Myoglobin and Hemoglobin: Structure & Function
1/34
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
35 Terms
Describe the structure of the heme unit.
Describe the structures of the myoglobin and hemoglobin molecules.
Explain how the globin chain decreases the CO binding to hemoglobin.
Describe the conformational changes in the quaternary structure of hemoglobin upon oxygen binding.
Explain myoglobin and hemoglobin´s oxygen binding curves and explain the factors that lower the affinity of hemoglobin for O2.
Discuss fetal and adult hemoglobins in terms of polypeptide chains and oxygen affinity.
Explain the mechanism(s) associated with sickle cell anemia and thalassemias.
Explain the importance of myoglobin and hemoglobin.
Describe allosterism and the Bohr effect and how they affect the affinity of hemoglobin for oxygen.
Describe the effect of 2,3-biphospho-glycerate (BPG) on hemoglobin´s oxygen affinity.
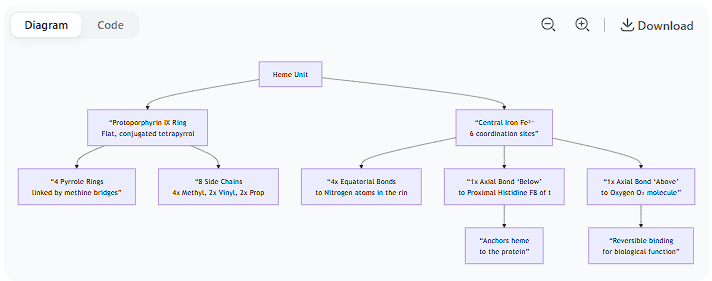
Describe the structure of the heme unit.
Here is a detailed description of the structure of the heme unit, breaking it down into its core components.
Overview
Heme is a complex, ring-like molecule that serves as the prosthetic group (the non-protein, cofactor component) in heme proteins such as hemoglobin, myoglobin, and cytochromes. Its primary function is to bind oxygen or participate in electron transfer reactions, a capability derived from its central iron atom.
The Core Components of Heme
The structure of heme can be described in four key parts:
1. The Organic Ring: Protoporphyrin IX
The foundation of heme is a large, flat, organic ring system called a porphyrin. Specifically, the heme in hemoglobin and myoglobin is Protoporphyrin IX. Its characteristics are:
Tetrapyrrole Ring: The ring is made up of four smaller pyrrole rings (five-membered rings containing one nitrogen and four carbon atoms). These are labeled Pyrrole Rings I, II, III, and IV.
Bridging Methine Groups: The four pyrrole rings are linked together by methine bridges (=CH– groups), forming a large, conjugated, and entirely planar macrocycle.
Conjugated System: The alternating single and double bonds in the ring create a conjugated pi-electron system. This is why heme is deeply colored (red/brown).
2. The Central Iron Atom (Fe)
At the very center of the porphyrin ring sits a single iron atom.
Oxidation State: In the deoxygenated state, it is Ferrous iron (Fe²⁺). This is essential for binding O₂. If oxidized to Ferric iron (Fe³⁺), it can no longer bind oxygen (as in methemoglobin).
Coordination Number: Iron has six coordination sites, meaning it can form bonds in six directions.
3. The Side Chains: What Makes it "Protoporphyrin IX"
The porphyrin ring has a specific pattern of side chains that define it as Protoporphyrin IX. There are a total of eight side chains:
4 Methyl groups (–CH₃)
2 Vinyl groups (–CH=CH₂)
2 Propionate groups (–CH₂–CH₂–COO⁻)
These side chains are attached to the pyrrole rings in the specific order that gives heme its chemical identity and solubility properties. The propionate groups, being negatively charged, help make heme water-soluble and are oriented towards the surface of the protein pocket.
4. The Coordination Bonds: How Iron is Held and Binds Ligands
This is the most functionally critical aspect of the structure. The central iron atom forms six coordination bonds:
Four In-Plane Bonds (Equatorial): The iron is bonded to the four nitrogen atoms at the center of each pyrrole ring. This holds the iron firmly within the plane of the porphyrin ring.
The Fifth Coordination Site (Axial): Below the plane of the ring, this site is occupied by a nitrogen atom from a histidine residue of the protein. This is known as the proximal histidine. It anchors the heme unit tightly to the protein.
The Sixth Coordination Site (Axial): Above the plane of the ring, this site is the binding site for oxygen (O₂). When oxygen binds, it completes the octahedral coordination geometry of the iron.
Visual Summary of the Structure
The diagram below illustrates the key structural features of the heme unit and its coordination bonds within a protein like hemoglobin:
Key Structural-Punctional Relationships
The Role of the Proximal Histidine: This bond not only anchors the heme but also is crucial for hemoglobin's cooperativity. When oxygen binds, it pulls the iron atom into the plane of the porphyrin ring. This tug moves the proximal histidine and its associated protein helix, triggering the conformational change from the T (tense) to the R (relaxed) state.
The Role of the Distal Histidine: Another histidine residue (the distal histidine, E7) is located on the oxygen-binding side. It does not bind the iron but plays a critical role in:
Stabilizing the bound O₂ molecule via hydrogen bonding.
Preventing the oxidation of Fe²⁺ to Fe³⁺ (which would irreversibly disable oxygen binding) by sterically hindering the approach of larger, oxidizing molecules like CO.
The Hydrophobic Pocket: The heme is nestled in a hydrophobic pocket within the protein. This excludes water, which would otherwise oxidize the Fe²⁺ ion and stabilize the unwanted ferric state.
In summary, the heme unit is a sophisticated molecular device where a protoporphyrin IX ring holds a ferrous iron atom in a specific geometry, allowing it to be anchored by a proximal histidine and to reversibly bind oxygen at its sixth coordination site, all within a protective protein environment.
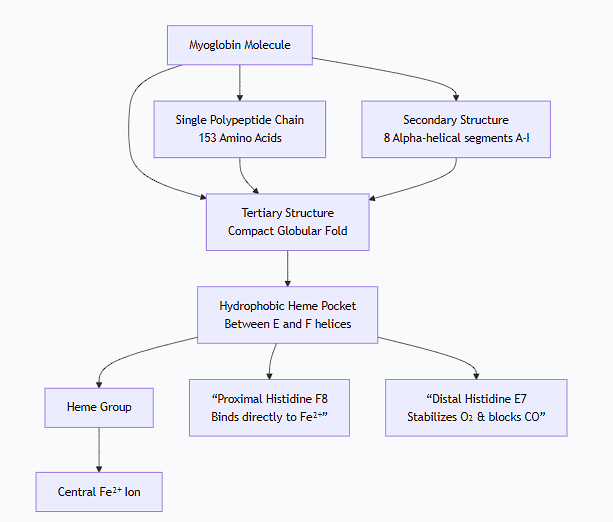
Describe the structures of the myoglobin and hemoglobin molecules
Here is a detailed description and comparison of the structures of myoglobin and hemoglobin, which are fundamental to understanding oxygen transport and storage in vertebrates.
Overarching Similarity and Difference
Similarity: Both are heme proteins. They contain a protoporphyrin IX ring with a central iron atom (Fe²⁺) that is the actual site of oxygen binding.
Difference: Their primary functional difference stems from their structural complexity:
Myoglobin is a monomeric, single-chain protein for oxygen storage.
Hemoglobin is a tetrameric, four-chain protein for oxygen transport.
1. Myoglobin (Mb) - The Oxygen Reservoir
Myoglobin's structure is optimized for holding onto oxygen tightly and releasing it only when oxygen levels become very low.
A. Primary Structure
The sequence is a single, relatively short polypeptide chain of 153 amino acids.
B. Secondary Structure
It is composed predominantly of alpha-helices. The chain folds into 8 alpha-helical segments (labeled A through H), connected by short, non-helical segments.
C. Tertiary Structure
The molecule folds into a compact, globular, three-dimensional structure, often described as a "muscle globin" fold.
The heme group is nestled in a deep, hydrophobic (water-repellent) pocket or cleft between the E and F helices. This pocket protects the iron from being permanently oxidized to the Fe³⁺ state, which cannot bind O₂.
Key amino acids in the pocket:
Proximal Histidine (His F8): This histidine residue is the 8th amino acid in the F helix. It binds directly to the iron atom from below the heme plane, anchoring the heme to the protein.
Distal Histidine (His E7): This histidine residue is the 7th amino acid in the E helix. It sits on the oxygen-binding side of the heme. It does not bind the iron but stabilizes the bound O₂ and prevents the binding of other small molecules like carbon monoxide (CO) as effectively.
In summary, myoglobin is a single polypeptide chain, folded into a compact globular structure with a protected heme pocket, designed for high-affinity oxygen binding.
The following diagram illustrates the key structural features of the myoglobin molecule:
2. Hemoglobin (Hb) - The Oxygen Transporter
Hemoglobin's structure is a masterpiece of evolution, designed for cooperative oxygen binding and efficient transport. Its quaternary structure is the key to its function.
