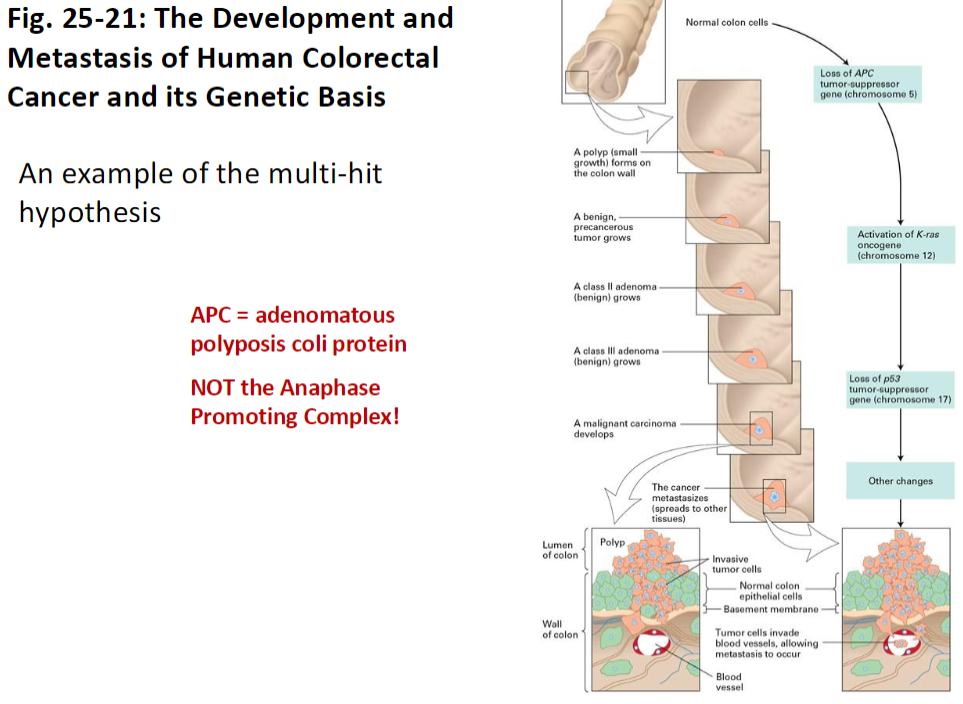
Colorectal Cancer Development – APC Loss
Normal Colon Cells
Cells grow and function normally, maintaining tissue architecture
Polyp Formation
A polyp (small growth) forms on the colon wall
APC Gene Loss
Loss of APC (adenomatous polyposis coli protein, chromosome 5)
(Not the Anaphase Promoting Complex)
APC function: suppresses cell growth via cell-to-cell contact and signal transduction pathways
Signaling Reminder
Signaling by plasma-membrane-attached proteins involves a signaling cell and an adjacent target cell
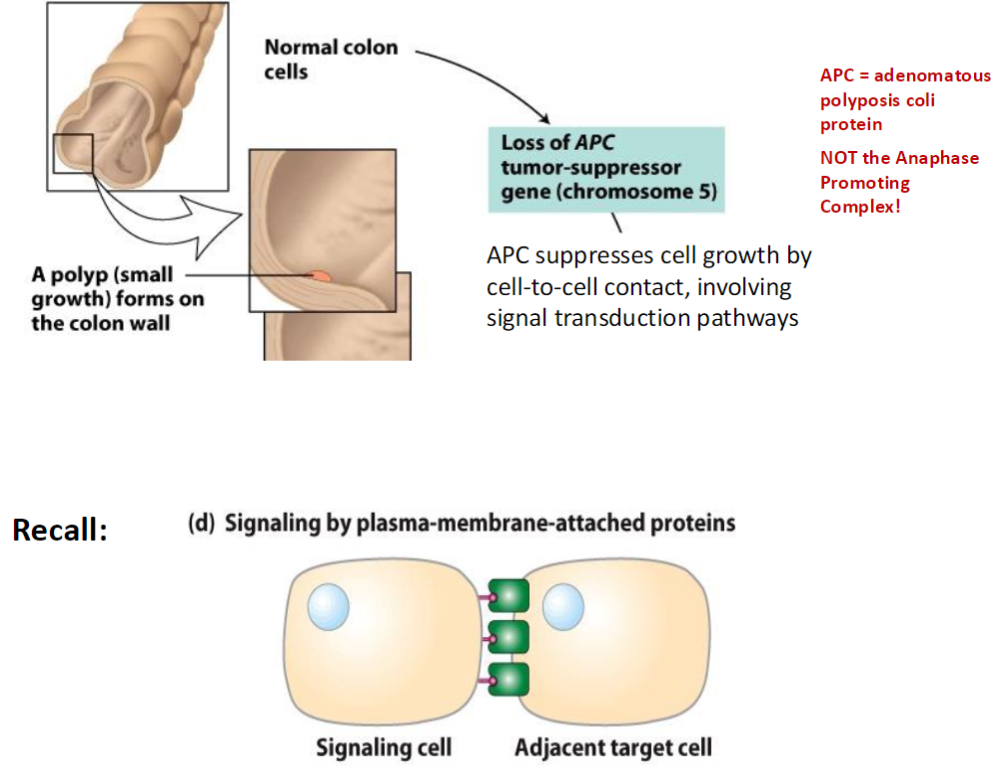
Colorectal Tumor Progression – K-ras Activation
Polyp Formation
A polyp (small growth) forms on the colon wall
Develops into a benign, precancerous tumor
