!! DNA Sequencing
1/38
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
39 Terms
Human genome project
1990-2003.
Goal:
It was a global scientific research project whose primary goal was to generate the first sequence of the human genome.
To determine the base pairs that make up human DNA and identify, map, and sequence all of the genes of the human genome from both a physical and functional standpoint.
Finding:
Sequenced 92% of the human genome with fewer than 400 gaps.
About 23,000 genes.
Significantly more segmental
duplication.
Major impact:
Personalized health care.
Cancer research.
Cost:
$3 billion over 15 years.
Sequencing pipeline
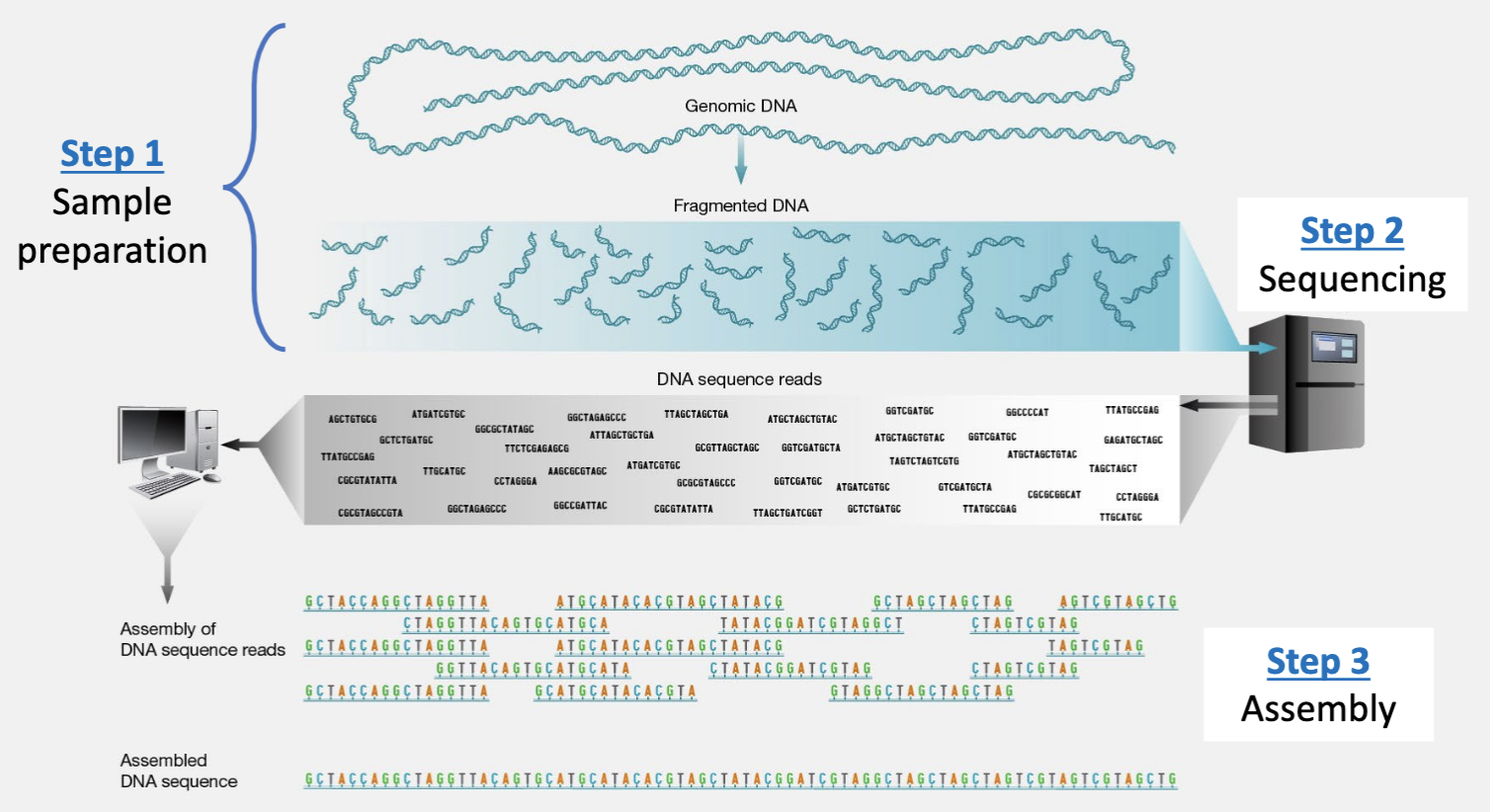
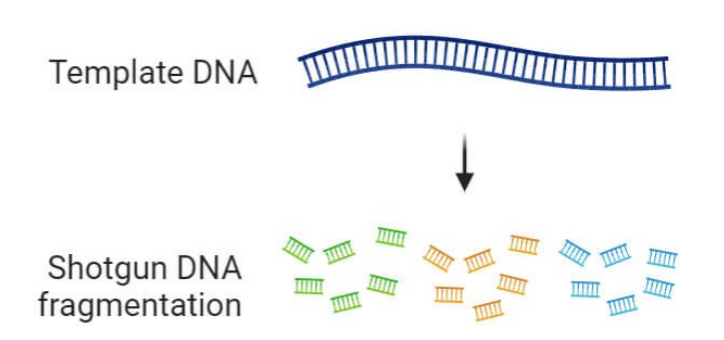
Step 1: sample preparation
Shotgun sequencing:
Randomly fragment the entire DNA sample into millions of small, overlapping pieces.
Random reads.
Step 2: sequencing
First generation: Sanger sequencing.
Second generation (next generation): Illumina.
Third generation: Oxford nanopore.
Sanger sequencing
A chain termination method that reads DNA sequence one fragment at a time.
Low throughput.
It is high-cost, making it less cost-effective for large projects.
Sanger sequencing process
Sample Preparation: A single-stranded copy of the DNA segment to be sequenced is prepared.
The Sequencing Reaction: The reaction mixture contains DNA polymerase, a primer, standard deoxynucleotides (A, T, G, C), and a small amount of chain-terminating dideoxynucleotides (ddNTPs). The ddNTPs are key because they stop DNA synthesis when incorporated into a growing DNA strand.
A binds to T.
G binds to C.
The number of short segments depends on how many binding base pairs are present.
For example, if there are three Ts and the DNA segment is A, then there will be three short segments.
