PSYC 304 Exam 4
1/104
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
105 Terms
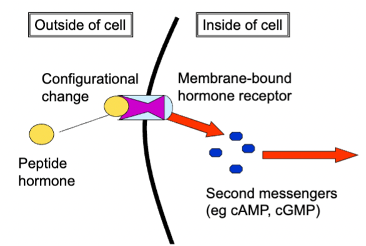
peptide hormones
chemical signalling molecules composed of 10-100 amino acids
water-soluble
effect endocrine system
found only on the cell surface
ex. insulin and glucagon
amine hormones
single amino acid signalling molecules
water or fat-soluble
found on cell surface or intracellular
ex. epinephrine and norepinephrine
amino acid functions
alters protein synthesis
alters metabolism of cells
alters neural activity (affecting ion channels)
act quickly (second - minute timescale
steroid hormones
derived from cholesterol, fat soluble
can go inside the cells and bind to DNA
Increase gene transcription/protein synthesis
results take longer to show and are more long-lasting
changes the architecture of cells
different proteins can be generated from one steroid hormone due to receptor co-factor
types of hormones
corticosteroids
sex steroids (androgens and estrogens)
gonadal/sex hormones
most common androgen = testosterone
dihydrotestosterone (DHT)
most common estrogen = estradiol
sex hormone cycle in the body
increased release triggered by hypothalamus → anterior pituitary releases them → gonads: male testes release testosterone and female ovaries release estradiol
where do sex hormones come from?
released by sex glands
ovaries: release more estrogens than androgens
testes: release more androgens than estrogens
regulated by negative feedback mechanisms
adrenal cortex also releases small amounts of sex hormones
sexual determination in utero
two types of chromosome combinations: XX (female), XY (male)
sex is not as simple as the chromosomes
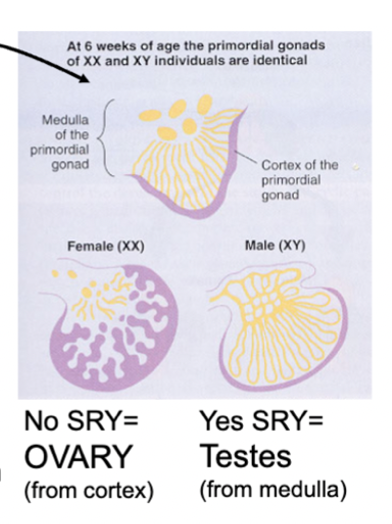
gonadal sexual development in utero
undifferentiated until 6 weeks of development - after, if an SRY gene is present, the Y chromosome begins testes development
if SRY gene is not present, gonads will develop as female
gonads made of medulla (inside) and cortex (outside) - cortex forms ovaries and medulla forms testes
gonadal ducts
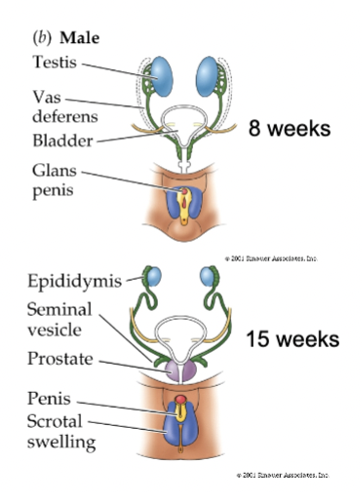
Wolffian = male: develops into epididymis, vas deferens seminal vesicles
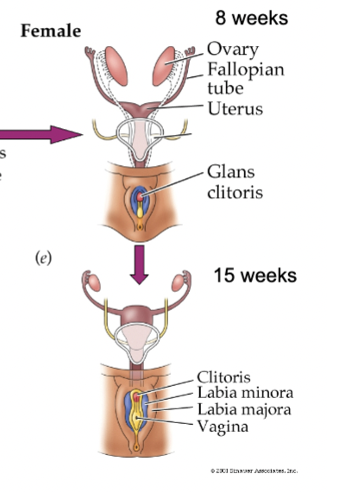
Mullerian = female: develops into fallopian tubes, uterus, inner vagina
female (XX) sexual development
no Y chromosome = no testosterone
Wolffian ducts shrink away and Mullerian ducts grow due to no T (around week 7)
male (XY) sexual development
SRY gene = SRY protein = Y chromosome
testes produce testosterone and anti-mullerian hormone (AMH) - causes Mullerian ducts to shrink and T causes Wolffian ducts to grow
T masculinizes other structures (aided by DHT, which is converted from T by 5 alpha-reductase) - prostate gland, scrotum, penis
what controls if a fetus develops into a male or female?
