The mitochondrial genome
1/34
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
35 Terms
General structure of mtDNA
Double-stranded circular DNA, 16.6 kb in length, approximately 15,000 times smaller than chromosome 1. Consists of Heavy (H) and Light (L) strands. Multicopy per cell (10–100,000 copies).
Circularity protects against telomere-related instability.
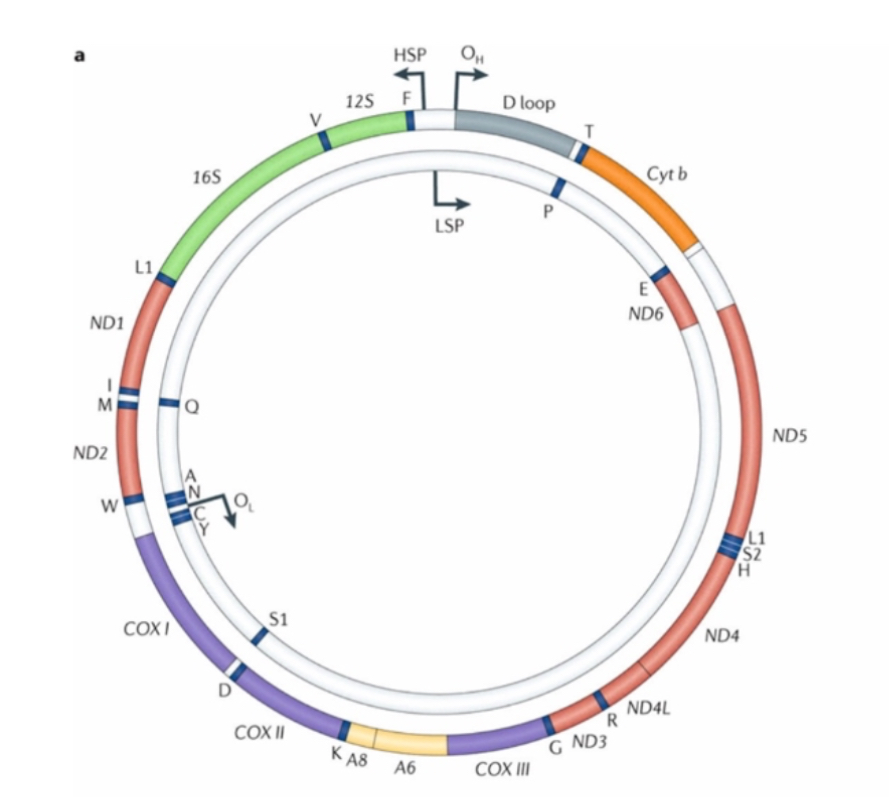
Gene content of mtDNA
37 genes: 13 OXPHOS subunits, 22 tRNAs, 2 rRNAs (12S and 16S). No introns.