K-ras Oncogene Activation
K-ras (chromosome 12) becomes activated, promoting growth of a class II adenoma (benign)
Ras Signaling Pathway
Ras transmits growth signals from growth factors via RTK (receptor tyrosine kinase) receptors
GRB2 binds activated receptor
Sos promotes dissociation of GDP from Ras
GTP binds Ras, activating it and allowing it to dissociate from Sos
Key Concept
Activated Ras drives cell proliferation, contributing to tumor growth
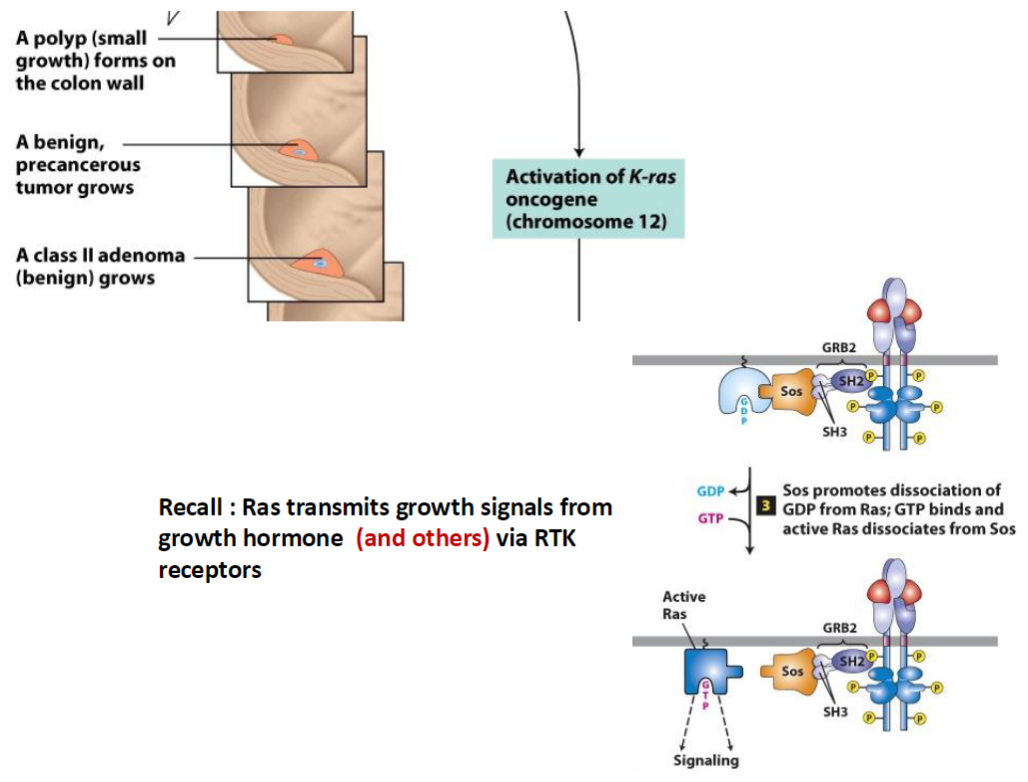
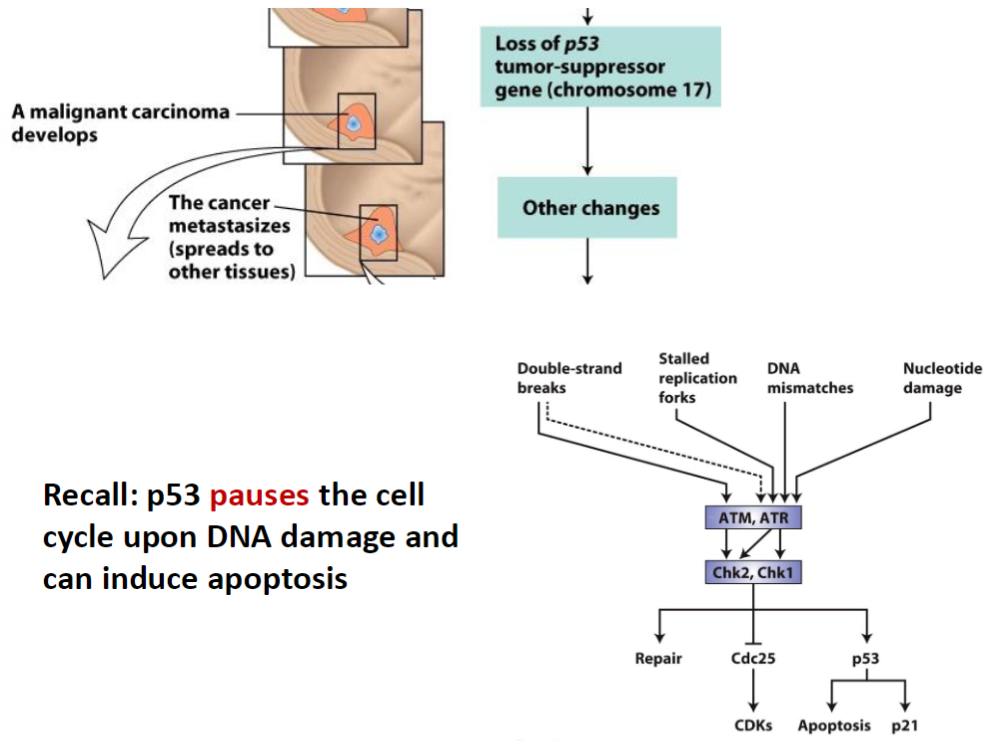
Colorectal Cancer Progression – Malignancy and Metastasis
Malignant Transformation
A malignant carcinoma develops from the benign adenoma
Metastasis
The cancer spreads to other tissues, establishing secondary tumors
Genetic Changes