A. Primary Structure
Adult hemoglobin (HbA) is a tetramer composed of four polypeptide chains: two identical alpha (α) chains and two identical beta (β) chains (α₂β₂).
Each chain is similar in length and 3D fold to a myoglobin molecule (a testament to a common evolutionary ancestor), but their amino acid sequences are different.
B. Secondary and Tertiary Structure
Like myoglobin, each individual subunit (α and β) is composed primarily of alpha-helices (7 in α, 8 in β) and folds into a similar globin fold with a hydrophobic heme pocket.
Each subunit contains its own proximal and distal histidine and binds one heme group, meaning one hemoglobin molecule can bind four O₂ molecules.
C. Quaternary Structure - The Critical Difference
This is the most important level of structure for hemoglobin's function. The four subunits are arranged in a tetrahedral pattern, forming a roughly spherical molecule.
Subunit Interactions: The subunits are held together by extensive non-covalent interactions (salt bridges, hydrogen bonds, van der Waals forces), primarily at the α₁β₁ and α₂β₂ interfaces, which are relatively stable.
The Critical Contact: The α₁β₂ Interface: The contact between the α₁ chain of one dimer and the β₂ chain of the other dimer is crucial. This interface is mobile and changes shape when oxygen binds. This single feature is the structural basis for cooperativity.
Structural Basis for Cooperativity: The T and R States
Hemoglobin exists in equilibrium between two primary conformational states:
1. The T (Tense) State
Prevalent in Deoxyhemoglobin: This is the low-affinity state.
Structure: The subunits are held tightly together by additional salt bridges (e.g., between the C-termini of the β chains).
The Heme: In the T state, the iron atom is pulled slightly out of the heme plane towards the proximal histidine. The heme itself is slightly "domed." This makes it harder for an oxygen molecule to bind.
2. The R (Relaxed) State
Prevalent in Oxyhemoglobin: This is the high-affinity state.
Structure: Binding of O₂ breaks the stabilizing salt bridges of the T state.
The Heme: When O₂ binds to a subunit in the T state, it pulls the iron atom into the plane of the heme ring. This small movement (about 0.4 Å) tugs on the proximal histidine, which in turn shifts the position of the entire alpha-helix it's attached to.
3. The Mechanism of Cooperativity
The shift in the helix at the α₁β₂ interface forces the other subunits to change their conformation slightly.
This disrupts the salt bridges and stabilizes the R state in the remaining subunits.
The other subunits now have their heme groups in a higher-affinity conformation, making it easier for the next oxygen molecules to bind.
This "molecular breathing" is the reason for hemoglobin's sigmoidal oxygen-binding curve. The binding of one O₂ positively influences the binding of the next.
Comparison Table
Feature | Myoglobin | Hemoglobin |
|---|---|---|
Biological Role | Oxygen Storage in muscle | Oxygen Transport in blood |
Oligomeric State | Monomer (single subunit) | Tetramer (α₂β₂ - four subunits) |
Number of Heme Groups | 1 | 4 |
Structure Level | Up to Tertiary | Up to Quaternary |
O₂ Binding Curve | Hyperbolic (normal, non-cooperative) | Sigmoidal (cooperative) |
O₂ Affinity | High (binds O₂ tightly even at low pressure) | Lower (releases O₂ readily at low pressure) |
Key Regulatory Molecule | None | 2,3-Bisphosphoglycerate (BPG), which binds to the T state and stabilizes it, decreasing O₂ affinity. |
In conclusion, myoglobin is a simple storage unit with a stable structure, while hemoglobin is a complex, dynamic molecular machine whose sophisticated quaternary structure allows it to efficiently load oxygen in the lungs and unload it in the tissues.
Describe the structures of the myoglobin and hemoglobin molecules (summary table)
Summary Table: Myoglobin vs. Hemoglobin
Feature | Myoglobin | Hemoglobin |
|---|---|---|
Biological Role | Oxygen storage in muscle | Oxygen transport in blood |
Oligomeric State | Monomer (1 subunit) | Tetramer (α₂β₂, 4 subunits) |
Heme Groups | 1 | 4 |
Quaternary Structure | None | Yes; complex interactions between subunits |
O₂ Binding Curve | Hyperbolic | Sigmoidal (S-shaped) |
O₂ Affinity | High (binds O₂ tightly) | Lower (releases O₂ easily) |
Cooperativity | No | Yes; binding of one O₂ facilitates binding of others |
Response to 2,3-BPG | No effect | Binds to T-state, stabilizing it and decreasing O₂ affinity |
Explain how the globin chain decreases the CO binding to hemoglobin.
This is an excellent example of how a protein structure fine-tunes the chemistry of its bound cofactor for a specific biological purpose.
The globin chain decreases CO binding to hemoglobin through a mechanism known as steric hindrance and distal pocket engineering. While CO has an inherently high affinity for free heme (~25,000 times higher than O₂), the protein environment of hemoglobin reduces this advantage to only about 200-250 times that of O₂. This is a crucial safety feature, as it prevents trace amounts of CO from completely incapacitating hemoglobin.
Here’s a detailed, molecular-level explanation:
1. The Innate Problem: Heme's High Affinity for CO
In a free heme group (not in a protein), the iron atom has six coordination sites.
Both O₂ and CO bind to the sixth coordination site.
Due to its electronic structure, CO can form a very strong, linear bond (Fe–C≡O) with the iron, making it a much more potent ligand than the bent Fe–O–O bond of oxygen.
2. The Globin Solution: Strategic Placement of a Distal Histidine
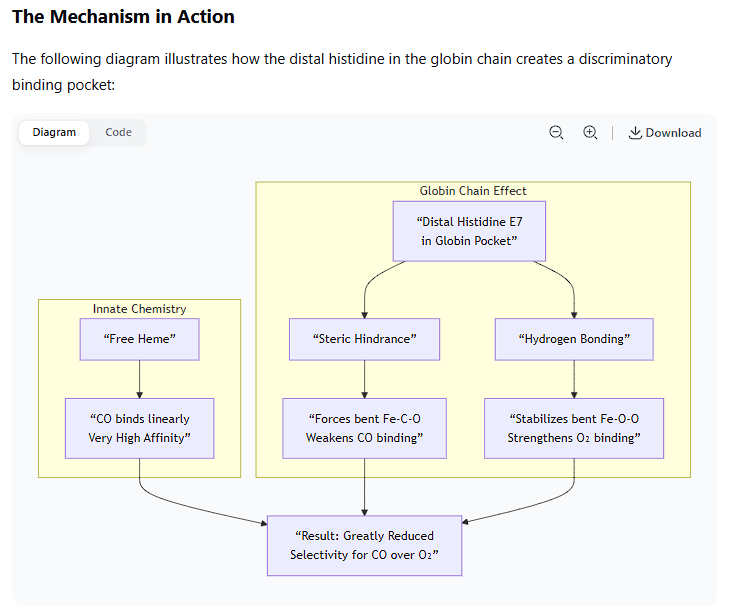
The key player is a specific amino acid in the globin chain called the distal histidine (Histidine E7). It is located on the side of the heme plane where O₂ and CO must bind.
This histidine serves two critical protective functions:
A. Steric Hindrance: Forcing a Suboptimal Bond Angle
The distal histidine side chain is positioned too close to the iron atom to allow for the thermodynamically favored linear Fe–C–O geometry.
To bind, the CO molecule is forced to bend at an angle. This distorted geometry is energetically less favorable and weakens the bond between the iron and CO.
B. Polar Stabilization of the Correct Ligand
The distal histidine also has a nitrogen-hydrogen (N–H) group in its imidazole ring.
This N–H group can form a hydrogen bond with the bound oxygen molecule (O₂). This hydrogen bond stabilizes the preferred, bent Fe–O–O geometry and strengthens the binding of O₂.
Carbon monoxide (C≡O) cannot form this hydrogen bond effectively. Its oxygen atom is less basic and is part of a triple bond, making it a very poor hydrogen bond acceptor.
This creates a discriminatory environment: the protein pocket penalizes the preferred binding mode of CO while rewarding the preferred binding mode of O₂.
The Mechanism in Action
The following diagram illustrates how the distal histidine in the globin chain creates a discriminatory binding pocket:
Comparison to Myoglobin
This same mechanism operates in myoglobin. However, because myoglobin's sole function is oxygen storage, it needs to bind O₂ very tightly all the time. The steric hindrance in myoglobin is even slightly more effective, reducing CO's relative affinity compared to hemoglobin.
In hemoglobin, this discrimination is part of a larger, cooperative mechanism. When hemoglobin is in its low-affinity T-state, the steric constraints on the heme pocket are even greater, further reducing CO binding in the tissues where oxygen needs to be released.
Why This Matters Biologically
If hemoglobin bound CO with the same affinity as free heme does (~25,000x that of O₂), even minuscule amounts of CO in the air would be instantly fatal. The structural adaptation of the globin chain reduces this threat, making life possible in an environment where trace amounts of CO are naturally occurring.