Compact organisation causes high susceptibility to functional effects of mutations.
It encodes proteins for oxidative phosphorylation
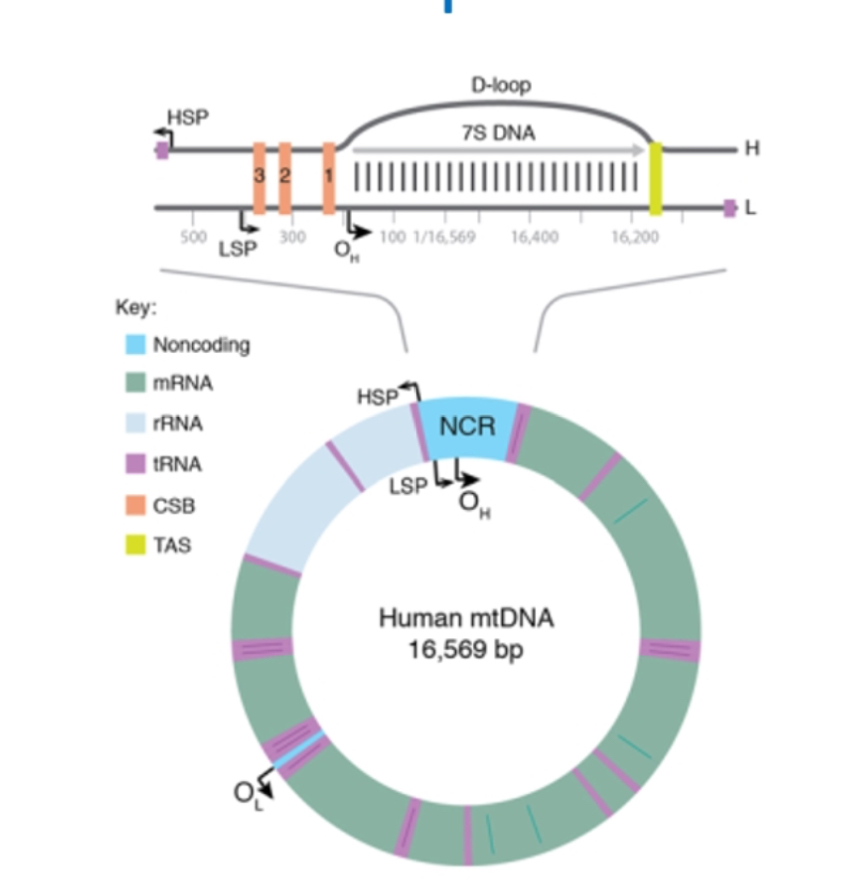
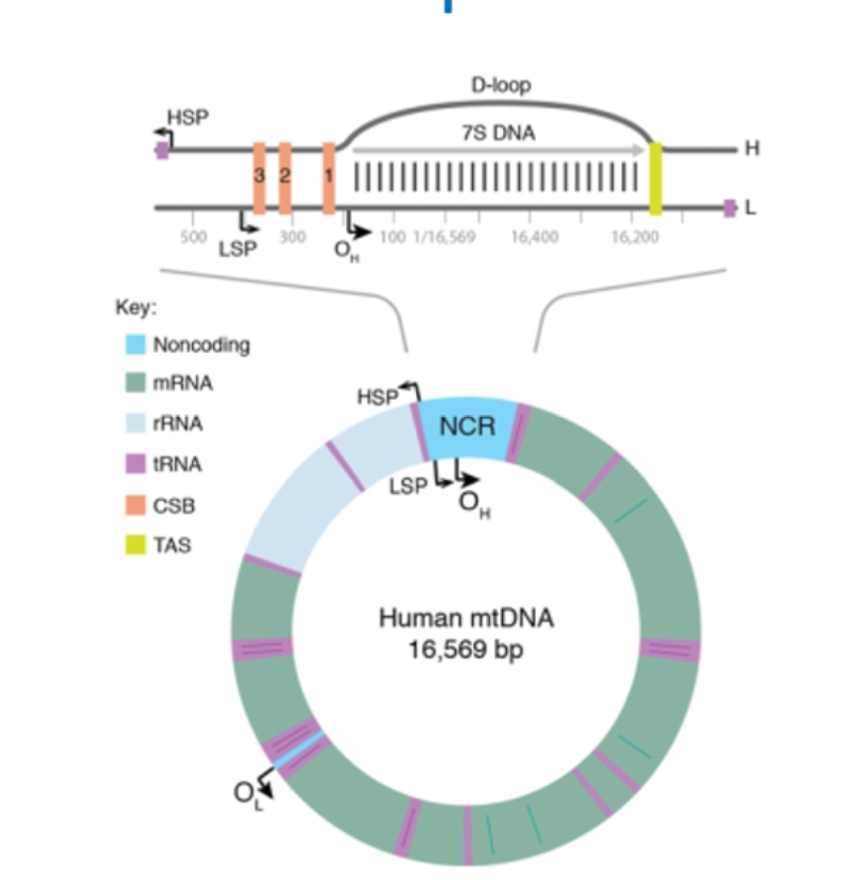
Non-coding D-loop region
It’s a non coding region
Contains regulatory sequences for replication and transcription initiation.
Most common region for mtDNA mutations and heteroplasmy drift.
Maternal inheritance
mtDNA is inherited almost entirely from the mother. No recombination.
Paternal inheritance is rare but increasingly documented.