Loss of p53 (tumor-suppressor gene, chromosome 17)
Other genetic and epigenetic changes accumulate
p53 Function
p53 pauses the cell cycle in response to DNA damage
Can induce apoptosis to prevent propagation of damaged cells
Key Concept
Loss of p53 removes a critical growth checkpoint, allowing malignant progression and metastasis
Cancer-Inducing Mutations
Normal Genes
Proto-oncogenes and tumor suppressor genes are normal genes that regulate cell growth and survival
Proto-Oncogenes
Examples: HER2, Ras, Myc, Fos
Promote cell survival
Suppress apoptosis
Mutation converts them into oncogenes
Can be gain-of-function or hyperactivating mutations
Tumor Suppressor Genes
Examples: p53, Rb, APC
Pause or slow down the cell cycle
Induce apoptosis
Mutation reduces or eliminates their function
Can be gene deletions or loss-of-function mutations
Key Concept
Cancer arises when mutations disrupt the balance of cell proliferation and death, enabling uncontrolled growth

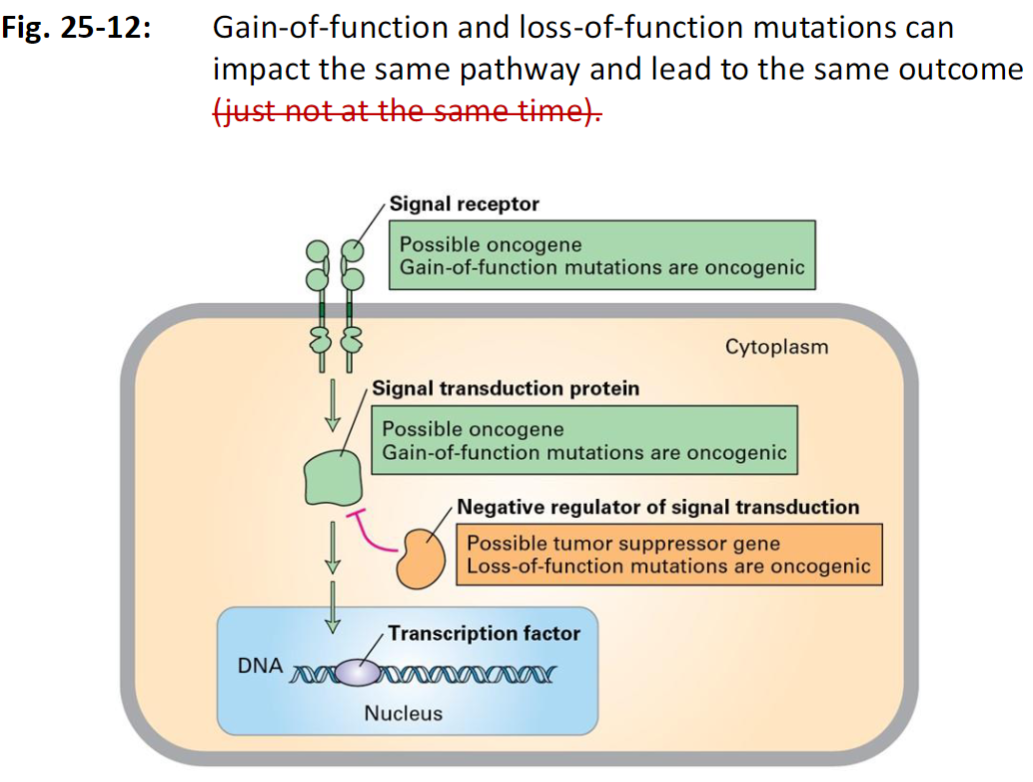
Mutations Affecting the Same Pathway
Signal Pathway Overview
Mutations in different components of a signaling pathway can lead to similar oncogenic outcomes
Gain-of-Function Mutations
Occur in proto-oncogenes such as:
Signal receptors
Signal transduction proteins
These mutations are oncogenic, causing hyperactive signaling
Loss-of-Function Mutations
Occur in negative regulators of signal transduction (tumor suppressor genes)
These loss-of-function mutations remove inhibitory control, also resulting in oncogenic signaling
Outcome