Creating Fragments: The reaction continues until all strands of DNA have undergone this reaction. Because the ddNTPs are in a low concentration, DNA polymerase will make copies of the DNA segment that terminate at every possible position where a specific nucleotide (A, T, C, or G) is found.
Separating the Fragments: The resulting fragments are then separated by size using electrophoresis. The longer DNA fragments move more slowly than the shorter ones.
Heavy = slow.
Light = short.
Reading the Sequence: The sequence is then read directly from the gel, starting with the shortest fragments and ending with the longest. This process determines the order of the nucleotides and thus the DNA sequence.
Illumina sequencing pipeline
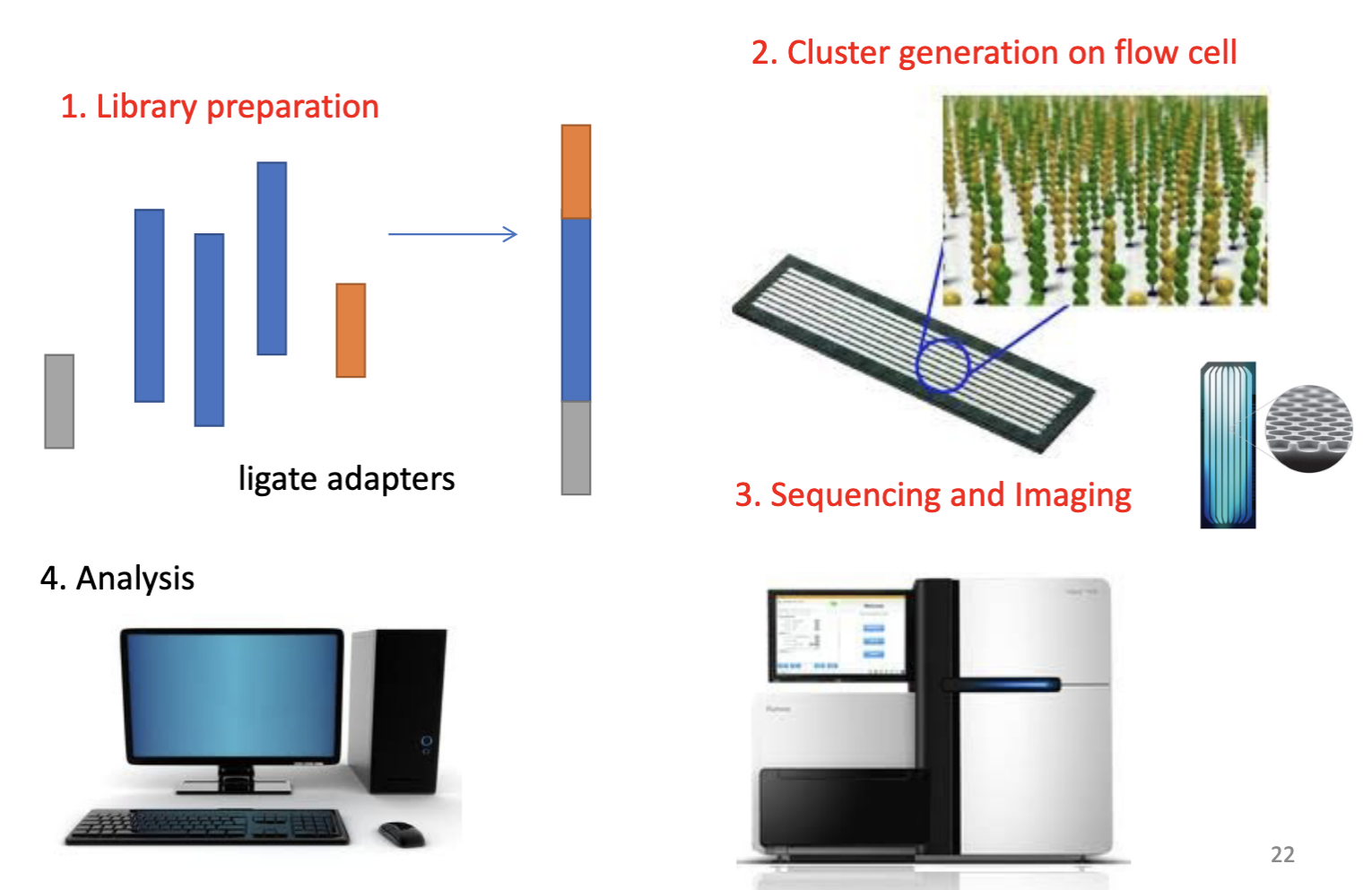
Step 1 - Illumina sequencing
Step 1 = library preparation.
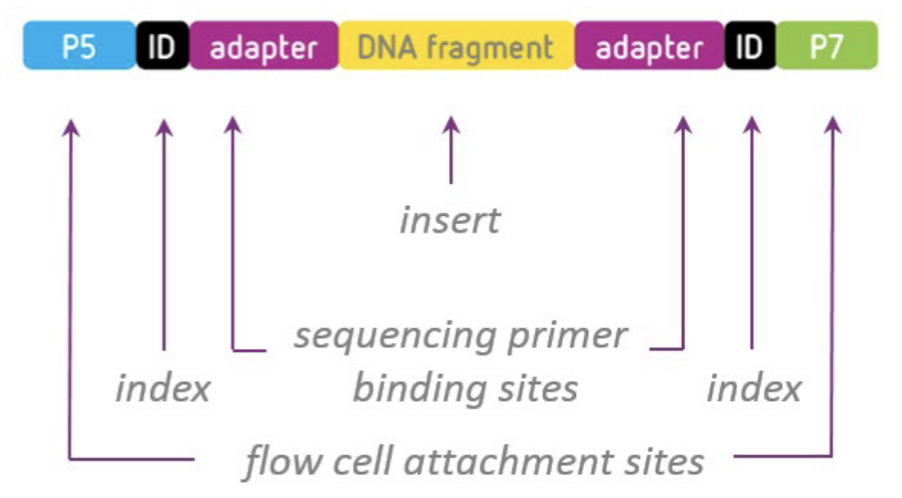
Adapters attach to DNA fragments to provide priming sites for cluster amplification and sequencing on the flow cell.
Adapters are a pair of annealed oligonucleotides that facilitate clonal amplification and sequencing reactions. Identical duplex adapters are ligated to both ends of the library fragments so that oligos on the flow cell can recognize them for sequencing.