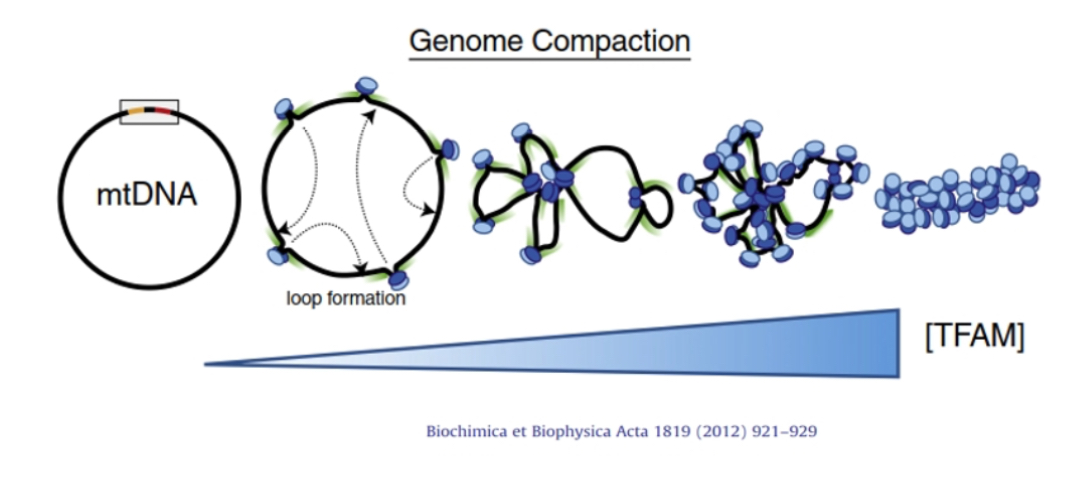
mtDNA packaging
Organised into nucleoids containing 1–2 copies each. TFAM acts like a histone equivalent to compact DNA and regulate transcription.
Deviation from universal genetic code
AUA and AUG = methionine; UGA = tryptophan; AGA and AGG = stop codons.
Reflects evolutionary divergence from bacterial ancestors.
mtDNA Haplogroups
Lineage-specific mtDNA variants used in ancestry, evolution, and migration studies.
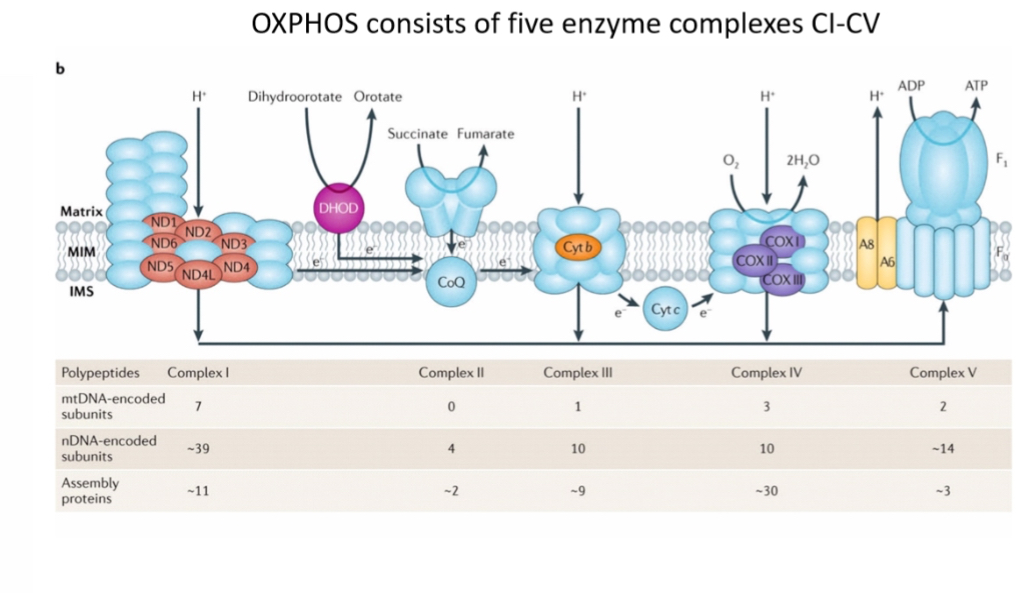
Dual genomic dependence
Mitochondria require both nuclear- and mtDNA-encoded proteins. Only 13 proteins from mtDNA; over 1,000 mitochondrial proteins total derived mainly from nuclear DNA.
mDNA encodes 13 proteins of OXPHOS
But OXPHOS requires >100 proteins
To make the 13 OXPHOS proteins mtDNA must be:
Replicated
Transcribed
Translated
All proteins involved in replication, transcription and translation of mDNA are encoded by nuclear genes and imported into mitochondria
In total >1000 mitochondrial proteins but only 13 made by mtDNA, all others made by nuclear genes!!