presence or absence of testosterone
not by estrogen
sex chromosome = sex of the gonad
gonadal hormones = sex of rest of body
androgen insensitivity syndrome
genetic defect that causes no presence of functional androgen receptors
no effect of T during development despite XY chromosomes - testes form but remain internalized
AMH causes mullerian ducts to shrink, but Wolffian ducts do not develop
Female external genitals develop and estrogen present causes female secondary sex characteristics
look and behave like an XX woman, but no menstruation or body hair
androgenital syndrome
caused by congenital adrenal hyperplasia which results in fetuses not producing enough cortisol = higher than normal levels of T (cortisol blocks androgen receptors)
T masculinizes XX fetus
Primordial gonads still form ovaries, Wolffian and Mullerian ducts grown, external genitalia are intersex
Many act/look like males at puberty
Turner’s syndrome
only have one sex chromosome - a single X
person has recognizable ovaries, but they are underdeveloped
Non-functioning 5𝝰-reductase
T cannot be converted to DHT
XY fetus will develop testes/male internal reproductive structures, but external genitalia will not masculinize fully
Androgen production increases at puberty and a penis (smaller than normal) will develop
rat sexual behaviour
starts with a female (ovulate every 4-5 days)
Step 1: female displays proceptive behaviours (darting, hopping, ear wiggling) during ovulation
Step 2: male begins to mount receptive female
Step 3: female arches back and moves tail to one side (lordosis)
Step 4: male then intromits (inserts penis) and thrusts
Step 5: repeat steps 2-4 until ejaculation
rat sexually stereotyped behaviours
females: lordosis, darting, hopping, ear wiggling
males: mounting, intromissions
primary measures of whether sexual brain has developed more male or female-like
how do sex hormones regulate sexual behaviour
organizational effects
activational effects
organizational effects
permanent/non-reversible changes in body/brain occurring during development
hormones exert these effects during critical periods of development - in utero and puberty
activational effects
on body/brain - behaviour after development
typically after sexual maturity (puberty)
transient effect, varies with amount of hormones in bloodstream
neural sexual development in rats
development of parts of brain responsible for sexual behaviour continues to occur after birth - regulated by presence/absence of T
can do hormone studies on rats because their brains are still developing
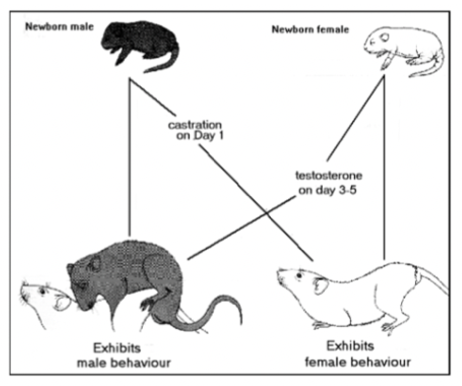
altering male rat sexual behaviour
castrating males in 1st week after birth = de-masculinizes and feminizes behaviour in adulthood (not observed if rats are castrated after initial critical period)
replacement T in adulthood doesn’t reinstate male-like behaviours (past critical period)
giving estrogens to adult male rats castrated neonatally can induce female behaviours
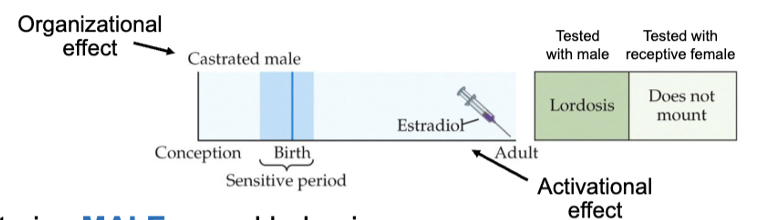
altering sexual behaviour in female rats
give T to female rat ~ 1st week after birth = defeminizes and masculinizes sexual behaviour in adulthood - not observed if given after puberty
T given to these females in adulthood induces male sexual behaviours
giving more estrogen to a neonatal female can also cause male behaviours
aromatization hypothesis
aromatase enzymes convert T to estradiol in the brain
early exposure to estradiol is what masculinizes the rat brain
why doesn’t estrogen in the brain masculinize female fetuses?