In summary, the globin chain uses the strategic placement of a distal histidine residue to sterically hinder CO's preferred linear binding geometry and to hydrogen-bond with O₂, thereby dramatically decreasing the relative affinity of hemoglobin for carbon monoxide compared to oxygen.
Describe the conformational changes in the quaternary structure of hemoglobin upon oxygen binding.
The conformational changes in hemoglobin's quaternary structure upon oxygen binding are the fundamental basis for its cooperative behavior. This process is elegantly described by the Two-State Model (Tense and Relaxed States).
Here is a step-by-step description of these changes.
Overview: The Two-State Model
Hemoglobin exists in an equilibrium between two primary quaternary structures:
The T (Tense) State: The deoxygenated form; a low-affinity conformation.
The R (Relaxed) State: The oxygenated form; a high-affinity conformation.
Binding oxygen shifts the equilibrium from the T state to the R state.
Step-by-Step Conformational Changes1. Starting Point: The T State (Deoxyhemoglobin)
Quaternary Structure: The tetramer is held in a "tense" configuration by a network of salt bridges and hydrogen bonds between the subunits, particularly between the carboxyl-terminal residues of the four chains.
Key Interactions: Notable salt bridges include:
Between the α-chain amino groups and the α-chain carboxyl groups of the opposite α-chain.
Between His HC3 (the C-terminal histidine) on each β-chain and Asp FG1 on the opposite β-chain and Lys C5 on the α-chain.
The Heme Group: In the T state, the iron atom is pulled slightly out of the heme plane (towards the proximal histidine). The heme itself is slightly "domed." This high-spin state has a larger ionic radius, so it doesn't fit perfectly in the plane of the porphyrin ring. This makes it harder for an oxygen molecule to bind because the binding site is sterically less accessible.
2. The Trigger: Oxygen Binding to a Heme
When an oxygen molecule finally binds to the iron atom in one subunit (e.g., an α-subunit), it forms a coordinate covalent bond.
This bond causes the electrons of the iron to redistribute. The iron atom shrinks slightly (transition to a low-spin state) and can now fit perfectly into the center of the porphyrin ring.
Consequently, the iron atom moves about 0.4 Å (angstroms) into the plane of the heme.
3. The Tertiary Change: A Pull on the Protein
The movement of the iron atom pulls the proximal histidine (His F8) with it.
This histidine is part of the F helix. The pull on His F8 causes the entire F helix to shift and move.
This is a tertiary structural change within the oxygenated subunit.
4. The Key Quaternary Change: Breaking the α₁β₂ Interface
The movement of the F helix is transmitted to the interface between the α₁ and β₂ subunits (and equivalently, the α₂ and β₁ subunits). This interface is the "switch" for the quaternary change.
In the T state, this interface is tight, with specific residues in close contact.
The shift from the F helix causes these contact points to break and reform into a new, more stable arrangement.
This process breaks the critical salt bridges that were stabilizing the T state. For example, the bond between His HC3 on the β-chain and Asp FG1 on the opposite β-chain is severed.
5. The Final State: The R State (Oxyhemoglobin)
As the α₁β₂ (and α₂β₁) interfaces shift, the entire tetramer undergoes a dramatic quaternary change.
The two αβ dimers rotate ~15 degrees relative to each other and slide closer together.
The protein now settles into the R (Relaxed) State. In this state:
The salt bridges are broken.
The protein has a more compact, relaxed conformation.
The remaining unliganded subunits now have their heme groups in a high-affinity conformation. The iron is now in a geometry that makes it easier for the next oxygen molecules to bind.
The "Cascade" of Cooperativity
This mechanism explains the cooperative (sigmoidal) oxygen-binding curve:
The first O₂ binding event (to a T-state hemoglobin) is difficult and has low affinity.
This binding triggers the T → R quaternary transition.
The remaining subunits are now in the R state and have a much higher affinity for oxygen.
Therefore, the second, third, and fourth O₂ molecules bind much more readily.
Summary of the Changes
Feature | T State (Deoxy) | R State (Oxy) |
|---|---|---|
Overall Stability | Stabilized by salt bridges | Salt bridges are broken |
Subunit Rotation | Two αβ dimers are further apart | Dimers rotate ~15° and slide closer |
Central Cavity | Larger; perfect for BPG binding | Smaller; BPG is expelled |
Heme Geometry | Domed; Fe²⁺ is out of plane | Planar; Fe²⁺ is in the plane |
O₂ Affinity | Low | High |
In conclusion, the binding of oxygen induces a small tertiary change (iron and helix movement) within a single subunit, which is amplified into a major quaternary change (subunit rotation and salt bridge breakage) across the entire tetramer. This allosteric transition is the key to hemoglobin's efficient function as an oxygen transport protein.
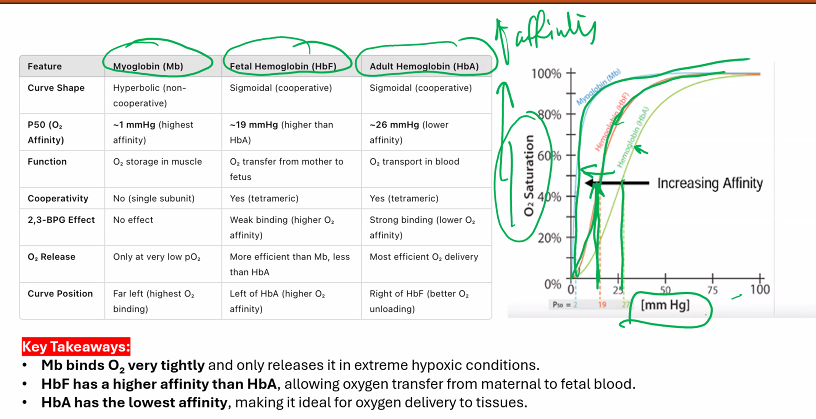
Explain myoglobin and hemoglobin´s oxygen binding curves and explain the factors that lower the affinity of hemoglobin for O2.
This is a central concept in respiratory physiology. Let's break down the oxygen-binding curves and the factors that regulate hemoglobin's affinity.
1. Oxygen-Binding Curves: Myoglobin vs. Hemoglobin
The oxygen-binding curve is a plot of the percentage of heme groups bound to oxygen (Y) versus the partial pressure of oxygen (pO₂).
Myoglobin (Mb): Hyperbolic Curve
Shape: Hyperbolic.
Interpretation:
This shape is characteristic of simple, non-cooperative binding.
Myoglobin has a very high affinity for oxygen at low pO₂. It becomes saturated with oxygen even at relatively low oxygen pressures (e.g., in muscle tissue).
It acts as an oxygen reservoir, holding onto oxygen tightly and only releasing it when the pO₂ drops to very low levels (as during strenuous exercise).
Physiological Role: Oxygen storage in muscle tissue, ensuring a supply for mitochondria when demand is high.
Hemoglobin (Hb): Sigmoidal Curve
Shape: Sigmoidal (S-shaped).
Interpretation:
This shape is the hallmark of positive cooperativity.
The binding of the first oxygen molecule to a hemoglobin subunit induces a conformational change (T state → R state) that makes it easier for the next oxygen molecules to bind.
At low pO₂ (in tissues): The affinity is low, facilitating oxygen unloading.
At high pO₂ (in lungs): The affinity becomes very high, facilitating oxygen loading.
The steep part of the curve in the middle is where small changes in pO₂ (as in capillaries) result in large changes in oxygen delivery.
Physiological Role: Oxygen transport from the lungs to the tissues.
The following diagram illustrates the key differences between these two curves:
2. Factors That Lower the Affinity of Hemoglobin for O₂ (The Bohr Effect)
To efficiently release oxygen where it's needed, hemoglobin's affinity must decrease in the peripheral tissues. Three key factors accomplish this, and they often occur together in metabolically active tissues.
1. Decreased pH (Increased [H⁺]) - The Bohr Effect
Mechanism: Protons (H⁺) bind to specific amino acid residues in hemoglobin (e.g., His β146), stabilizing the T (deoxy) state through the formation of additional salt bridges.
Physiological Context: In active tissues (e.g., muscle), CO₂ production and lactic acid buildup lower the pH.
Result: The oxygen-binding curve shifts to the RIGHT. At any given pO₂, hemoglobin has a lower affinity for O₂ and releases more oxygen.
2. Increased Carbon Dioxide (CO₂)
CO₂ exerts its effect in two ways:
1. Direct Effect: CO₂ can bind reversibly to the N-terminal amino groups of hemoglobin to form carbamates. This carbamate formation stabilizes the T state and decreases O₂ affinity.
2. Indirect Effect (via pH): CO₂ is hydrated in red blood cells by the enzyme carbonic anhydrase: CO₂ + H₂O ⇌ H₂CO₃ ⇌ H⁺ + HCO₃⁻. This reaction generates H⁺, which then acts as described above.
Physiological Context: Tissues producing CO₂ from metabolism.
Result: The oxygen-binding curve shifts to the RIGHT, enhancing O₂ unloading.