Adapter ligation is a method of attaching synthetic oligonucleotides with known sequences
It can also incorporate index barcodes that allow multiplexing of multiple samples in a single run.
On the 8-lane flow cell:
Each spot has an attached oligo.
Oligo = oligonucleotide = single-stranded fragment of DNA or RNA.
DNA fragments need to bind these oligos to attach to the surface.
Add adaptors to fragments.
PCR (polymerase chain reaction) is a molecular biology technique used to make many copies (amplify) of small sections of DNA or a gene.
Using PCR, we can generate thousands to millions of copies of a particular section of DNA from a minimal amount of DNA so that we can visualize them.
Nowadays, Illumina also provides an option with PCR-free sequencing.
Indexes, also known as barcodes, are used to identify which read corresponds to which sample after sequencing bioinformatically
Step 2 - Illumina sequencing
Step 2 = cluster generation (bridge amplification).
How it works:
DNA fragments are prepared with adapters that attach to oligos (short DNA molecules) on the flow cell surface.
The fragments bind to these oligos on the flow cell to create a “bridge.”
DNA polymerase copies the stand, and the new strand attaches to nearby oligos, creating clusters of identical DNA molecules.
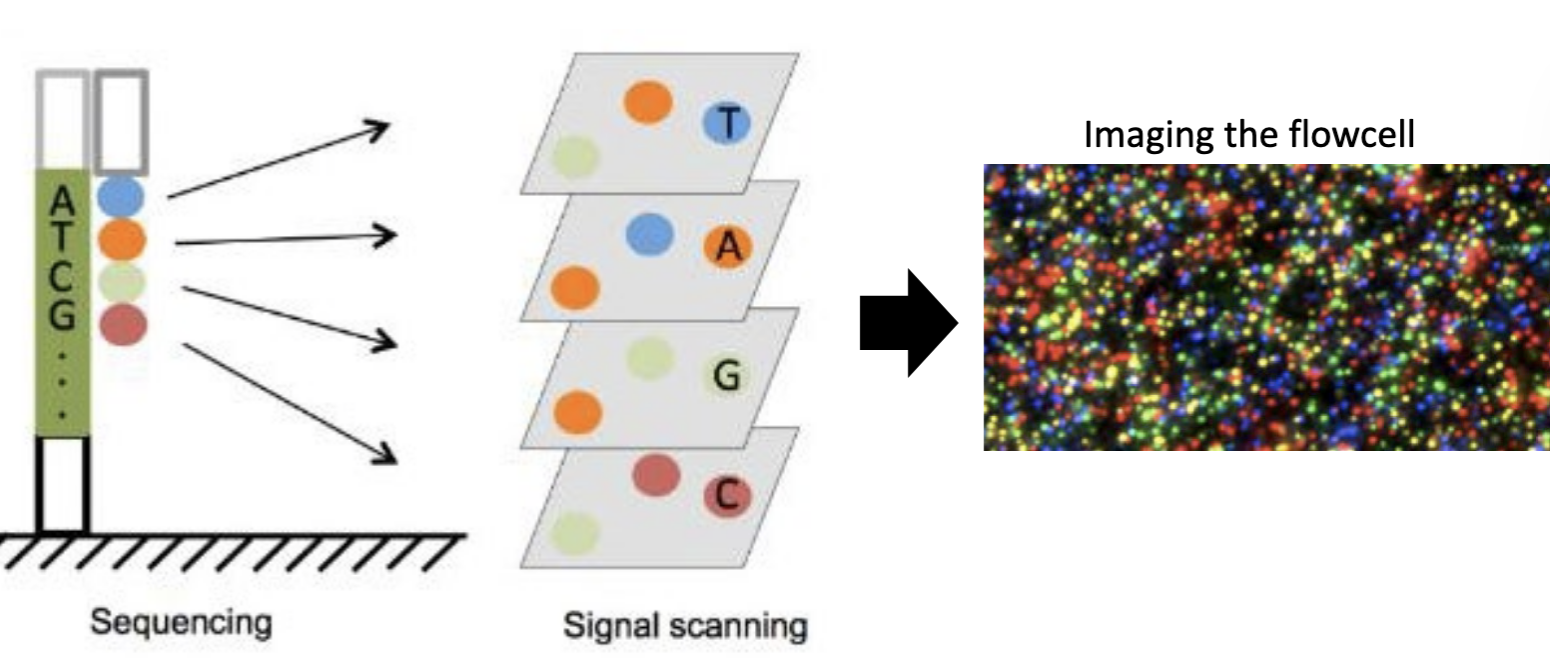
Step 3 - Illumina sequencing
Step 3 = sequencing through imaging.
The process sequences native DNA in real-time with single-molecule resolution.
It utilizes tiny nanopores on the machine's surface to measure changes in electric current (color) as single-stranded DNA passes through.
Each base has a unique color that the camera records.
Short reads
DNA fragments are typically longer than what can be fully sequenced.
Reads are Short DNA sequences (A, T, G, C) from individual DNA fragments.
Illumina sequencing reads are typically between 50 and 300 base pairs (bp).
Depending on the sequencing platform and the number of cycles selected.
Single read:
Sequence a DNA fragment from only one end.
Generate one continuous stretch of bases (typically 50 to 300 bp) per fragment.
Paired-end read
Random DNA fragment with an approximately known size.
Both ends of the fragments are sequenced.