𝝰-Fetoprotein is in blood of male and female neonates
binds to free floating estradiol and prevents its entry into the brain
it does not bind to androgens - T can enter brain and be converted to estradiol
when too much estrogen is injected into female rats, the bloodstream is flooded with estrogen and ends up in the brain
evidence for aromatization hypothesis
neonatal estradiol masculines behaviour - in males, this saturates all 𝝰-fetoprotein molecules in bloodstream, extra estradiol can pass into brain
Aromatase is present in neonates
Blocking aromatase/estrogen receptors disrupts masculinizing effects of T
Female 𝝰-fetoprotein knockout mice show more male/less female like behaviour
DHT cannot be converted to estradiol, has no masculinizing effects when given early in development in rats
neural sexual development in primates
Sex hormone binding globulin (SHBG) protects fetal primate brain from estrogens
can bind to androgens and estrogens
𝝰-fetoprotein is not responsible
early treatment with either estrogens or androgens in pregnant primates can masculinize behaviour of female offspring (unlike rats)
some synthetic estrogens that are not blocked can also masculinize primate brain
changes in brain during sexual development
preoptic area of hypothalamus
spinal nucleus of bulbocavernosus in spinal cord
sex differences with the preoptic area of hypothalamus
this are specifically important for sexual behaviour
nucleus is larger in males than females
T injections in females = bigger nucleus
castration in males = smaller nucleus
agrees with aromatization hypothesis
sex differences with the spinal nucleus of bulvocavernosus (SNB)
rats have more motor neurons in the area because they need more muscles for the penis
muscles in females die out early
androgen injection in females spares some SNB motor neurons
castration in males causes them to die too
contains androgen receptors
retained in human females until adulthood because it helps constrict vaginal opening
sexual orientation vs. sexual development
they are different - developmental basis of both not well understood
sexual orientation
determined early in development, due to biological factors
males: 52% of monozygotic, 22% of dizygotic twins both homosexual
females: 46% monozygotic, 16% dizygotic twins both homosexual
prevalence of homosexuality
2-10% of pop in Western countries
hormones and sexual orientation
homo- and heterosexuals do not differ in hormone levles
in animals, castration or perinatal T can cause same sex preference in many species
the more older brothers a boy has, the more likely he is to be gay - maternal immune response to neutralize Y proteins in the body
neural differences and sexual orientation
some structural differences potentially
anterior hypothalamus differences
differences in regions related to body perception
lesbians tend to show markers indicative of fetal androgen exposure (longer ring fingers)
activational effects of hormones in male sex behaviour
removing T effects reduces sex drive (need some T to have it) - replacement T will bring it back to normal levels
for castrated males too
relative sex drive and T levels are uncorrelated - T injections do not increase sex drive, they just have to have some androgen to have it
takes a while to set in - cells becoming receptive to T
other factors appear to control sex drive
neural regulation of male sex behaviour
sufficient levels of T in bloodstream primes certain brain region to be receptive to sexual stimuli - specifically mPOA and medial amygdala
does not cause sexual behaviour but is important for it
activational effect
copulatory behaviour to ventral midbrain → projects to basal ganglia for mounting behaviours → spinal cord (stimulates erections)
evidence for T/Estradiol regulation of male sexual behaviour
mPOA/medial amygdala lesions disrupt sexual beahviour
mPOA stimulation initiates sexual behaviour
in castrated males - administration of T or estradiol into just the mPOA reinstates sexual behaviour
hormone regulation of female sex behaviour
in rats, estradiol increases ~2 days before ovulation (causes brain to make progesterone receptors) (cycle length = ~4-5 days)
when progesterone and estrogen peak, ovulation occurs (fertility and sexually stereotyped behaviours)
estrogen peaks → then progesterone peaks
neural regulation of female sex behaviours
ventromedial hypothalamus (VMH) monitors changes in hormonal values, constantly detecting hormone levels in blood
estradiol causes VMH and periaqueductal gray to have modifying ability, produce proteins necessary for lordosis
hormones hitting their peaks activates multisynaptic pathways that include motor areas
signals output through spinal cord → lordosis
evidence for estradiol regulation of female sexual behaviour
VMH/PAG lesions disrupt lordosis
Implantation of estrogens into VMH reinstates lordosis in ovarectomized females
hormonal pathway for sexual behaviour in female rats
sexual signals: pheromones/odours
olfactory circuits reach the hypothalamus, hormones induce estrus
tactile stimuli from male
lordosis is activated
presentation of the vagina
sex begins
hormones and human female sexual behaviour
estrogen: sexual motivation/behaviour is not as tightly linked to estrogens released during menstrual cycle - ovariectomy does not have reliable effects on them (important for ovulation)
androgens: T levels can correlate with measures of sexual motivation - following ovariectomy, replacement T rekindles sexual motivation
menstrual cycle and female sexual behaviour