3. Increased 2,3-Bisphosphoglycerate (2,3-BPG)
Mechanism: 2,3-BPG is a highly negative molecule found in red blood cells. It binds tightly to a specific central cavity in the T-state of hemoglobin, forming salt bridges with the β-chains. This binding stabilizes the T state and makes it much harder for hemoglobin to switch to the high-affinity R state.
Physiological Context:
High Altitude/Anemia: The concentration of 2,3-BPG increases as an adaptation to low oxygen availability, forcing hemoglobin to release more O₂ to the tissues.
Fetal Hemoglobin (HbF): HbF has a higher O₂ affinity than adult hemoglobin (HbA) because it has a γ-chain that binds 2,3-BPG less effectively.
Result: The oxygen-binding curve shifts dramatically to the RIGHT.
Summary of the Bohr Effect
Factor | Change in Active Tissues | Effect on Hb-O₂ Affinity | Shift in Curve |
|---|---|---|---|
pH | Decreases (↑ [H⁺]) | Decreases | Right |
pCO₂ | Increases | Decreases | Right |
[2,3-BPG] | Increases (in chronic low O₂) | Decreases | Right |
Temperature | Increases | Decreases | Right |
These factors work synergistically. For example, in a working muscle, the rise in CO₂ and H⁺, along with increased temperature, ensures that hemoglobin unloads a maximal amount of oxygen precisely where and when it is needed for aerobic respiration.
Explain myoglobin and hemoglobin´s oxygen binding curves and explain the factors that lower the affinity of hemoglobin for O2.
Summary Table: Factors Causing a Right Shift
Factor | Change | Physiological Signal For | Effect on O₂ Affinity | Result |
|---|---|---|---|---|
pH | Decrease (Acidosis) | High metabolic activity | ↓ Decreased | Enhanced O₂ unloading |
pCO₂ | Increase | High respiratory activity | ↓ Decreased | Enhanced O₂ unloading |
[2,3-BPG] | Increase | Altitude, Anemia | ↓ Decreased | Enhanced O₂ unloading |
Temperature | Increase | Exercise, Fever | ↓ Decreased | Enhanced O₂ unloading |
Discuss fetal and adult hemoglobins in terms of polypeptide chains and oxygen affinity.
The differences between fetal and adult hemoglobin are a fascinating example of molecular adaptation to ensure oxygen delivery to a developing fetus.
Overview
The key difference lies in their structure, specifically the polypeptide chains that make up the hemoglobin tetramer. This structural difference leads to a critical functional difference: fetal hemoglobin (HbF) has a higher oxygen affinity than adult hemoglobin (HbA), which is essential for transferring oxygen from the mother's bloodstream to the fetus.
1. Polypeptide Chain Composition
Feature | Adult Hemoglobin (HbA) | Fetal Hemoglobin (HbF) |
|---|---|---|
Formula | α₂β₂ | α₂γ₂ |
Chains | Two alpha (α) chains and two beta (β) chains. | Two alpha (α) chains and two gamma (γ) chains. |
The α-chain is common to both HbA and HbF.
The critical difference is the replacement of the β-chain (in HbA) with a γ-chain (in HbF). The genes for the β-chains and γ-chains are part of the same globin gene cluster, but they are expressed at different developmental stages.
2. Oxygen Affinity: Why HbF Binds Oxygen More Tightly
HbF has a significantly higher affinity for oxygen than maternal HbA. This means that at any given oxygen partial pressure (pO₂), HbF is more saturated with oxygen than HbA.
This is visually represented by a leftward shift of the oxygen dissociation curve for HbF compared to HbA.
The Molecular Mechanism: Interaction with 2,3-Bisphosphoglycerate (2,3-BPG)
The primary reason for the difference in oxygen affinity is how each hemoglobin interacts with 2,3-BPG, a potent allosteric regulator that decreases oxygen affinity.
Action of 2,3-BPG in HbA (α₂β₂):
2,3-BPG is a highly negative molecule.
It binds tightly in the central cavity of deoxyhemoglobin (the T-state).
It forms specific salt bridges with positively charged residues on the β-chains (namely, His143, Lys82, and His2).
This binding stabilizes the T-state (deoxy state), making it harder for oxygen to bind, thereby decreasing oxygen affinity.
Action of 2,3-BPG in HbF (α₂γ₂):
The γ-chain in HbF has a key amino acid substitution: a serine residue replaces the histidine at position 143.
This single change is crucial. Histidine is positively charged and contributes to 2,3-BPG binding, but serine is neutral.
Because of this substitution, the γ-chain has a lower positive charge and binds 2,3-BPG less effectively.
Consequently, 2,3-BPG binds less tightly to HbF.
Since 2,3-BPG is less effective at stabilizing the T-state, HbF is more prone to switch to the high-affinity R-state, resulting in a higher oxygen affinity.
In essence: HbF is less sensitive to the inhibitory effect of 2,3-BPG because its γ-chains don't bind it as well as the β-chains in HbA do.
3. Physiological Significance: The "Placental Oxygen Transfer System"
This difference in oxygen affinity is not just a biochemical curiosity; it is vital for survival. The fetus depends entirely on the mother for oxygen, and their blood interfaces at the placenta, but does not mix directly.
The following diagram illustrates how the difference in oxygen affinity enables efficient oxygen transfer across the placenta:
This system creates a powerful "pull" for oxygen from the maternal circulation to the fetal circulation. Without this affinity difference, the fetus would not receive adequate oxygen.
Summary Table
Feature | Adult Hemoglobin (HbA) | Fetal Hemoglobin (HbF) |
|---|---|---|
Structure | α₂β₂ | α₂γ₂ |
O₂ Affinity | Lower | Higher |
2,3-BPG Binding | Binds strongly, stabilizing the T-state. | Binds weakly due to Ser143 in γ-chain. |
Physiological Role | Transport O₂ from lungs to tissues in adults. | Transport O₂ from maternal blood to fetal tissues. |
Postnatal Presence | Predominant hemoglobin from a few months after birth throughout life. | Predominant hemoglobin during fetal development and the first few months after birth. |
In conclusion, the substitution of β-chains with γ-chains in fetal hemoglobin creates a molecule with a higher oxygen affinity, a simple yet elegant evolutionary solution to the unique challenge of intrauterine oxygen delivery.
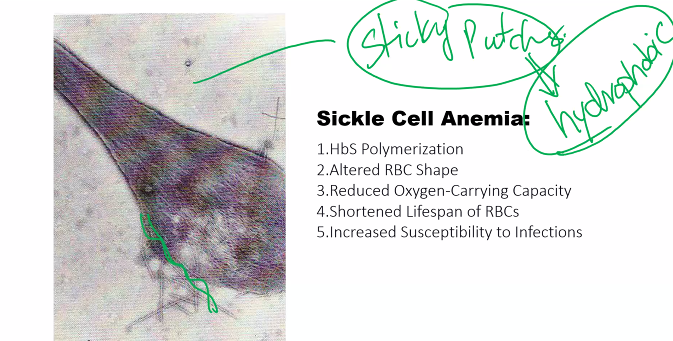
Explain the mechanism(s) associated with sickle cell anemia and thalassemias.
Sickle cell anemia and thalassemias are both inherited genetic disorders affecting hemoglobin, but they have fundamentally different mechanisms. Sickle cell is a qualitative disorder (the hemoglobin molecule is structurally abnormal), while thalassemia is a quantitative disorder (the production of normal hemoglobin is reduced).
1. Sickle Cell Anemia
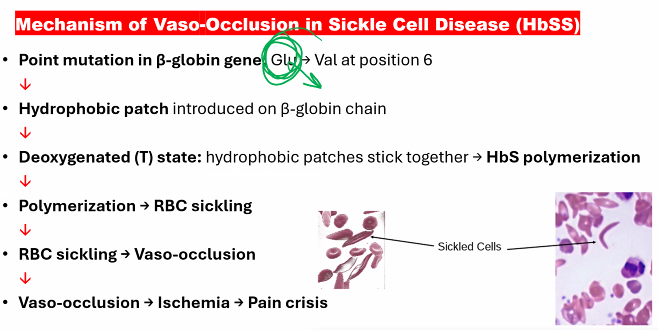
Sickle cell anemia is a hemoglobinopathy caused by a single point mutation in the gene encoding the β-globin chain.
A. Molecular Mechanism: The Glutamic Acid to Valine Swap
Genetic Defect: A single nucleotide substitution (A → T) in the 6th codon of the β-globin gene.
Amino Acid Change: This changes the codon from GAG (which codes for glutamic acid) to GUG (which codes for valine).
Resulting Hemoglobin: The abnormal hemoglobin is called Hemoglobin S (HbS).