Mitochondrial DNA polymerase
Polymerase γ (POLγ):
• POLγA: catalytic subunit with 3’→5’ exonuclease proofreading
• POLγB: accessory subunits that increase processivity and binding
mitochondrial DNA is copied by DNA polymerase γ (Pol γ), and its accuracy and speed depend on how its subunits work together.
Explanation:
Mitochondrial DNA polymerase γ is the only DNA polymerase that replicates mtDNA. It is a heterotrimer, meaning it has three subunits. One is the catalytic subunit (POLγA), which actually synthesises new DNA by adding nucleotides to the growing strand. POLγA also contains a 3′→5′ exonuclease domain, which proofreads the newly made DNA by removing incorrectly added nucleotides, increasing replication fidelity. The other two subunits are accessory subunits (POLγB). These do not catalyse DNA synthesis themselves; instead, they stabilise the polymerase on the DNA template, strengthen DNA binding, and increase processivity, meaning POLγA can copy long stretches of mtDNA without falling off. Together, this complex ensures mtDNA is replicated efficiently and with low error rates inside mitochondrial nucleoids.
Helicase TWINKLE
Hexamer(6 TWINKLE subunits) that unwinds ds-mtDNA during replication.
Mitochondrial single stranded binding protein(mtSSBP)
Binds displaced single-stranded mtDNA, prevents degradation and secondary structure formation, enhances TWINKLE activity.
mtSSBP binds single-stranded mitochondrial DNA and keeps it stable so replication can continue.
Explanation (step-by-step):
mtSSBP (mitochondrial single-stranded DNA-binding protein) binds to mtDNA as soon as it becomes single-stranded during replication. Its main role is to prevent the exposed ssDNA from forming secondary structures or being degraded. By coating the ssDNA, mtSSBP keeps the template straight and accessible for enzymes like DNA polymerase γ and the Twinkle helicase. This stabilisation also helps coordinate leading-strand synthesis and ensures smooth, continuous replication within mitochondrial nucleoids.
DNA Replication initiation
Begins at OH for heavy strand in the Non-Coding Region. Heavy strand synthesis displaces parental H-strand which is coated with mtSSBP.
replication starts at Oₕ (origin of the heavy strand) in the non-coding region (D-loop).
Step-by-step (clear and corrected)
Initiation occurs at Oₕ in the non-coding control region of mtDNA.
The new heavy strand is synthesised using the light strand as the template.
As this new heavy strand is made, the original (parental) heavy strand gets displaced and becomes single-stranded.
That displaced single-stranded parental heavy strand is coated by mtSSBP, which protects and stabilises it while replication continues.
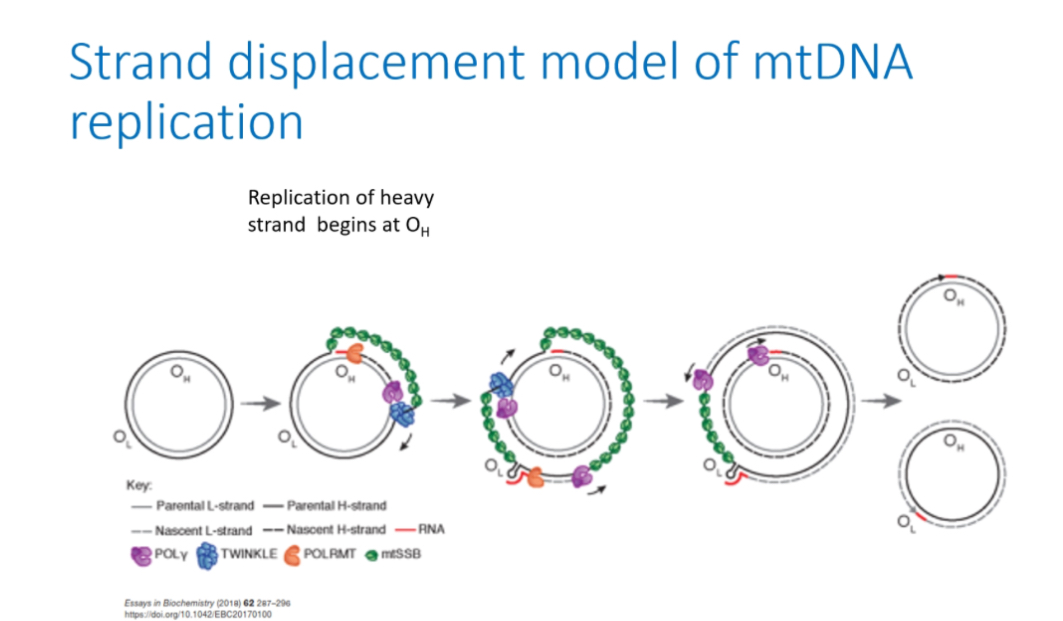
Strand displacement model
Step 1: Closed circular mtDNA
Mitochondrial DNA exists as a double-stranded circular molecule with two strands: the heavy (H) strand and light (L) strand. Both origins (Oₕ and Oₗ) are present but inactive.