Both types of mutations disrupt normal cell growth control
Leads to abnormal proliferation and potential tumor formation
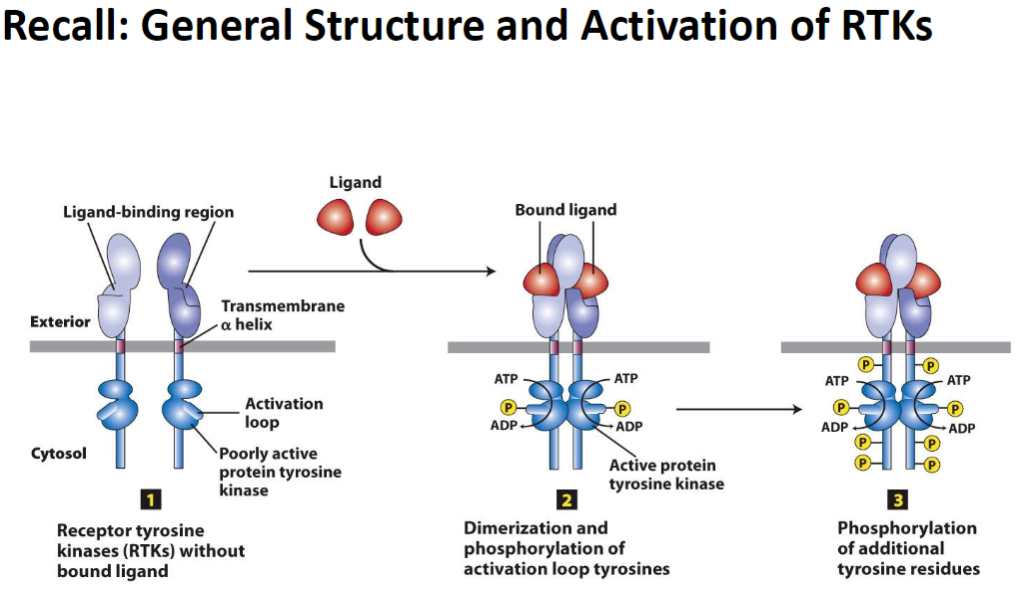
Receptor Tyrosine Kinases (RTKs) – Structure and Activation
Inactive State
RTKs exist without a bound ligand
ATP is present but the kinase is inactive
Activation Step 1: Dimerization
Ligand binding induces dimerization of RTKs
Phosphorylation of activation loop tyrosines occurs
ATP → ADP, transferring phosphate groups (P)
Activation Step 2: Tyrosine Phosphorylation
Additional tyrosine residues in the receptor are phosphorylated
Creates docking sites for downstream signal transduction proteins
Key Concept
Activated RTKs initiate intracellular signaling pathways that regulate cell growth, survival, and differentiation