behaviour patterns change during cycle - greater probability of having intercourse/achieving orgasm as ovulation approaches
women appear to be more attracted to stranger’s smell than their partner’s during ovulation
4 major stages of reproductive behaviour
sexual attraction
appetitive behaviour
copulation
post-copulatory behaviour
sexual attraction stage
traits that are the products of sexual selection pressures
pair-bonds (stay together after copulation) or sexual-bonds (separate after copulation)
men seem to overestimate women’s interest
appetitive behaviour stage
if mutually attracted, display species-specific behaviour that establish, maintain, promote sexual interaction
females display initial proceptive behaviours (approaching males, staying close to them)
copulation stage
if both animals display appetitive behaviours
involves one or more intromissions into females
depends on female’s sexually receptive behaviour/in heat
all mammals employ internal fertilization (fusion of gametes = zygote)
post-copulatory behaviour stage
will not mate again until refractory period has elapsed (can be minute to months long)
many mammals will mate sooner if provided a new partner (Coolidge effect)
some animals will be in copulatory lock where their penis swells so much that they cannot remove it from the female
pheromones
chemical signals that communicate info between animals to help coordinate reproductive activities
special structures in animals: vomeronasal organ (VNO), accessory olfactory bulb
pheromone structural path
VNO specialized receptor cells near olfactory epithelium → accessory olfactory bulb → medial amygdala → mPOA (integrates hormonal and sensory info and coordinates motor patterns of copulation)
male sexual response patterns
always have an absolute refractory period after orgasm - often followed by renewed arousal
activity seen in subcortical areas, penile stimulation activates right insula and secondary somatosensory cortex
female sexual response patterns
show a lot more variation than men - absolute refractory after orgasm and no renewed arousal, renewed arousal, never reach orgasm, etc.
clitoral stimulation and orgasm associated with hypothalamus, amygdala, cerebellum, cingulate, brain stem, basal forebrain
why do we have to eat?
to maintain energy levels of the body
how does the body use energy?
basal metabolism: ~60% of energy usage maintains body heat and other resting functions (life-sustaining functions) (varies with body weight)
active behavioural processes: ~25% is for behaviours other than rest (vary depending on activity level)
digestion of food: ~15% is to process food, break it down into molecules (varies with type of food)
remainder gets stored as energy reserves
rate of basal metabolism
kilocalories/day = 70 x weight0.75
basic nutrients
carbohydrates
amino acids
lipids (fats)
vitamins and minerals
carbohydrates
~4 kcal per gram, glucose is primary fuel of body, all other carbs get converted to glucose
short-term storable form is glycogen: stored in liver and muscles
amino acids
~4 kcal per gram - comes from proteins, basic building blocks for all cells
20 types, 9 cannot be produced by the human body (essential amino acids)
can be converted to glucose
lipids
~9 kcal per gram - long term energy source
can be converted to free fatty acids as an alt energy source for most cells of the body
can be stored long-term
use when blood sugar is low or under food deprivation as ketones
vitamins and minerals
needed to assist in bodily functions (digestion, cell building, homeostasis)
steps of digestion
chewing (mastication)
saliva (lubrication)
swallowing (getting there)
stomach - storage and breakdown (HCl, Pepsin)
duodenum - absorption
gall bladder and pancreas fluids further break down food in duodenum (proteins → amino acids/starch → simple sugars)
bile from liver (stored in gall bladder) breaks down fats
remaining water and electrolytes absorbed by large intestine or ejected via the anus
whole process takes 18-24 hours
glucose regulation
done by the pancreas
two main hormones: glucagon (converts glycogen into glucose - increases blood sugar) and insulin (decreases blood sugar)
negative feedback system
primary actions of insulin
promotes use of glucose as primary energy source: most cells need insulin to get glucose in cells, brain is one exception (used glucose without need for insulin)
converts bloodborne fuels to storable forms: glucose → glycogen (muscles/liver), glucose and fatty acids → adipose tissue (body fat), amino acids → protein (muscles)
mechanisms controlling insulin release
brain (via vagus nerve): sight/smell/taste/thought of food can trigger insulin release before food hits gut (cephalic phase)
other hormones in bloodstream released by gut during digestive phase
nutrients/glucose entering bloodstream, signal pancreas to release insulin (absorptive phase)
glucose detection by the brain
glucodetectors in liver → vagus nerve → nucleus of solitary tract (NST) → hypothalamus (informs brain of glucose levels and contributes to hunger)
diabetes mellitus (Type 1 [juvenile-onset] diabetes)
pancreas stops producing insulin, excess glucose in blood stream
brain cannot use all of it, and cells of body cannot use glucose without insulin, start using fatty acids
when left untreated, diabetes can lead to more eating that doesn’t satisfy hunger and paradoxical weight loss (brain thinks we are starving because we are not getting any glucose in the cells)
Type II diabetes = sensitivity to insulin (adult onset)
is insulin a satiety signal?