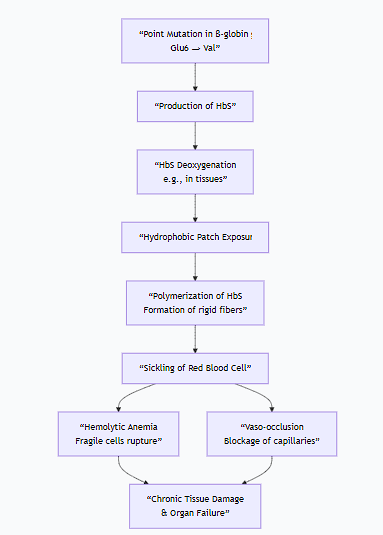
B. Pathophysiological Mechanism: Polymerization and Sickling
The replacement of a charged, hydrophilic amino acid (glutamate) with a non-polar, hydrophobic one (valine) has catastrophic consequences:
Deoxygenation and Hydrophobic Patch Exposure: When HbS releases oxygen, the valine at position 6 on the β-chain creates a hydrophobic "sticky patch" on the surface of the molecule.
Polymerization: This hydrophobic patch on a deoxy-HbS molecule fits into a complementary hydrophobic pocket on another deoxy-HbS molecule. This causes the HbS molecules to aggregate into long, rigid, rope-like fibers called polymers.
Erythrocyte Sickling: The formation of these long HbS polymers inside the red blood cell (RBC) distorts the cell's flexible, biconcave shape into a fragile, rigid, and sickle-shaped (crescent) form. This process is initially reversible but becomes permanent over time.
Vicious Cycle of Pathological Consequences: The sickled cells trigger a cascade of events:
Hemolytic Anemia: The sickled cells are fragile and rupture easily, leading to a chronic shortage of red blood cells.
Vaso-occlusion: The rigid, sickled cells are unable to flow smoothly through small capillaries. They get stuck, blocking blood flow and causing ischemic pain and organ damage (e.g., spleen, kidneys, bones, brain).
Inflammation: The blocked vessels and damaged tissues trigger severe inflammatory responses.
The following flowchart illustrates this vicious cycle:
2. Thalassemias
Thalassemias are caused by mutations that reduce or eliminate the synthesis of one of the globin chains (α or β). This leads to an imbalance in the α:β chain ratio.
A. Alpha (α)-Thalassemia
Genetic Defect: Deletions (most commonly) or mutations in one or more of the four α-globin genes (two from each parent).
Mechanism: Reduced synthesis of α-globin chains.
Consequence: Excess β-globin chains (in adults) or γ-globin chains (in newborns) accumulate.
Excess β-chains form unstable tetramers called Hemoglobin H (β₄), which precipitate and cause hemolytic anemia.
Excess γ-chains form tetramers called Hemoglobin Barts (γ₄), which have a very high oxygen affinity, preventing oxygen release to tissues.
Severity Spectrum:
Silent Carrier (1 gene deletion): Asymptomatic.
α-Thalassemia Trait (2 gene deletions): Mild microcytic anemia.
Hemoglobin H Disease (3 gene deletions): Moderate to severe hemolytic anemia.
Hydrops Fetalis (4 gene deletions): Fatal in utero or shortly after birth; no α-globin is produced, so no functional fetal or adult hemoglobin can be made.
B. Beta (β)-Thalassemia
Genetic Defect: Over 200 different point mutations (most commonly) in the two β-globin genes (one from each parent) that can reduce (β⁺) or eliminate (β⁰) β-globin production.
Mechanism: Reduced synthesis of β-globin chains.
Consequence: Excess α-globin chains accumulate.
Key Problem: Unpaired α-globin chains are extremely unstable and precipitate in the red blood cell precursors in the bone marrow.
This precipitation leads to ineffective erythropoiesis (premature death of RBC precursors), severe hemolytic anemia, and intramedullary destruction of the marrow.
Severity Spectrum:
β-Thalassemia Minor/Trait (1 mutated gene): Mild microcytic anemia.
β-Thalassemia Intermedia (2 mutated genes, but some residual β-chain production): Moderately severe anemia.
β-Thalassemia Major (2 severely mutated genes): Life-threatening, transfusion-dependent anemia presenting in the first year of life as the switch from fetal (γ) to adult (β) hemoglobin occurs.
Summary Table
Feature | Sickle Cell Anemia | Thalassemia |
|---|---|---|
Type of Defect | Qualitative (Abnormal Hb structure) | Quantitative (Reduced globin chain synthesis) |
Genetic Cause | Point mutation (Glu6Val in β-chain) | Mostly deletions (α) or point mutations (β) |
Primary Problem | Polymerization of deoxy-HbS | Imbalance of α:β globin chain ratio |
Key Pathological Event | Sickling of RBCs & Vaso-occlusion | Precipitation of unpaired globin chains & Ineffective Erythropoiesis |
Hallmark | Sickled cells on blood smear, painful crises | Severe microcytic, hypochromic anemia |
In conclusion, while both disorders disrupt hemoglobin function and cause anemia, sickle cell anemia is a disease of HbS polymerization and RBC shape leading to vaso-occlusion, whereas thalassemia is a disease of globin chain imbalance and precipitation leading to destruction of RBC precursors in the bone marrow.
Explain the importance of myoglobin and hemoglobin.
The importance of myoglobin and hemoglobin lies in their fundamental and complementary roles in oxygen management, which is essential for aerobic life in vertebrates.
In short: Hemoglobin is for oxygen transport in the blood, and myoglobin is for oxygen storage in muscle.
Here is a detailed breakdown of their distinct and critical importance.
Hemoglobin: The Essential Oxygen Transporter
Hemoglobin's importance cannot be overstated—without it, human life as we know it would be impossible.
1.Efficient Oxygen Delivery from Lungs to Tissues
High Capacity: Each hemoglobin molecule can carry four oxygen molecules, allowing blood to transport a vast amount of oxygen.
Cooperative Binding: Its sigmoidal oxygen-binding curve means it becomes very good at picking up oxygen in the high-oxygen environment of the lungs and very good at releasing it in the low-oxygen environment of the tissues. This is the basis for efficient loading and unloading.
2.Responsive Oxygen Unloading (The Bohr Effect)
This is a critical feature for meeting metabolic demand. Hemoglobin's structure allows it to sense the metabolic state of tissues and release oxygen precisely where it's needed most.
In active tissues, factors like low pH (high CO₂ and lactic acid) and high temperature cause hemoglobin's affinity for oxygen to decrease.
This results in a rightward shift of the oxygen dissociation curve, meaning more oxygen is released exactly to those tissues that are working hard and are most oxygen-starved.
3.Carbon Dioxide Transport
Hemoglobin also plays a vital role in the reverse process. It helps transport carbon dioxide (a waste product of metabolism) from the tissues back to the lungs in two ways:
As Carbamates: CO₂ binds directly to the N-termini of the globin chains.
By Buffering H⁺: The Bohr effect involves binding protons (H⁺), which are generated when CO₂ is converted to bicarbonate in the blood. This buffering action is crucial for maintaining blood pH.
Consequence of Failure: Without functional hemoglobin, oxygen cannot be delivered to tissues. This is fatal, as seen in severe carbon monoxide poisoning or certain genetic disorders like beta-thalassemia major.
Myoglobin: The Strategic Oxygen Reservoir
Myoglobin's importance is centered on supporting muscle function, especially under conditions of high demand or low supply.
1.Oxygen Storage for Muscle
Myoglobin acts as a short-term oxygen buffer within muscle cells. It binds oxygen tightly and holds onto it, creating a local reserve.
This ensures a readily available supply of oxygen inside the muscle cell, independent of momentary changes in blood flow.
2.Facilitates Oxygen Diffusion
By binding oxygen, myoglobin increases the effective solubility of oxygen in the muscle cytoplasm.
This creates a steep concentration gradient that facilitates the diffusion of oxygen from the cell membrane to the mitochondria, where it is used for energy production.
3.Sustains Aerobic Metabolism During Hypoxia
This is its most crucial role for diving mammals and during strenuous exercise in other mammals.
When blood flow is interrupted (e.g., a contracted muscle compresses its own blood vessels) or during breath-holding, the oxygen stored in myoglobin can be used to maintain aerobic ATP production for a critical period.
This prevents the muscle from having to rely solely on inefficient and acid-producing anaerobic glycolysis, thereby delaying fatigue.
Consequence of Failure: While not immediately fatal, a lack of myoglobin would impair muscle endurance. Muscles would become fatigued more quickly during sustained activity as they would be more dependent on immediate blood flow and anaerobic metabolism.
Summary Table: Their Complementary Roles
Feature | Hemoglobin | Myoglobin |
|---|---|---|
Primary Role | Oxygen Transport in blood | Oxygen Storage in muscle |
Location | Red Blood Cells | Muscle Cells (Cardiac & Skeletal) |
Structure | Tetramer (α₂β₂) - 4 heme groups | Monomer - 1 heme group |
O₂ Binding Curve | Sigmoidal (S-shaped) - Cooperative | Hyperbolic - Non-cooperative |
O₂ Affinity | Lower (Releases O₂ easily in tissues) | Very High (Holds onto O₂ tightly) |
Key Regulatory Mechanism | Bohr Effect (pH, CO₂, 2,3-BPG) | None; simple binding and release |
Evolutionary and Physiological Significance
Together, these two molecules form an elegant system:
Hemoglobin acts as a long-distance trucking system, picking up a massive cargo of oxygen in the lungs and making deliveries throughout the body.