Paired-end reads refer to the two ends of the same DNA molecule. You can sequence one end, then turn it around and sequence the other. The two sequences you get are paired-end reads or “mate-pairs”
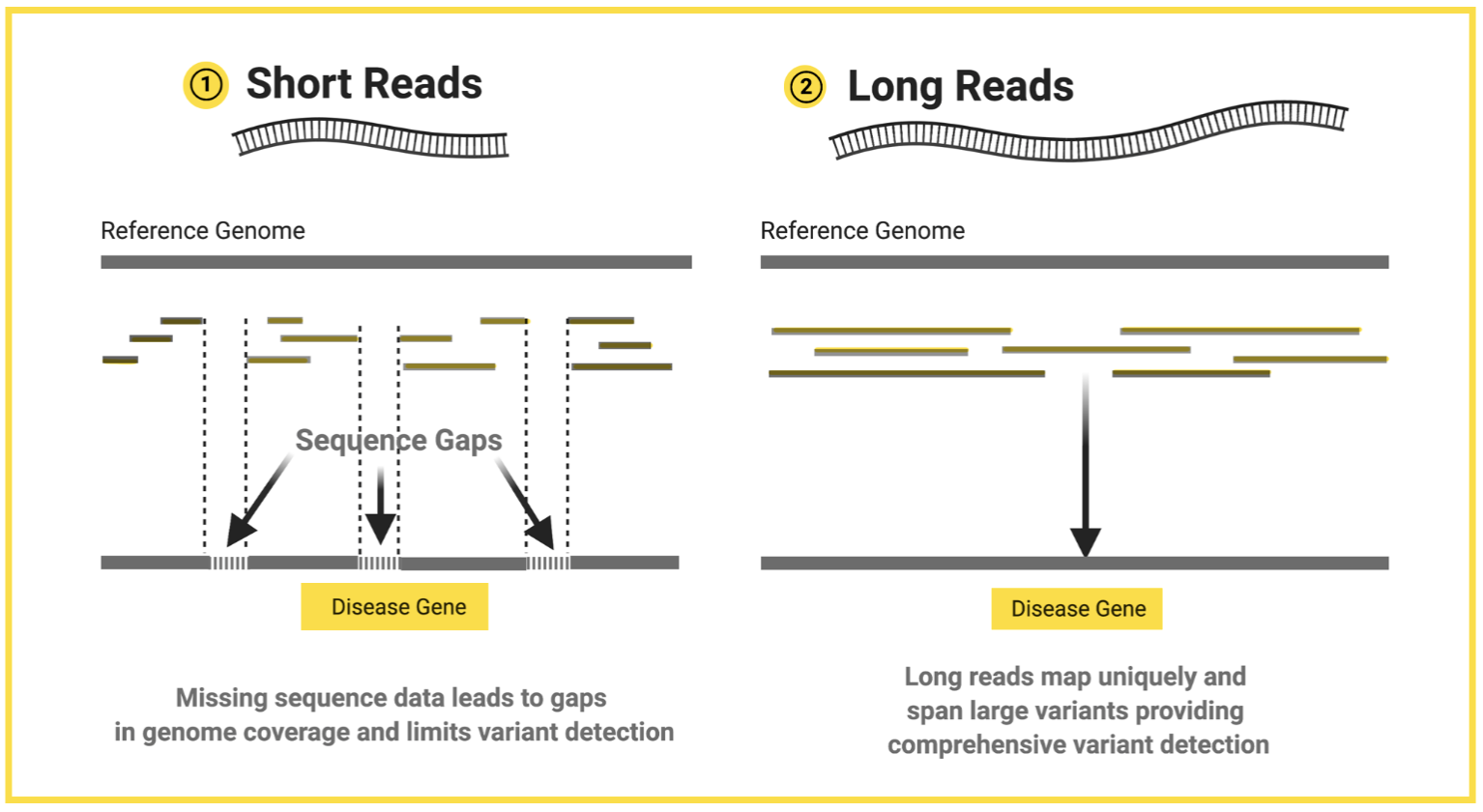
Limitations of short reads
Illumina platforms sequence short reads (up to 300 bps).
Limitations in identifying structure variations.
Short length makes it difficult to span large insertions, deletions, inversions, or repetitive regions, leading to ambiguous or incomplete mapping.
As they are short, it may not be possible to map reads to the specific region of the reference genome they came from. Complex genomic regions, structural variations, and large stretches of repetitive sequences can push short read sequencing methods to their limits, leading to gaps and ambiguities in the assembled sequences.
Long reads vs. short reads
Short reads = Illumina (second-generation).
Long reads = Oxford nanopore (third-generation).
!! Pacific Biosciences
Pacific Biosciences: ??
Long reads: on average, a few kbs.
what are kbs?
Rely on the signal from a single molecule.
High error rates: uniformly random errors, up to 15%
Does single-molecule, real-time sequencing (SMRT).
SMRT long-read sequencing
SMRT detects fluorescence events corresponding to adding one specific nucleotide by a polymerase tethered to the bottom of a tiny well.
Every well has a polymerase molecule attached inside.
Polymerase helps fill in nucleotides on a single-stranded piece of DNA.
Each time the polymerase adds a base, a camera takes a picture.
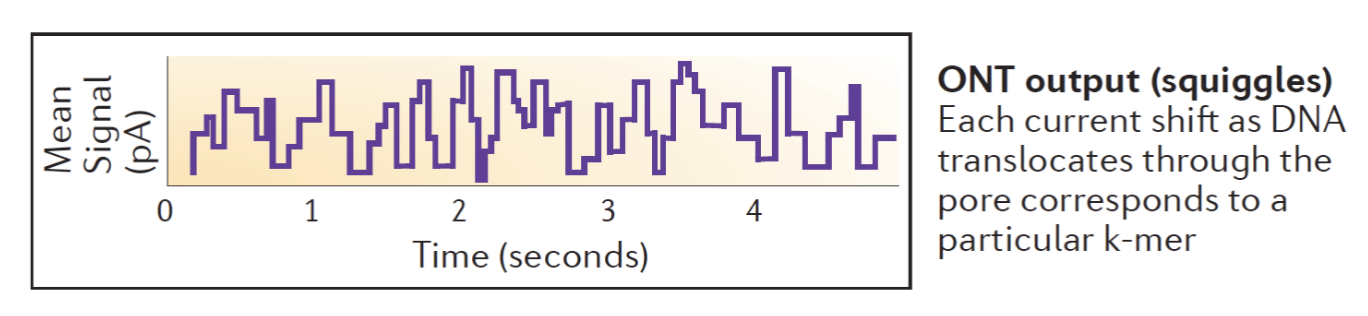
Oxford Nanopore technology
Each nucleotide base is a different size and has different electrical properties.
The wells of the machine measure the electrical current changes that occur when single-stranded DNA pass through tiny nanopores on the surface.
Each base has its own electrical signature that the machine measures and records.
Picture: output.