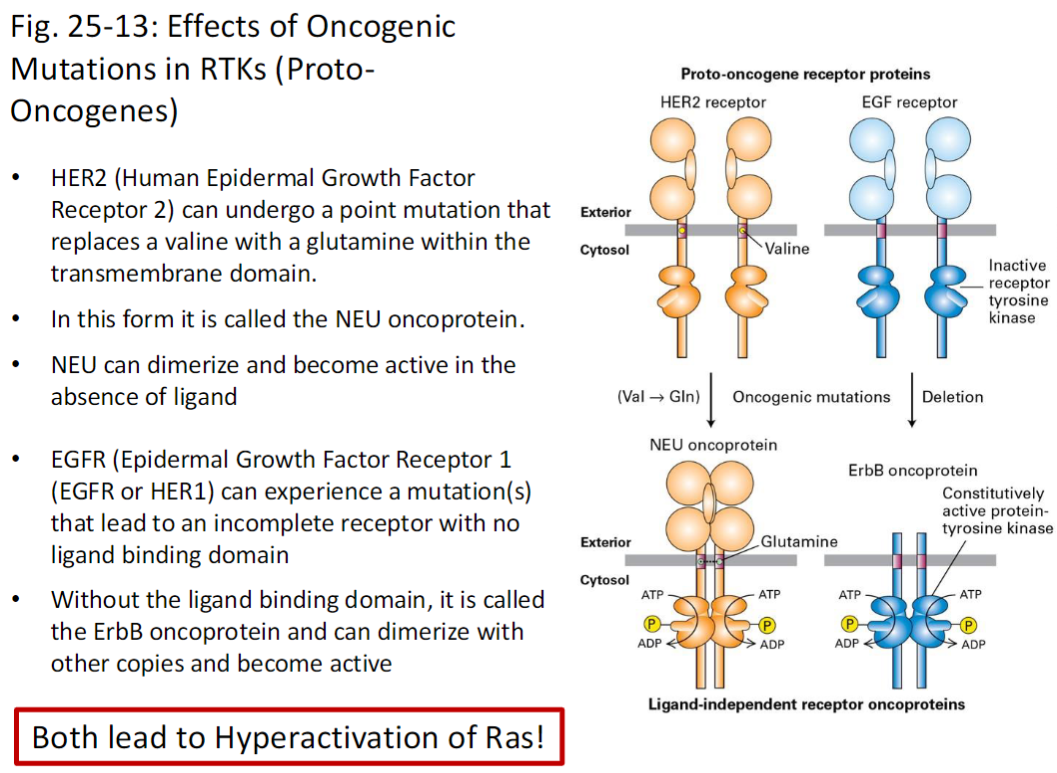
Oncogenic Mutations in RTKs (Proto-Oncogenes)
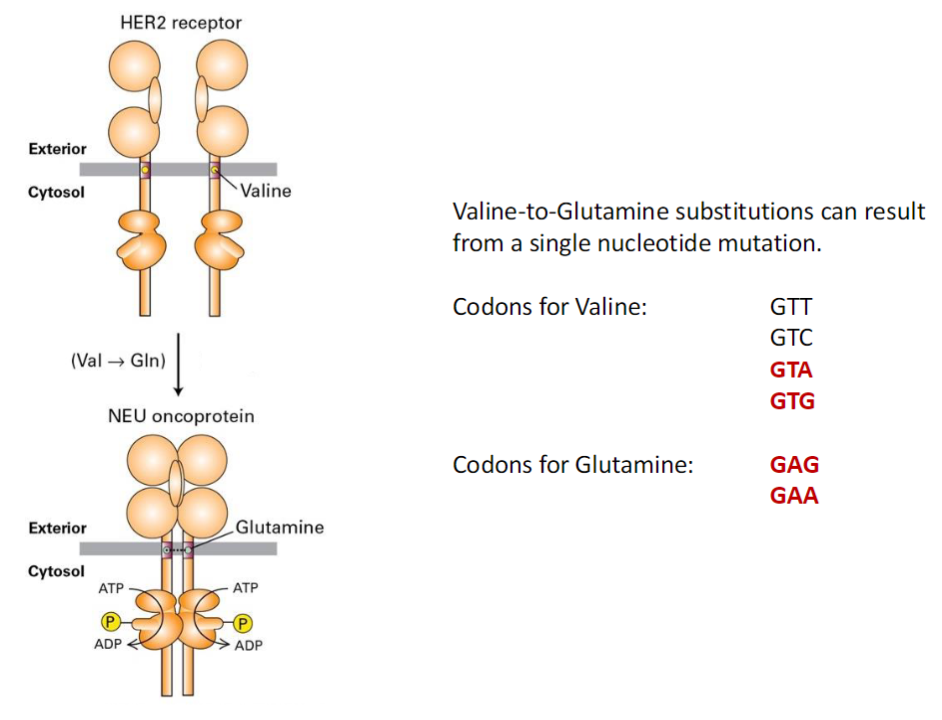
HER2 Mutation
HER2 (Human Epidermal Growth Factor Receptor 2) can undergo a point mutation replacing valine with glutamine in the transmembrane domain
Mutated form = NEU oncoprotein
NEU can dimerize and become active without ligand
EGFR Mutation
EGFR (Epidermal Growth Factor Receptor 1 / HER1) can mutate to produce an incomplete receptor lacking the ligand-binding domain
Mutated form = ErbB oncoprotein
Can dimerize with other copies and become active
Key Outcome
Both mutations lead to hyperactivation of Ras, driving cell proliferation and potential tumorigenesis
Valine-to-Glutamine Substitution
Genetic Basis