Step 2: Initiation at Oₕ
Replication starts at Oₕ (origin of the heavy strand) in the non-coding control region.
POLRMT(RNA pol)synthesises a short RNA primer at Oₕ.
Step 3: H-strand synthesis begins
DNA polymerase γ (POLγ) extends from the RNA primer and begins synthesising a new heavy strand, using the parental L strand as the template.
DNA polymerase γ does NOT copy the H strand.
Instead, it reads the parental L strand.
As POLγ moves along the parental L strand, it adds complementary bases:
• A pairs with T
• G pairs with C
This creates a new H strand (because it is complementary to the L strand).
As the new heavy strand is made, the parental heavy strand is physically displaced and becomes single-stranded.
Step 5: Stabilisation of displaced strand
The exposed single-stranded parental H strand is coated by mtSSBP, which:
• prevents degradation
• prevents secondary structure formation
• keeps the strand stable during replication
This is why it’s called strand displacement replication
Light strand replication
When newly synthesised heavy strand replication reaches OL, a stem loop prevents mtSSBP binding, POLRMT generates primer, POLγ synthesises the light strand.
Completion
Replication continues until both strands are fully synthesised, followed by daughter molecule segregation.
Lack of replication checkpoints increases mutation accumulation.
Mitochondrial diseases
Rare monogenic diseases
Affect between 1:2000 - 1:4000 individuals
Oxidative phosphorylation disorders are most common form of mitochondrial disease
Affect highly metabolic organs abundant in mitochondria
Can affect one (isolated) or several organ systems (multisystem).
Start at any age
Wide severity spectrum e.g.
Adult onset hearing loss
Affect ~1:2000–1:4000 individuals; highly metabolic tissues most affected (brain, heart, muscle). Age of onset varies.
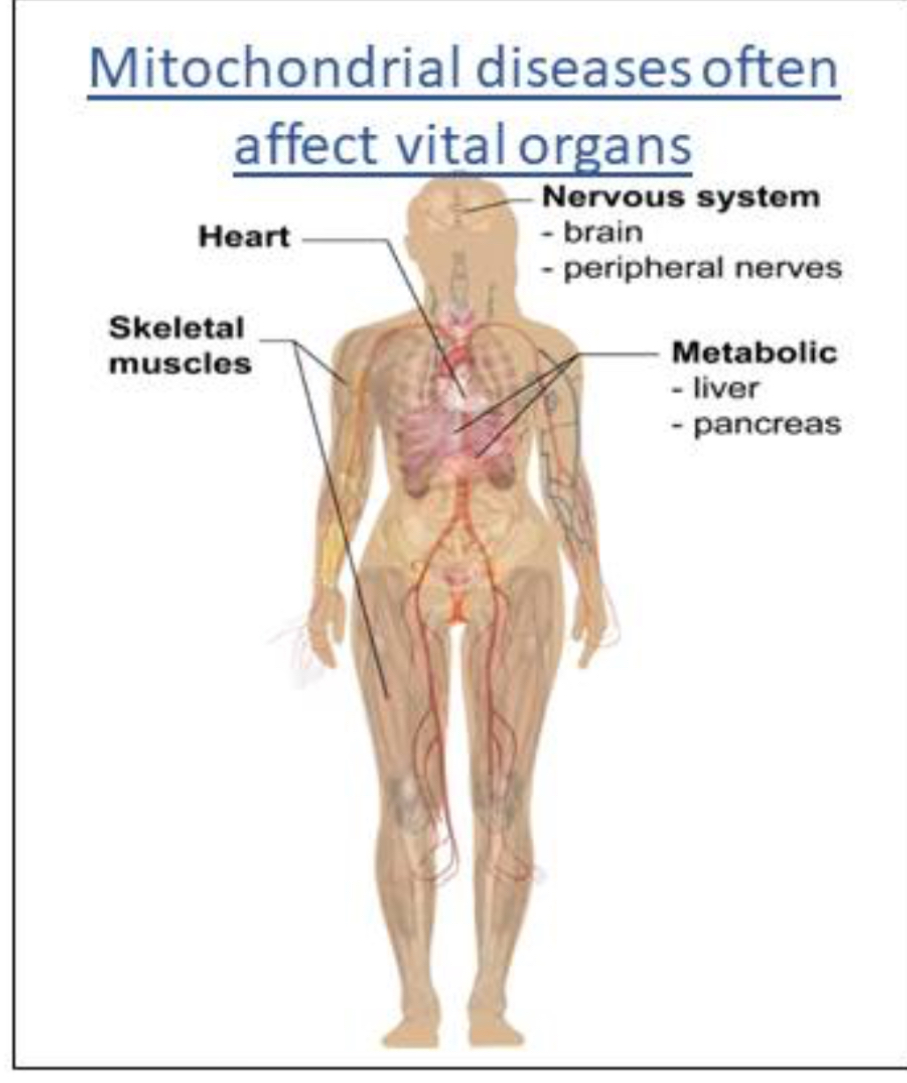
Clinical range
Isolated or multisystem involvement. Severity ranges from mild hearing loss to fatal cardiomyopathy.
Common mitochondrial syndromes
Leigh syndrome, LHON, KSS, MELAS, MERFF, NARP, MINGIE.
Multigenic etiologies often >80 genes per syndrome.
Diagnostic methods
Most mt disorders effect the brain
Clinical evaluation; biochemical markers (lactate, pyruvate); neuroimaging (MRI/MRS); muscle biopsy histochemistry; DNA testing.

Muscle biopsy hallmarks
COX-negative fibres and ragged-red/blue fibres reflect mitochondrial dysfunction.
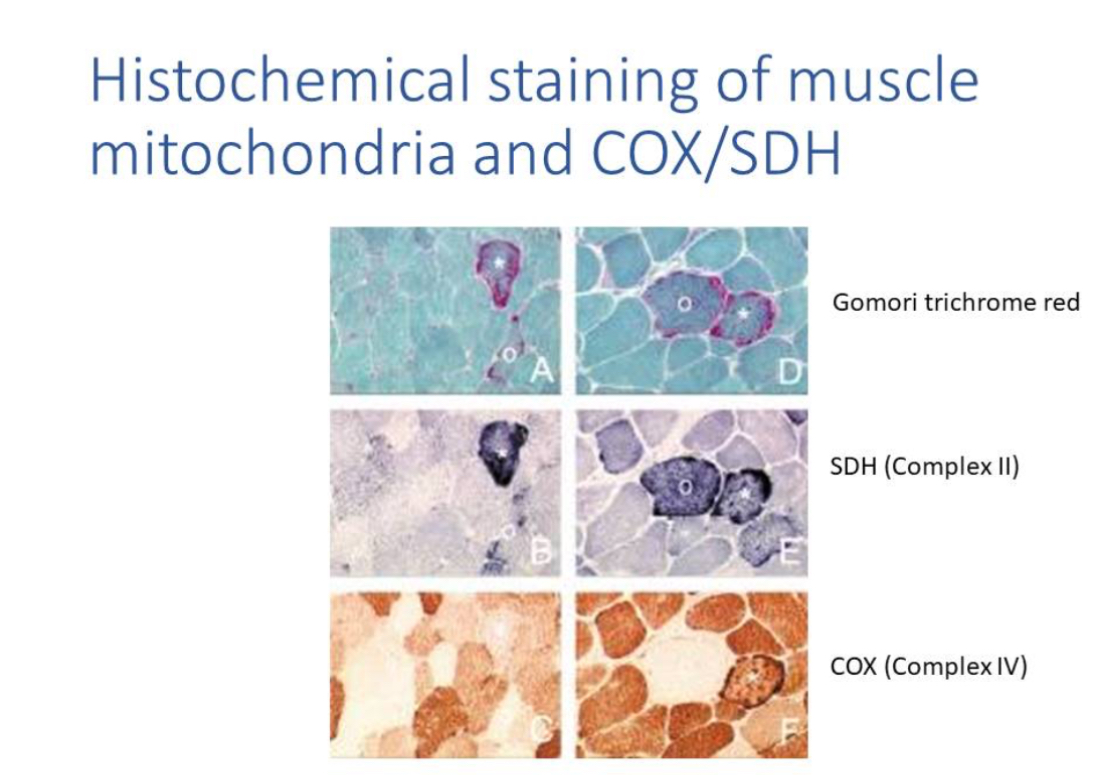
Spectrophotometry of OXPHOS activity
Spectrophotometry is an analytical technique that measures how much light a substance absorbs (or transmits) at specific wavelengths to determine its concentration or properties in a sample.