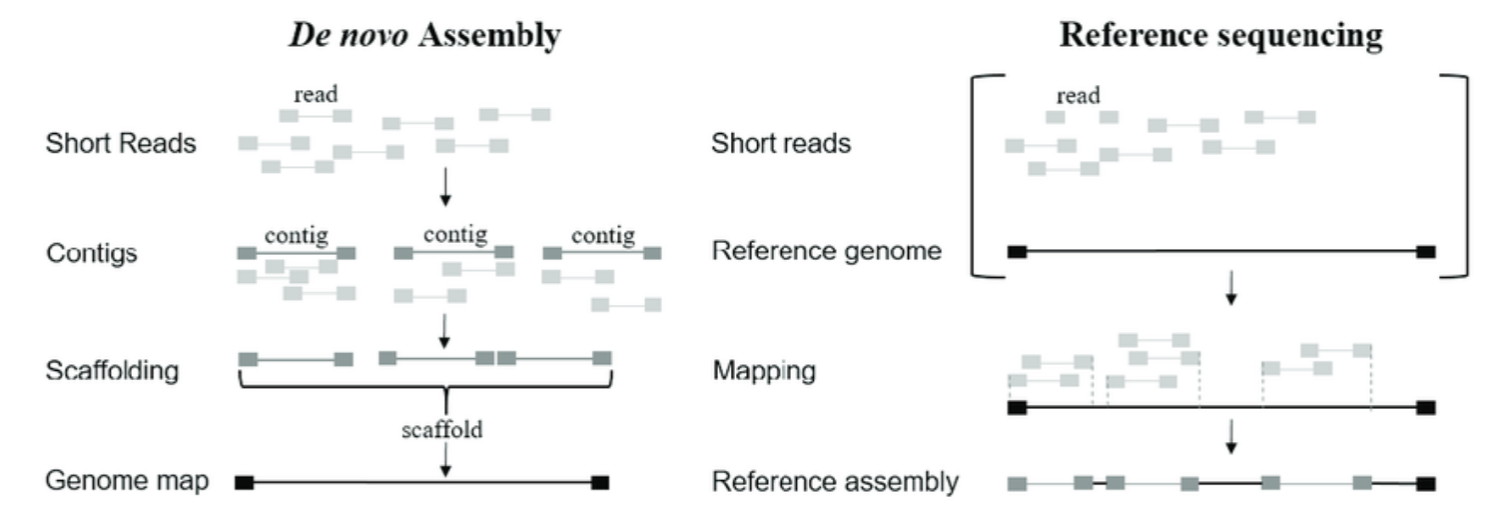
Genome assembly
Alignment and merging of reads to determine their original order and form a continuous representation of chromosomes.
De novo assembly: assemble from scratch without reference.
Reference-based assembly: map reads directly to an already assembled reference sequence.
!! Reference-based assembly
Reference genome: a digital nucleic acid sequence database, assembled by scientists as a representative example of the set of genes in one idealized individual organism of a species.
The reference genome provides a consensus sequence (coordinates) to which individuals’ data can be compared.
Reference genome examples:
hg19 (GRCh37.xx).
hg38 (GRCh38.xx).
HOW DO YOU KNOW WHICH ONE TO CHOOSE???
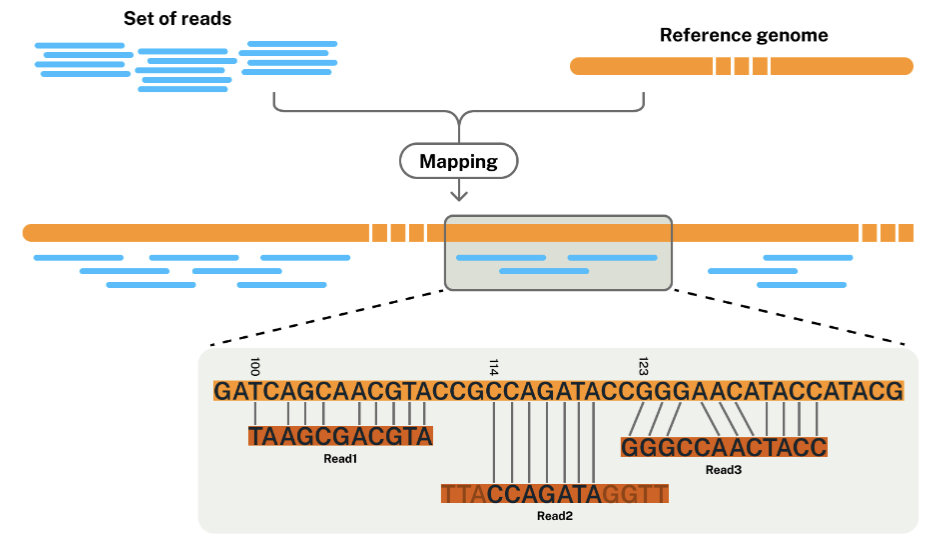
De novo assembly
Without an established reference genome.
Piecing together short reads into a complete genome by finding overlaps between them.
Like piecing together a puzzle without a picture.
Contigs: overlapping reads to form longer, continuous sequences.
Gaps in contigs arise due to technical limitations and biological complexities.
Prevent the assembler from joining all reads into a continuous sequence.
Scaffolds: Contigs are organized using additional information to provide a higher-order structure.
Composed of >=1 contigs, separated by gaps, with unknown sequence.
Technical limitations:
Short reads may not span repetitive regions or complex genomic structures, making it hard for the assembler to order and orient them correctly.