if you lower an animal’s insulin level, it becomes hungry and eats a large meal
but, give it a large amount of insulin - converts most glucose to fat, less glucose in bloodstream
brain detects glucose deficit, initiates hunger - animals will eat a large meal again
is glucose a satiety signal?
not entirely - untreated diabetes leaves a lot of glucose in bloodstream, but still hunger
glucose levels can stay relatively stable for days, but we still get hungry
multiple signals in addition to insulin and glucose contribute to hunger and satiety
why do we get hungry: set-point theory
idea that the body has a predetermined weight range that it aims to maintain
hunger as an energy deficit - negative feedback system maintaining homeostasis
two types: glucostatic and lipostatic
glucostatic set point theory
eating is controlled by deviations from a hypothetical blood glucose set-point - focused on blood sugar levels dropping or rising
lipostatic set point theory
eating is controlled by deviations from a hypothetical body fat set point
problems with set-point theories
evolution argues against it: food availability was once inconsistent, so it was better to eat large quantities and store calories
fails when tested: drinking high glucose/caloric drinks before meals doesn’t reduce eating significantly
ignores other factors that stimulate eating: different tastes of food (dessert after dinner), social factors increasing eating
positive-incentive theory
anticipated pleasure of eating is the main factor controlling feeding
evolved to crave food not because of a deficit, but because we like it
factors that determine what we eat
taste preferences/aversions: some tastes have high incentive values (sweet, salty, fatty = more glucose), others are learned from experience (like bitterness) (can condition taste aversion)
learning to eat vitamins and minerals: can learn to select what you are lacking in - rats will choose the high B1 food when they are low in it out of 10 different foods
factors that influence when we eat
pavlovian conditioning: environment cues associated with eating can elicit hunger/feeding
caused by expectation of food
pair food with a tone, rats will always eat when they hear the tone, even if they just ate
pre-meal hunger: time of day one usually eats at can trigger hunger - conditions brain/body to prepare for incoming food, stomach growls to prepare for food
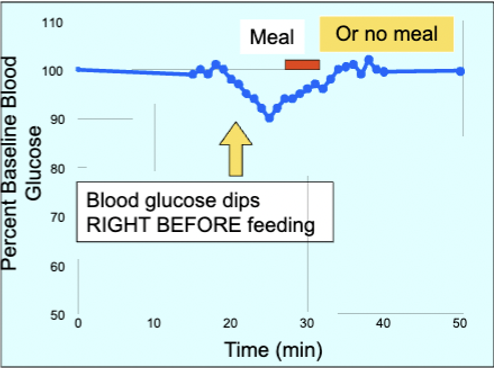
pre-meal hunger and changes in blood glucose
study found that when rats were provided with unlimited food, their glucose levels remained constant throughout the day except before meals were initiated - 10% drop in glucose
glucose not directly responsible for feeding - if food is removed, glucose levels return to previous homeostatic levels within 10-15 mins (like if the meal was consumed)
decline related to intention to eat - drop is preceded by increased insulin in anticipation for eating