Myoglobin acts as a local warehouse at the destination (the muscle), ensuring that the delivered oxygen is stored and available for immediate use, preventing shortages during periods of high demand.
This partnership allows large, complex, and active organisms to maintain the high metabolic rates required for endothermy (warm-bloodedness) and intense physical activity. The evolution of these efficient oxygen-handling proteins was a cornerstone for the development of advanced vertebrate life.
Describe allosterism and the Bohr effect and how they affect the affinity of hemoglobin for oxygen.
(i just put the picture on this side of the flashcard)
This is a central concept in understanding how hemoglobin is exquisitely tuned to deliver oxygen efficiently. Let's break down allosterism and the Bohr effect and how they work together to regulate hemoglobin's affinity for oxygen.
1. Allosterism in Hemoglobin
Definition: Allosterism (from Greek allos, "other," and stereos, "solid" or "shape") is the regulation of a protein's function by the binding of an effector molecule at a site other than the protein's active site.
How it Works in Hemoglobin:
The Two States: Hemoglobin exists in a dynamic equilibrium between two primary quaternary structures:
T (Tense) State: The deoxygenated form. This state is stabilized by salt bridges and has a low affinity for oxygen. It is "tense" because the subunits are more constrained.
R (Relaxed) State: The oxygenated form. This state has a high affinity for oxygen. It is "relaxed" because the salt bridges are broken and the subunits can move more freely.
The Mechanism:
The binding of an oxygen molecule to one heme group in the T-state triggers a conformational change in that subunit.
This change is transmitted to the other subunits, shifting the entire tetramer's equilibrium towards the R-state.
Once in the R-state, the remaining subunits have a much higher affinity for oxygen, making it easier for subsequent oxygen molecules to bind. This is positive cooperativity.
Key Allosteric Effectors: The molecules that bind to hemoglobin and stabilize one state over the other are:
O₂ itself: The primary ligand, binding to the heme.
H⁺ (Protons), CO₂, and 2,3-BPG (2,3-Bisphosphoglycerate): These are the classic allosteric effectors that bind to sites other than the heme and stabilize the T-state, thereby decreasing oxygen affinity.
In summary, allosterism is the "communication" between subunits that allows for cooperative binding and regulation by other molecules.
2. The Bohr Effect
Definition: The Bohr effect is a specific, physiological example of allosteric regulation. It is the decrease in hemoglobin's oxygen affinity caused by a decrease in blood pH (an increase in H⁺ concentration) or an increase in CO₂.
This effect ensures that hemoglobin unloads more oxygen precisely in the tissues that need it most—those that are metabolically active and producing acid and CO₂.
How it Works:
The Bohr effect has two interconnected components:
1. The Acid (H⁺) Component:
Mechanism: Protons (H⁺) bind to specific amino acid residues in hemoglobin (notably Histidine β146 and the N-termini of the α-chains).
Effect: This binding stabilizes the T-state by forming additional salt bridges that "lock" the deoxy conformation.
Result: A lower pH (more H⁺) causes a rightward shift of the oxygen dissociation curve, meaning more oxygen is released at any given partial pressure of oxygen (pO₂).
2. The CO₂ Component:
CO₂ exerts its effect in two ways:
Directly: CO₂ can bind to the N-terminal amino groups of hemoglobin to form carbamates. This carbamate formation stabilizes the T-state.
Indirectly (and more importantly): CO₂ is converted in red blood cells by the enzyme carbonic anhydrase into carbonic acid, which rapidly dissociates:
CO₂ + H₂O ⇌ H₂CO₃ ⇌ H⁺ + HCO₃⁻
This reaction generates H⁺, which then acts as described in the acid component above.
How They Work Together to Affect Oxygen Affinity
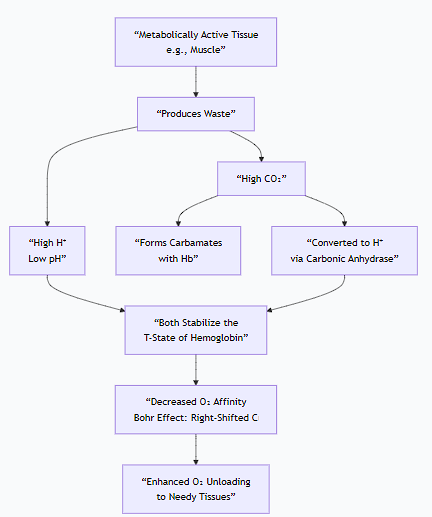
The following diagram illustrates how allosteric effectors like H⁺ and CO₂ promote oxygen release in metabolically active tissues:
Physiological Workflow in a Tissue Capillary:
Metabolic Activity: A working muscle produces CO₂ and lactic acid (H⁺).
Allosteric Binding: The CO₂ and H⁺ diffuse into the red blood cell. H⁺ binds directly to hemoglobin, and CO₂ forms carbamates with it.
Stabilization of T-state: The binding of these effectors stabilizes hemoglobin's low-affinity T-state.
Decreased Affinity: This stabilization makes it harder for hemoglobin to hold onto oxygen, significantly decreasing its oxygen affinity.
Enhanced Oxygen Release: More oxygen is released from hemoglobin than would be under normal pH/CO₂ conditions.
Reverse in the Lungs: In the lungs, the process reverses. High O₂ levels promote CO₂ and H⁺ release from hemoglobin (the Haldane Effect), which shifts the equilibrium back to the high-affinity R-state, ready to pick up oxygen again.
Summary of Effects
Effector | Binding Site on Hb | State Stabilized | Effect on O₂ Affinity | Physiological Role |
|---|---|---|---|---|
O₂ | Heme Iron (active site) | R-state | Increases | Cooperative binding |
H⁺ (Low pH) | Histidine residues | T-state | Decreases | Bohr Effect: Releases O₂ in active tissues |
CO₂ | N-terminus (carbamates) | T-state | Decreases | Bohr Effect: Releases O₂ and aids CO₂ transport |
2,3-BPG | Central cavity (β-chains) | T-state | Decreases | Promotes O₂ release in tissues; altitude adaptation |
In conclusion, allosterism is the general mechanism that allows hemoglobin's function to be regulated, and the Bohr effect is a specific, life-saving consequence of this mechanism, ensuring that oxygen delivery is dynamically matched to metabolic demand.
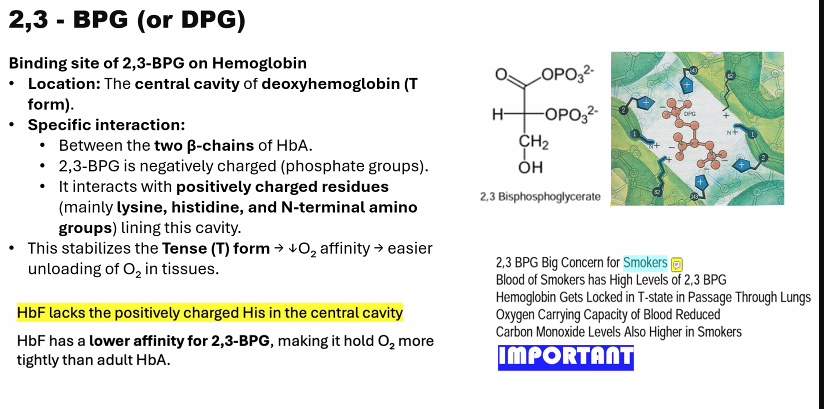
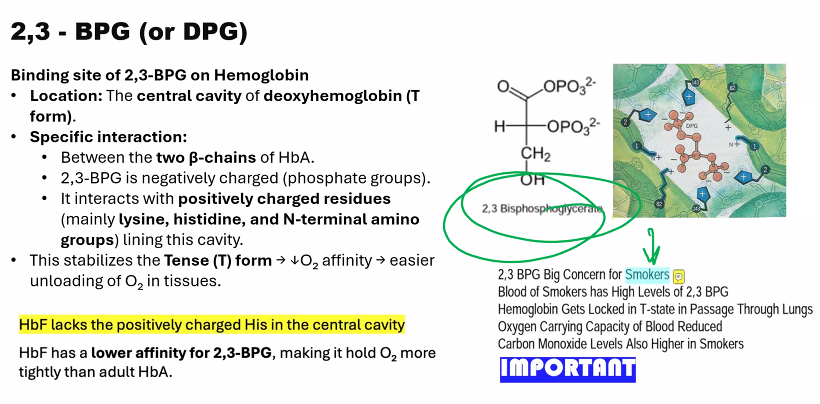
Describe the effect of 2,3-biphospho-glycerate (BPG) on hemoglobin´s oxygen affinity.
The effect of 2,3-Bisphosphoglycerate (BPG, or 2,3-DPG) on hemoglobin is a crucial allosteric mechanism that dramatically fine-tunes oxygen delivery to tissues.