Some regions of the genome are not well-covered by reads due to bias in sequencing (e.g., GC content bias), random sampling variability, poor-quality DNA,
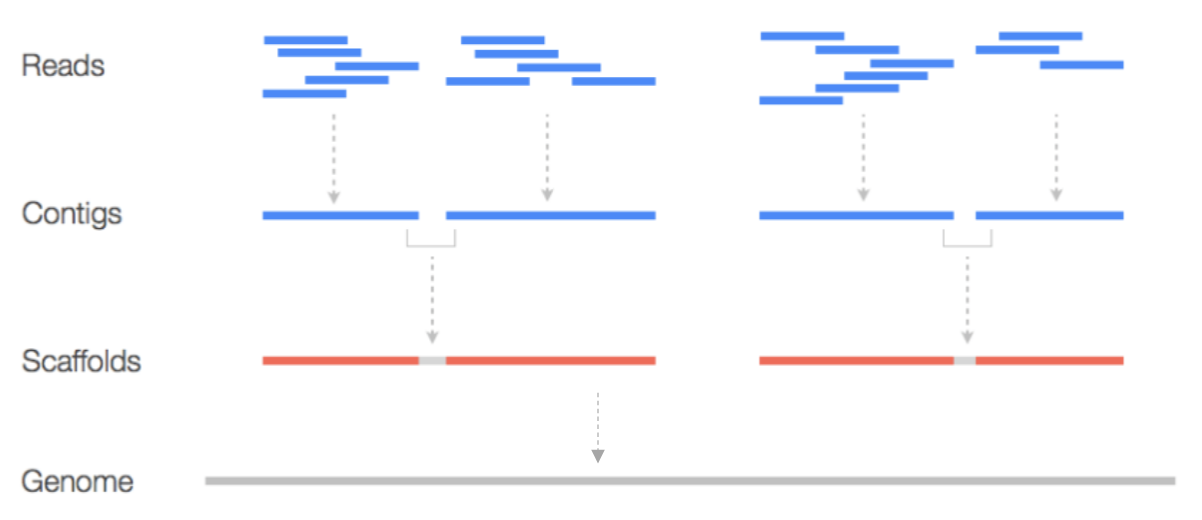
Genome assembly example
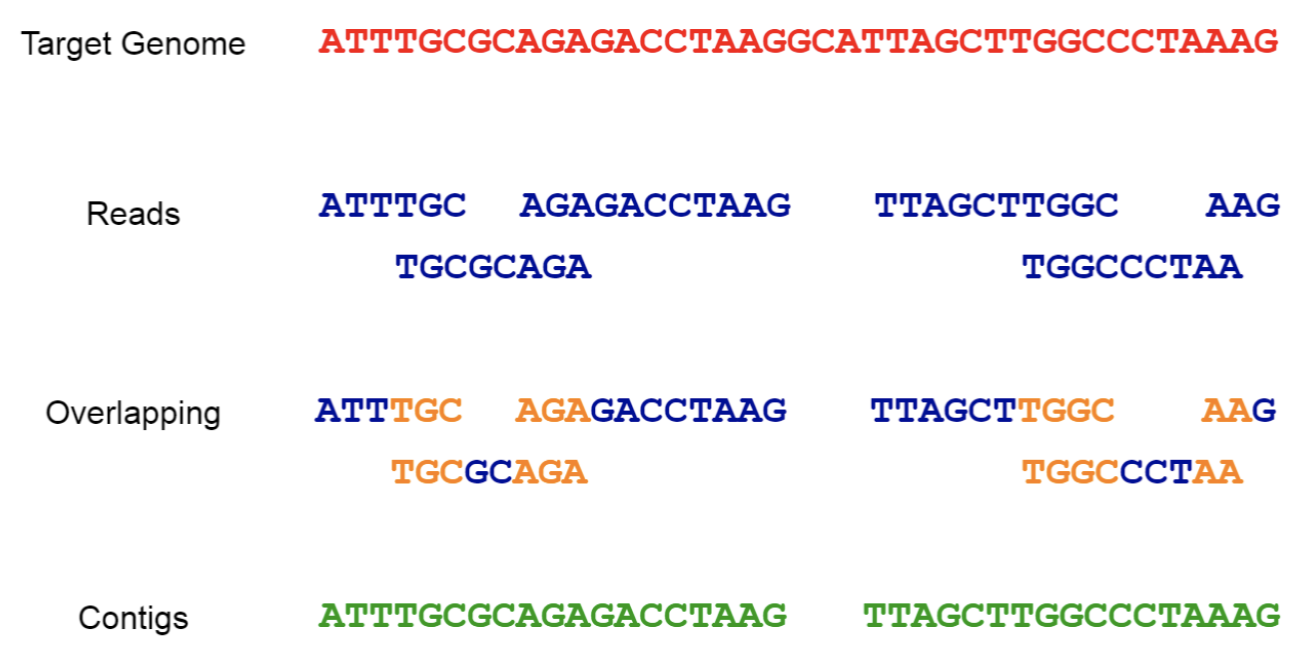
Paired-end reads for genome assembly
In reference-based assembly, paired reads improve alignment accuracy across repetitive or ambiguous regions.
In de novo assembly, they help bridge gaps and resolve repeats by linking contigs based on known insert sizes and orientations.
Paired-end sequencing might not necessarily provide sequencing data for the entire length of the fragment, but it can help bridge the gaps between contigs, as the distance between the paired reads is known.
!! Common approaches for de novo assembly
Overlap-layout-consensus (OLC) method:
Create a graph where nodes are reads and edges represent sequence overlaps.
Suitable for long and error-prone reads.
Inefficient for very large datasets.
De Bruijn graph (DBG) framework:
Decomposes reads into shorter k-mers and connects them into a graph based on overlaps.
WHAT ARE K-MERS?
Suitable for short, accurate reads.
De Bruijn graph construction
Choose a value of k.
For each k-mer that exists in any sequence create an edge with one node labeled as the prefix and one node labeled as the suffix.
Glue all nodes that have the same label.
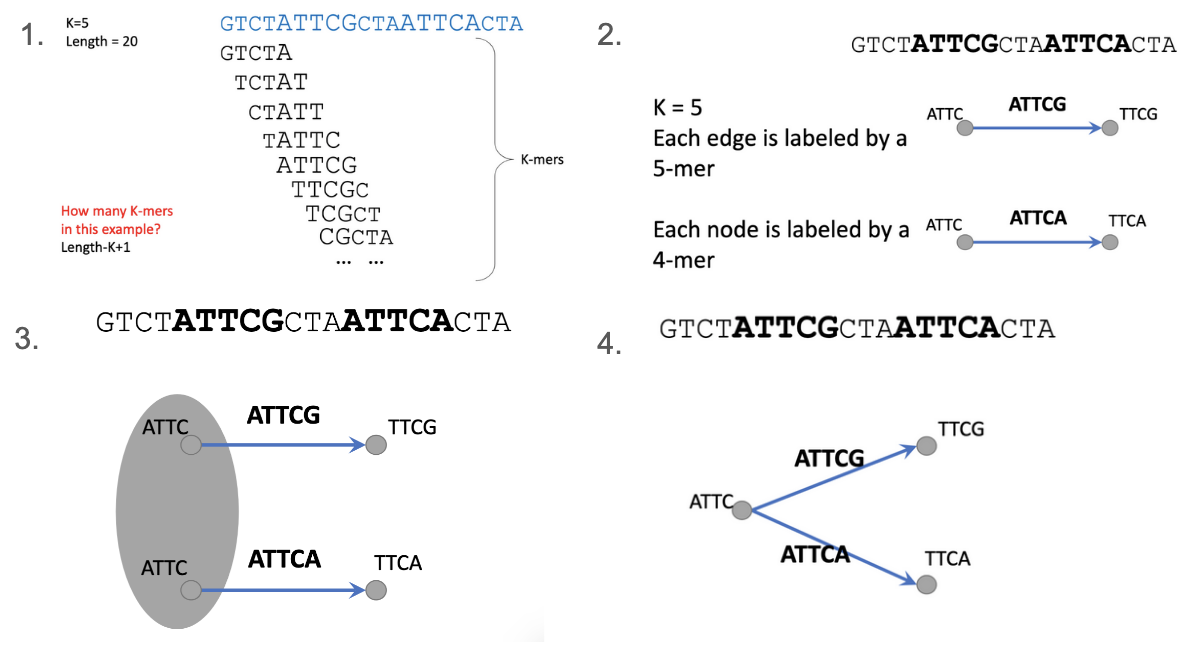
!! De Brujin graph example
Bulges = undireted cycles.