factors that influence satiety
social influences: humans and animals eat more in groups vs. alone (take bites together, eat similar amounts of food)
sensory specific satiety: humans and animals eat more calories if they are given varied diet
new taste = more consumption
physiology of hunger and satiety
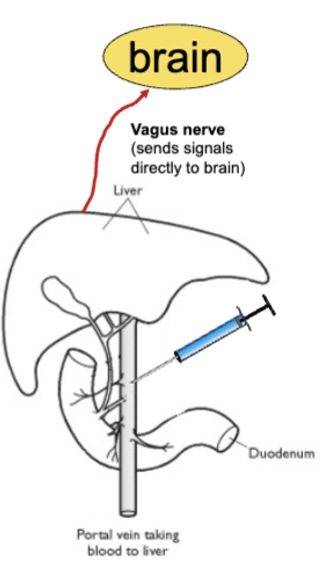
liver: signals brain about what’s in the bloodstream via vagus nerve - receives blood from small intestine and has detectors for glucose and fatty acids (everything you eat will first pass through the liver)
decides if food is right to go into general circulation
can trick liver into thinking glucose/fat levels are low (2-DG = competes with glucose for absorption, methyl palmoxirate = disrupts metabolism of fatty acids) - causes increase in feeding, takes nutrients from intestines instead
cutting vagus nerve abolishes effect of drug injection
satiety/hunger signals
use multiple hormones to signal brain to start/stop eating
signals can suppress hunger before food is fully digested
arcuate nucleus of hypothalamus (ANH) contains appetite controllers governed by circulating levels of hormones
only way gut can communicate with brain is through hormones in blood
hunger signals from stomach
CCK, bombesin, somatostatin
peptide Ghrelin levels remain high during fasting and drop during meal - acts on NPY cells in ANH
hunger signals from liver
detects changes in blood glucose, direct input to brain via vagus nerve
hunger signals from pancreas
insulin
hunger signals from intestines
PYY and GLP-1 (decrease appetite and rewarding aspects of food, oppose ghrelin)
fast-acting - PYY acts on NPY cells in ANH, GLP-1 acts on POMC cells in ANH
hunger signals from fat cells
leptin, gives continuous feedback on body’s energy stores (removal of fat gets rid of this satiety signal and increases hunger)
defects in production can lead to overeating
arcuate nucleus of hypothalamus appetite system
first-pass appetite control center
5 main hunger hormones: insulin (pancreas), leptin (fat cells), GLP1/PYY (intestines), ghrelin (stomach)
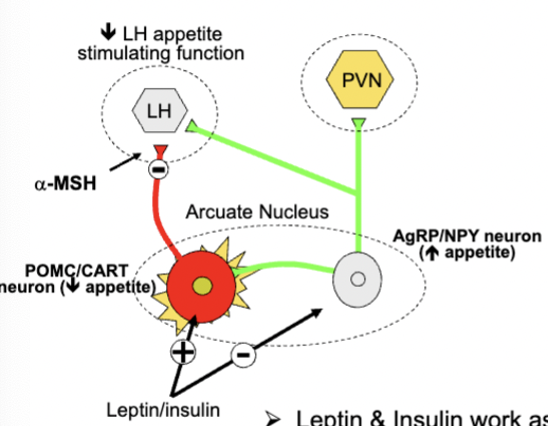
relies on two sets of proteins: POMC and CART neurons (inhibit appetite and increase metabolism), NPY and AgRP neurons (produce neuropeptide Y, stimulate appetite and inhibit POMC neurons, AgRP competes with 𝝰-MSH for receptors)
neural basis of hunger and satiety
ventromedial hypothalamus: lesions to the nucleus cause animals to become obese, starts with massive consumption that achieves new weight → then maintenance of that weight
do stop eating eventually - become finicky eaters (give lesioned rats less palatable foods, they will show minimal weight gain)
VMH not the satiety centre - instead it regulates energy metabolism
VMH lesions increase insulin levels - decreases breakdown of body fat into usable forms
Lateral hypothalamus: lesion caused rats to stop eating - force feeding rats will get them to start eating again