A single nucleotide mutation can convert valine to glutamine
Codons for Valine (Val)
GTT, GTC, GTA, GTG
Codons for Glutamine (Gln)
GAG, GAA
Key Concept
This point mutation in the HER2 gene can create the NEU oncoprotein, leading to ligand-independent activation and hyperactive Ras signaling
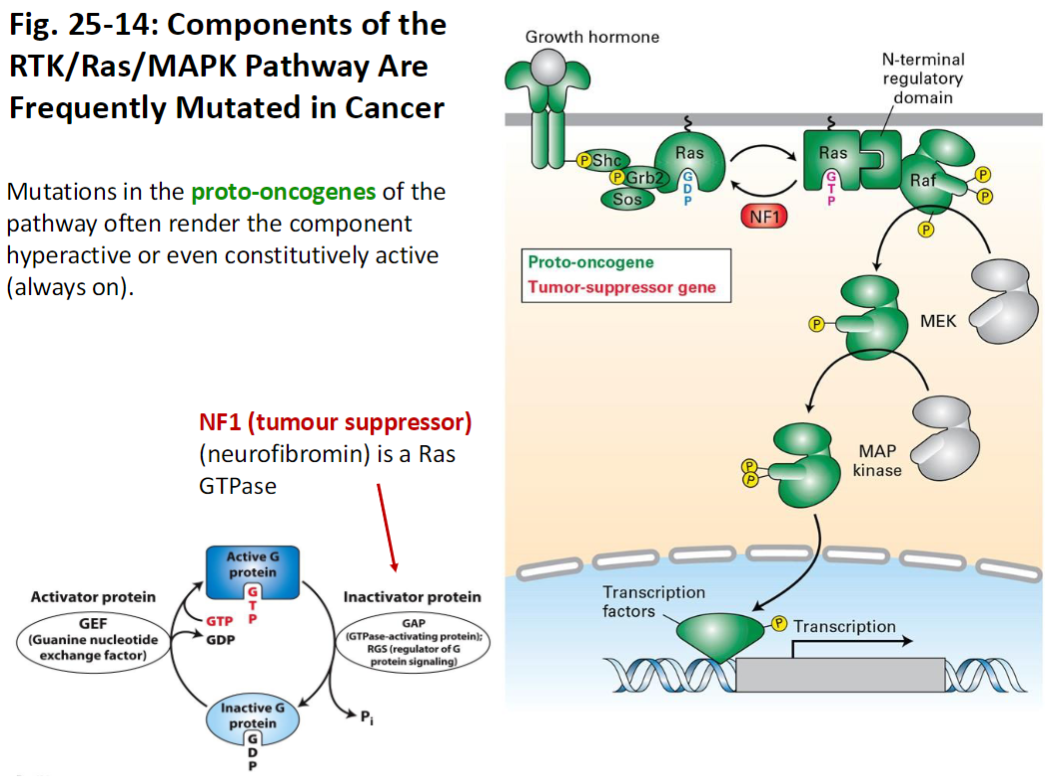
RTK/Ras/MAPK Pathway in Cancer
Pathway Components
Includes RTKs, Ras, MAPK, and regulators like NF1 (tumor suppressor, neurofibromin)
NF1 functions as a Ras GTPase, inactivating Ras
Mutations
Mutations in proto-oncogenes of this pathway often make the component hyperactive or constitutively active (always on)
Key Concept
Constitutive activation of the RTK/Ras/MAPK pathway drives uncontrolled cell proliferation, a hallmark of cancer