Overview
2,3-BPG is a potent allosteric effector that significantly decreases the oxygen affinity of hemoglobin. This ensures that hemoglobin releases its bound oxygen more readily in the peripheral tissues.
1. The Molecular Mechanism: How BPG Binds
The mechanism is a perfect example of stereospecific, allosteric regulation:
Structure of BPG: BPG is a highly negatively charged molecule.
Binding Site: BPG binds specifically and reversibly to a single, positively charged pocket in the center of the deoxygenated hemoglobin (T-state) tetramer. This pocket is located between the two β-globin chains.
Ionic Interactions: Upon binding, BPG forms salt bridges (ionic bonds) with positively charged residues on the β-chains, including:
The N-terminal amino groups
Histidine 2
Lysine 82
Histidine 143
Stabilization of the T-State: By forming these strong, cross-linking ionic bonds, BPG acts as a "molecular clamp" that stabilizes the T-state (deoxy) conformation of hemoglobin.
2. The Functional Consequence: Lowered Oxygen Affinity
The T-state has a naturally low affinity for oxygen.
By stabilizing this state, BPG makes it more difficult for hemoglobin to undergo the conformational switch to the high-affinity R-state.
As a result, a higher concentration of oxygen (a higher pO₂) is required to achieve the same level of saturation. In other words, hemoglobin holds onto oxygen less tightly and releases it more easily at the oxygen partial pressures found in tissues.
This effect is visually represented by a rightward shift of the oxygen dissociation curve.
3. Physiological Importance and Regulation
The body actively regulates BPG levels to adapt to different physiological and pathological conditions.
Condition | Change in [BPG] | Effect & Rationale |
|---|---|---|
High Altitude / Hypoxia | Increases | A compensatory mechanism to enhance oxygen unloading to tissues when oxygen availability is low. |
Anemia | Increases | Helps to maximize the oxygen delivery from the reduced number of red blood cells. |
Chronic Lung Disease | Increases | Improves oxygen release in the face of impaired lung function. |
Blood Storage | Decreases | BPG levels fall in stored blood over several weeks. Transfused blood with low BPG has a very high O₂ affinity, which can initially impair oxygen delivery to tissues until the recipient's body regenerates BPG. |
4. The Critical Difference in Fetal Hemoglobin (HbF)
This mechanism also explains a key difference between adult (HbA) and fetal hemoglobin (HbF).
HbF has a γ-globin chain instead of the β-globin chain.
A key mutation in the γ-chain: Histidine 143 (present in β-chain) is replaced by Serine.
Serine is neutral and does not carry a positive charge.
This single change reduces the positive charge of the BPG-binding pocket, meaning BPG binds less tightly to HbF.
Consequence: Because BPG binds less effectively, HbF is less stabilized in the T-state and has a higher oxygen affinity than maternal HbA. This creates the essential "pull" of oxygen from the mother's bloodstream, across the placenta, and to the fetus.
Summary
Molecule: 2,3-Bisphosphoglycerate (BPG)
Action: Allosteric inhibitor of hemoglobin.
Mechanism: Binds tightly to and stabilizes the deoxygenated T-state.
Effect: Decreases oxygen affinity, promoting oxygen unloading in tissues.
Importance: A vital adaptive response to hypoxia and a key differentiator between fetal and adult hemoglobin.
![<p>The effect of <strong>2,3-Bisphosphoglycerate (BPG, or 2,3-DPG)</strong> on hemoglobin is a crucial allosteric mechanism that dramatically fine-tunes oxygen delivery to tissues.</p><p>Overview</p><p class="ds-markdown-paragraph"><strong>2,3-BPG is a potent allosteric effector that significantly decreases the oxygen affinity of hemoglobin.</strong> This ensures that hemoglobin releases its bound oxygen more readily in the peripheral tissues.</p><div data-type="horizontalRule"><hr></div><p>1. The Molecular Mechanism: How BPG Binds</p><p class="ds-markdown-paragraph">The mechanism is a perfect example of stereospecific, allosteric regulation:</p><ul><li><p class="ds-markdown-paragraph"><strong>Structure of BPG:</strong> BPG is a highly negatively charged molecule.</p></li><li><p class="ds-markdown-paragraph"><strong>Binding Site:</strong> BPG binds specifically and reversibly to a single, positively charged pocket in the <strong>center of the deoxygenated hemoglobin (T-state) tetramer</strong>. This pocket is located between the two β-globin chains.</p></li><li><p class="ds-markdown-paragraph"><strong>Ionic Interactions:</strong> Upon binding, BPG forms <strong>salt bridges</strong> (ionic bonds) with positively charged residues on the β-chains, including:</p><ul><li><p class="ds-markdown-paragraph">The N-terminal amino groups</p></li><li><p class="ds-markdown-paragraph"><strong>Histidine 2</strong></p></li><li><p class="ds-markdown-paragraph"><strong>Lysine 82</strong></p></li><li><p class="ds-markdown-paragraph"><strong>Histidine 143</strong></p></li></ul></li><li><p class="ds-markdown-paragraph"><strong>Stabilization of the T-State:</strong> By forming these strong, cross-linking ionic bonds, BPG acts as a "molecular clamp" that <strong>stabilizes the T-state (deoxy) conformation</strong> of hemoglobin.</p></li></ul><p>2. The Functional Consequence: Lowered Oxygen Affinity</p><ul><li><p class="ds-markdown-paragraph">The T-state has a naturally low affinity for oxygen.</p></li><li><p class="ds-markdown-paragraph">By stabilizing this state, BPG makes it <strong>more difficult for hemoglobin to undergo the conformational switch to the high-affinity R-state</strong>.</p></li><li><p class="ds-markdown-paragraph">As a result, a higher concentration of oxygen (a higher pO₂) is required to achieve the same level of saturation. In other words, hemoglobin holds onto oxygen less tightly and <strong>releases it more easily</strong> at the oxygen partial pressures found in tissues.</p></li></ul><p class="ds-markdown-paragraph">This effect is visually represented by a <strong>rightward shift of the oxygen dissociation curve</strong>.</p><p>3. Physiological Importance and Regulation</p><p class="ds-markdown-paragraph">The body actively regulates BPG levels to adapt to different physiological and pathological conditions.</p><table style="min-width: 75px;"><colgroup><col style="min-width: 25px;"><col style="min-width: 25px;"><col style="min-width: 25px;"></colgroup><tbody><tr><th colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.12); font: 500 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; border-top: none; padding: 10px 16px 10px 0px; text-align: left;"><p><strong>Condition</strong></p></th><th colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.12); font: 500 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; border-top: none; padding: 10px 16px; text-align: left;"><p><strong>Change in [BPG]</strong></p></th><th colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.12); font: 500 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; border-top: none; padding: 10px 16px; text-align: left;"><p><strong>Effect & Rationale</strong></p></th></tr><tr><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 16px 10px 0px;"><p><strong>High Altitude / Hypoxia</strong></p></td><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 16px;"><p><strong>Increases</strong></p></td><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 0px 10px 16px;"><p>A compensatory mechanism to enhance oxygen unloading to tissues when oxygen availability is low.</p></td></tr><tr><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 16px 10px 0px;"><p><strong>Anemia</strong></p></td><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 16px;"><p><strong>Increases</strong></p></td><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 0px 10px 16px;"><p>Helps to maximize the oxygen delivery from the reduced number of red blood cells.</p></td></tr><tr><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 16px 10px 0px;"><p><strong>Chronic Lung Disease</strong></p></td><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 16px;"><p><strong>Increases</strong></p></td><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 0px 10px 16px;"><p>Improves oxygen release in the face of impaired lung function.</p></td></tr><tr><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 16px 10px 0px;"><p><strong>Blood Storage</strong></p></td><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 16px;"><p><strong>Decreases</strong></p></td><td colspan="1" rowspan="1" style="border-bottom: 1.06667px solid rgba(0, 0, 0, 0.1); font: 400 15px / 25px quote-cjk-patch, Inter, system-ui, -apple-system, BlinkMacSystemFont, "Segoe UI", Roboto, Oxygen, Ubuntu, Cantarell, "Open Sans", "Helvetica Neue", sans-serif; min-width: 100px; max-width: max(30vw, 320px); padding: 10px 0px 10px 16px;"><p>BPG levels fall in stored blood over several weeks. Transfused blood with low BPG has a very high O₂ affinity, which can initially impair oxygen delivery to tissues until the recipient's body regenerates BPG.</p></td></tr></tbody></table><p>4. The Critical Difference in Fetal Hemoglobin (HbF)</p><p class="ds-markdown-paragraph">This mechanism also explains a key difference between adult (HbA) and fetal hemoglobin (HbF).</p><ul><li><p class="ds-markdown-paragraph"><strong>HbF</strong> has a <strong>γ-globin chain</strong> instead of the β-globin chain.</p></li><li><p class="ds-markdown-paragraph">A key mutation in the γ-chain: <strong>Histidine 143 (present in β-chain) is replaced by Serine</strong>.</p></li><li><p class="ds-markdown-paragraph">Serine is neutral and does not carry a positive charge.</p></li><li><p class="ds-markdown-paragraph">This single change <strong>reduces the positive charge</strong> of the BPG-binding pocket, meaning BPG binds <strong>less tightly</strong> to HbF.</p></li></ul><p class="ds-markdown-paragraph"><strong>Consequence:</strong> Because BPG binds less effectively, HbF is less stabilized in the T-state and has a <strong>higher oxygen affinity</strong> than maternal HbA. This creates the essential "pull" of oxygen from the mother's bloodstream, across the placenta, and to the fetus.</p><p>Summary</p><ul><li><p class="ds-markdown-paragraph"><strong>Molecule:</strong> 2,3-Bisphosphoglycerate (BPG)</p></li><li><p class="ds-markdown-paragraph"><strong>Action:</strong> Allosteric inhibitor of hemoglobin.</p></li><li><p class="ds-markdown-paragraph"><strong>Mechanism:</strong> Binds tightly to and stabilizes the deoxygenated T-state.</p></li><li><p class="ds-markdown-paragraph"><strong>Effect:</strong> <strong>Decreases oxygen affinity</strong>, promoting oxygen unloading in tissues.</p></li><li><p class="ds-markdown-paragraph"><strong>Importance:</strong> A vital adaptive response to hypoxia and a key differentiator between fetal and adult hemoglobin.</p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/e7549c1c-38bd-4ee4-8b47-5f339c881f85.png)
A) hemoglobin has a LOWER p50, so it has a HIGHER AFFINITY.