Whirls = directed cycles.
!! EXPLAIN BULGES AND WHIRLS BETTER
They occur because of sequencing errors or repeats in the genome.
How to assess assembly
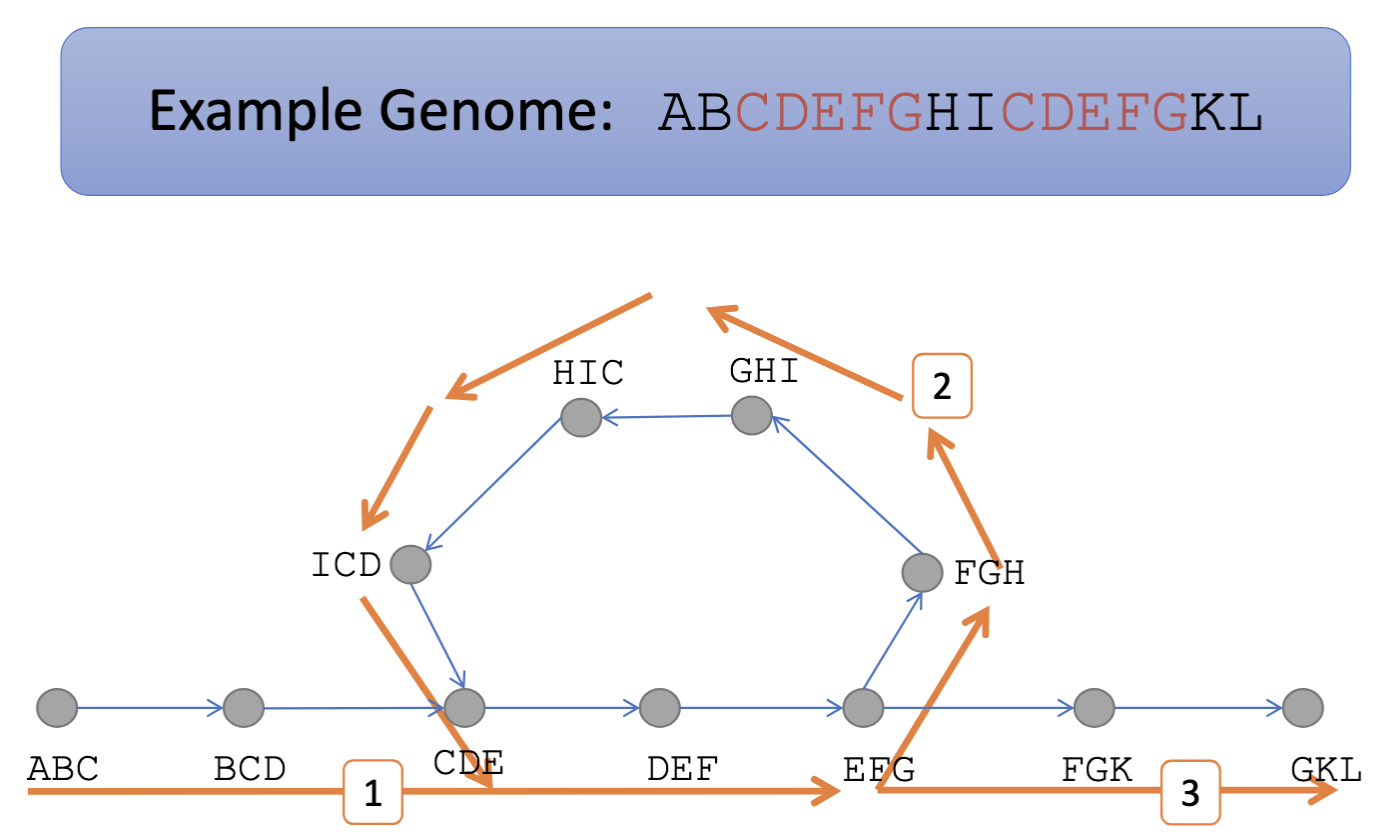
Bioinformaticians use metrics to assess an assembly's completeness, contiguity, and accuracy. For example, the commonly used N50 metric indicates the assembly's contiguity.
N50: the length of the contig where over 50% of the total assembled sequences are contained in contigs of that length or larger.
Step 1: Calculate the total length of all contigs and order them by length.
Step 2: Calculate 50% of the total length.
L50 = L/2.
Depth/coverage
Coverage in DNA sequencing is the number of unique reads that incldue a given nucleotide in the reconstruced sequence.
Higher depth = more confidence in assembly.
Low depth = risk of missing regions or errors.
Sequencing depth varies across the platforms and depends on the application goals.

Variant calling
Identify the differences in an individual’s genome compared to a reference genome.
Single Nucleotide Polymorphisms (SNPs).
Insertions/deletions (InDels).
Structural variants (SVs).
To account for the various types of error in the data, we only call variants at locations that have multiple reads.
Mapping to the reference assembly helps scientists identify single-nucleotide polymorphisms (SNPs) and small variations in sequences by comparing reads to known genomes.
Data files
FASTQ: sequence reads and quality control.
SAM & BAM: alignment to the genome.
BAM specifically does alignment cleanup.
VCF: variant calling.
FASTQ files
FASTQ files to store sequence reads.
An extension of the old FASTA format.
Includes both sequence and quality scores.
Four lines to represent each read.
Picture:
Line 1 begins with the ‘@’ character, followed by a sequence identifier and an optional description. It can contain flow cell IDs, lane numbers, and information on read pairs.