causes problems with the actions of eating, not hunger
many regions including other hypothalamic subregions, amygdala, frontal cortex are also involved in hunger and satiety
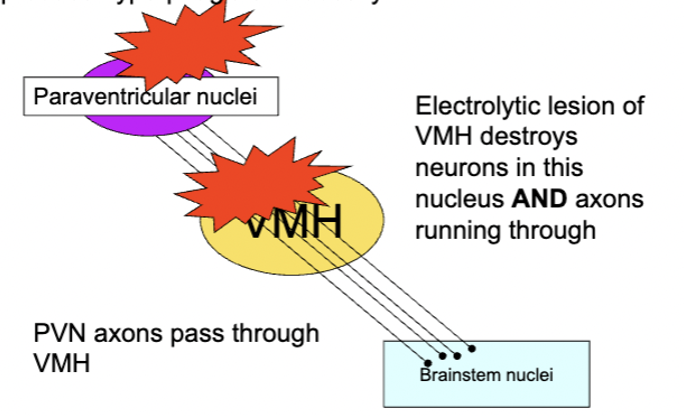
VMH lesions
destroy axons projecting from paraventricular nuclei of the hypothalamus - lesions of the fibres along produces hyperphagia and obesity
arcuate neural circuitry and appetite - long term appetite control
activate POMC/CART neurons and inhibit AgRP/NPY neurons
POMC/CARRT neurons inhibit lateral hypothalamus (LH) using 𝝰-melanocyte stimulating hormone
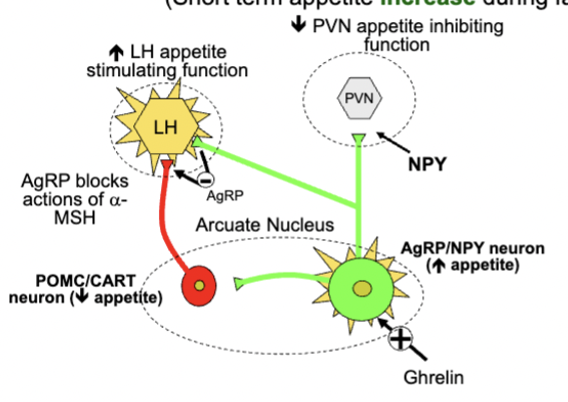
arcuate neural circuitry and appetite - short term appetite increase
inhibits PVN cells using NPY - release AgRP in LH and block 𝝰-melanocyte stimulating hormone, leading to increased LH activity
ghrelin released by stomach when empty stimulates AgRP and NPY
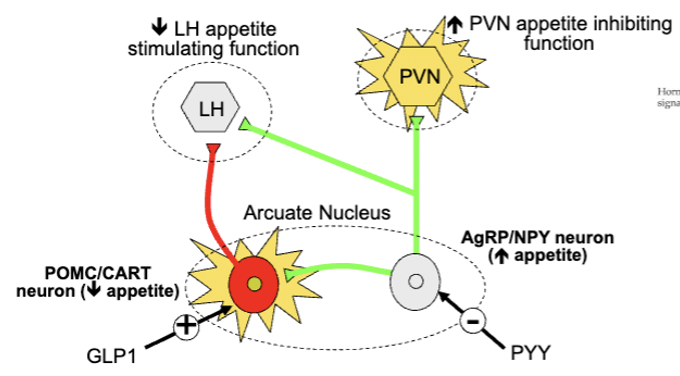
arcuate neural circuitry and appetite - short term appetite decrease
PYY inhibits AgRP/NPY neurons, which disinhibits (increases) PVN activity
GLP 1 stimulates POMC/CART neurons, which inhibits LH activity
PYY/GLP-1 released from intestines in response to a meal
bypassing the hypothalamic feeding circuit
prefrontal cortex and amygdala also can recognize feeding cues - leisions to these areas, disconnection of amygdala-LH path abolishes conditioned increases in feeding (only disrupt cue-induced feeding
neurochemistry of hunger and satiety
serotonin - major satiety signal
5-HT treatment can decrease feeding and amount of food consumed per meal, but not number of meals per day
also shift food preference away from fatty foods - acts as a short term satiety signals associated with meal consumption
inhibits release of NPY in the PVN of hypothalamus - disinhibits PVN neurons to promote satiety
anorexia nervosa
obsession with body weight/image and food - ~1% of population (mostly women)
self-induced starvation leading to significant low body weight in context of age, sex
often presents with BMI of <15 kg/m2
fear of gaining weight
distorted view of body - lack of recognition of seriousness of weight
obsessed with food, show higher than normal insulin release to food anticipation, often disgusted with sweet/fatty foods
anorexia treatments
very few effective ones
less than 30% show long term recovery
~10% die of starvation/suicide
anorexia causes
psychological: positive-incentive value for food goes up during starvation, meals are a disruptive event on the body, can cause conditioned taste aversions to food
biological: info processed in certain brain regions altered
abnormal frontal activation patterns
abnormal increases in amygdala activity to palatable tastes
feeding hormone levels reduced (AgRP, NPY, leptin)
5-HT abnormalities
obesity
excessive adipose tissue
adult males = >25% and females = >30% fat content
obesity = BMI > 30