what is the relationship between partial pressure and affinity?
INVERSE.
if the curve shifts to the LEFT (affinity increases) as partial pressure DECREASES.
two phases
t state: “tense”
r state: “relaxed”
when oxygen is bound to hemoglobin, hemoglobin has a HISTIDINE146 in the BETA CHAIN

you need to know the mechanism of hemoglobin.
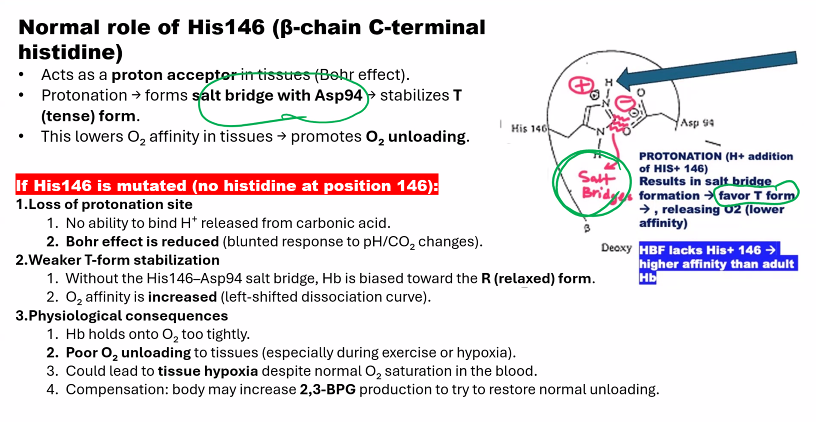
MEMORIZE THIS.
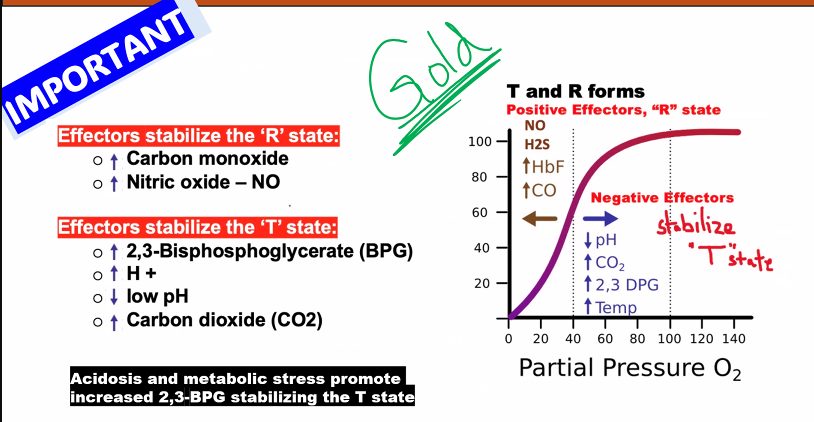
C)

D)
in fact, all of these, except A, favor the T state.
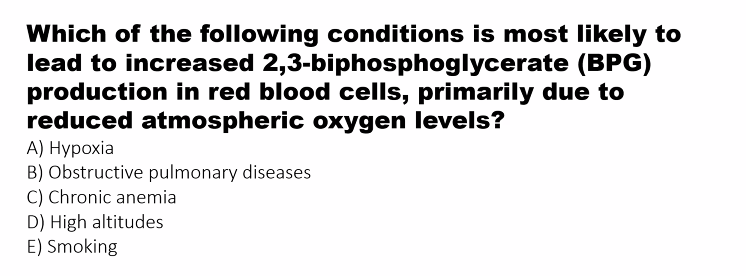
B)
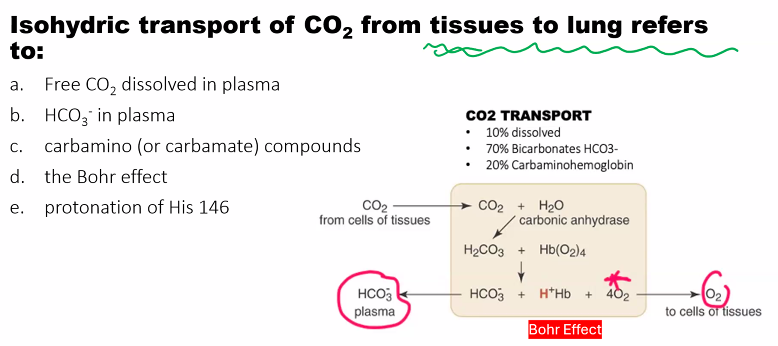

B)
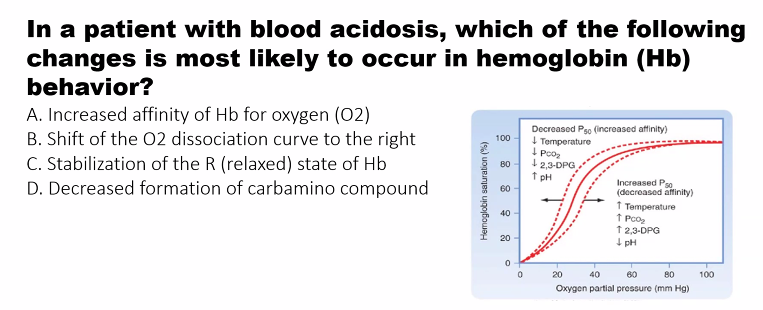
A)

B)
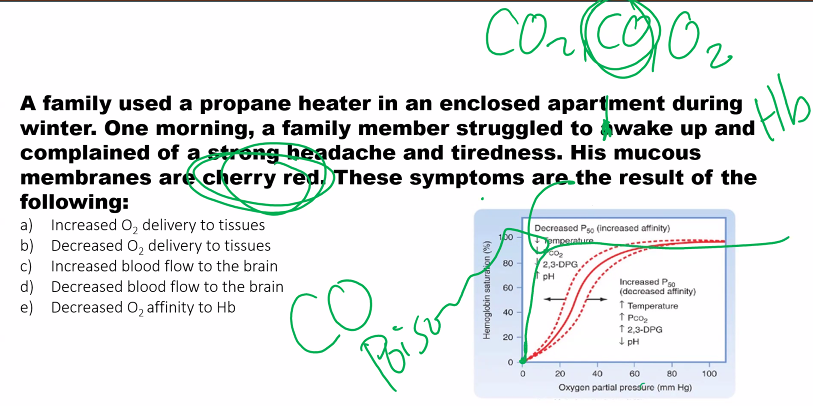
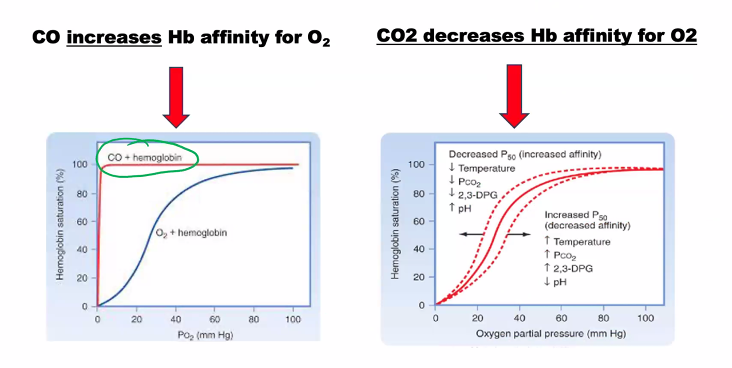

B)


B)