Line 2 is the sequence letters. of
Line 3 begins with a ‘+’ character; it marks the end of the sequence and is optionally followed by the same sequence identifier again in line 1.
Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence. Each letter corresponds to a quality score. A standard in the field is to use “Phred quality scores”.

SAM & BAM files
SAM (Sequence Alignment Map) files for reads aligned to the reference genome.
BAM files are compressed SAM files.

VCF files
VCF files record and store genetic variants found at specific locations within a DNA sequence.

Functional elements of DNA
Segments of DNA with a defined biological role.
Protein-coding genes.
Non-coding regulatory elements.
Promoters: Regions near genes where transcription begins.
Enhancers that control gene expression.
Noncoding RNA genes with direction functions themselves.
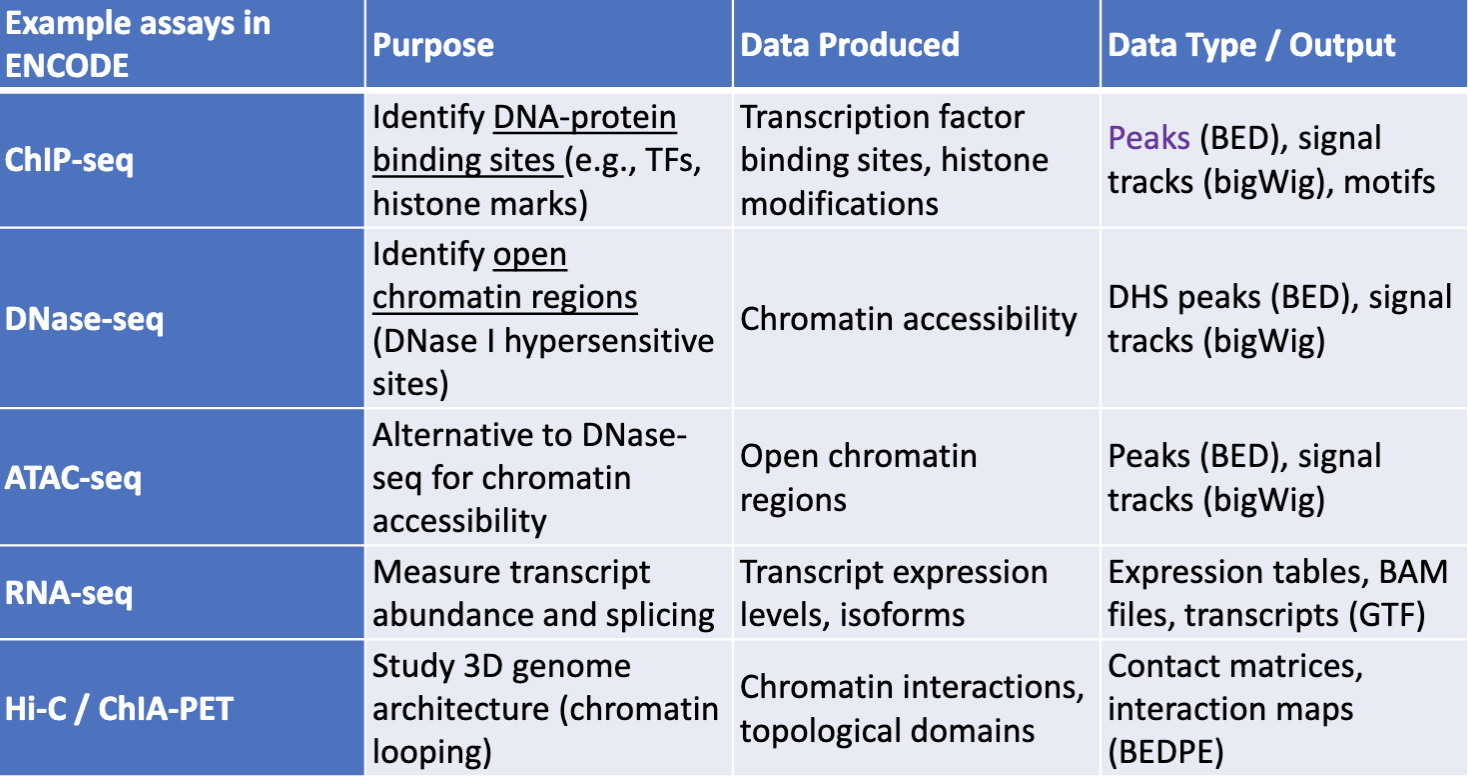
Encyclopedia of DNA Elements (ENCODE)
Build a comprehensive list of functional elements in the human genome → also known as genome annotation.
ENCODE genome annotation is tissue-specific.
ENCODE’s genome annotation is tissue-specific because the functional elements of the genome, such as which genes are active or inactive, vary between different cell and tissue types.
Help understand how gene activity is regulated in a tissue-specific manner.
GenBank
A collection of publicly available annotated nucleotide sequences.
250,000 organisms in total.
A primary database → updated only by submitters.
To get information on the files in GenBank, look at the presentation.
RefSeq
Reference sequence: a genomic sequence that has been chosen as the basis for annotations such as genes and sequence variations.
Genomic: gene sequence.
Transcript: sequence of mRNAs after alternative splicing.
Protein: sequence of downstream protein products corresponding to these genes.
It is a curated collection of DNA, RNA, and protein sequences built by NCBI.
There is only one example of each natural biological molecule for major organisms.
4,000 organisms in total.
A derivative database → continually updated by NCBI; uses information from the GenBank.
Functional divisions
Sequence tagged sites (STS):
Relatively short sequence (200 to 500 bp)
Occurred only once in the genome and
whose location and base sequence are
known.
Expressed sequence tags (ESTs):
A subset of sequence tagged site (STS)
located within coding region of a gene.
Genome survey sequence (GSS) and
High-throughput Genomic (HTG):Unfinished and partial genomic DNA
sequences.Will be moved to their respective divisions once complete.
International Nucleotide Sequence Database Collaboration (INSDC)
International collaboration:
DNA Data Bank of Japan (DDBJ).
European Nucleotide Archive (ENA).
GenBank.
Data sharing.
No use restriction.
Permanently accessible.
A unique identifier: accession number.
AAC37594.
Change in sequences:
Version number AAC37594.1.
Visualization of the genome
Use the UCSC genome browser.
For directions on how to use the browser, look at the presentation.