Spectrophotometry and high-resolution respirometry(measures O2 consumption) detect complex defects.
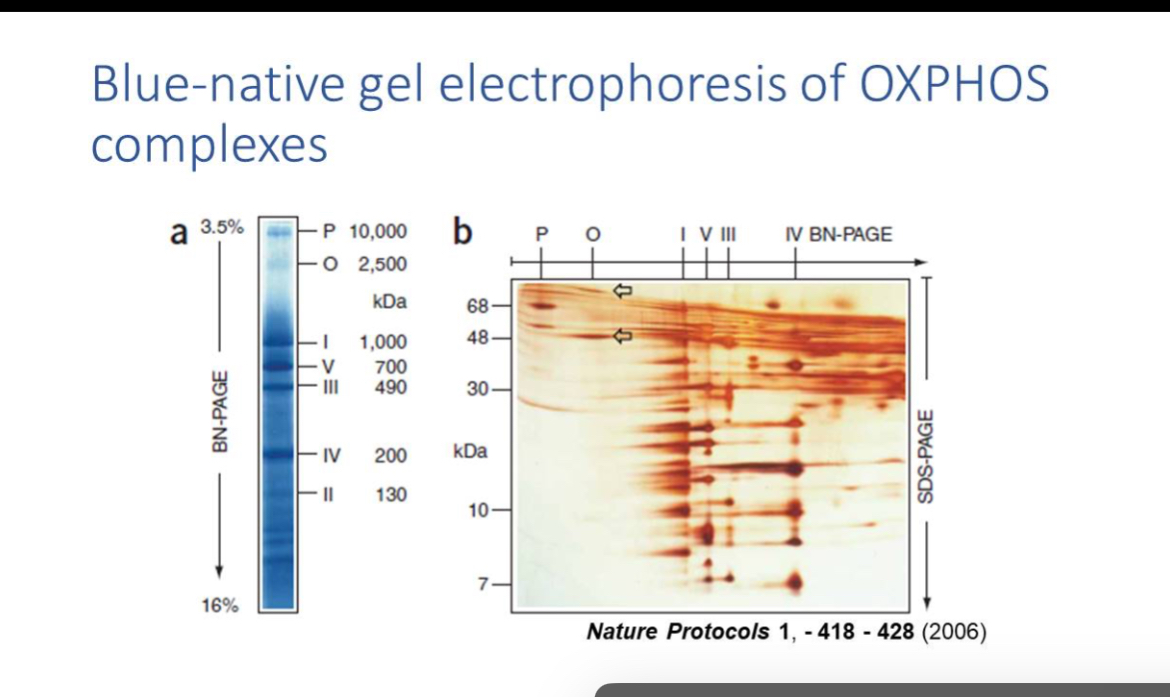
BN-PAGE and EM
When there are defects in OXPHOS this can also be assayed using Blue-native gel which resolves OXPHOS complexes; EM visualises structural abnormalities like paracrystalline inclusions.
NGS for mtDNA sequencing
Detects SNVs, deletions, duplications; tissue choice critical for heteroplasmic mutations (e.g. muscle for MELAS A3243G).
Cellular populations of mtDNA
A single cell can contain lots of copies of mitochondria
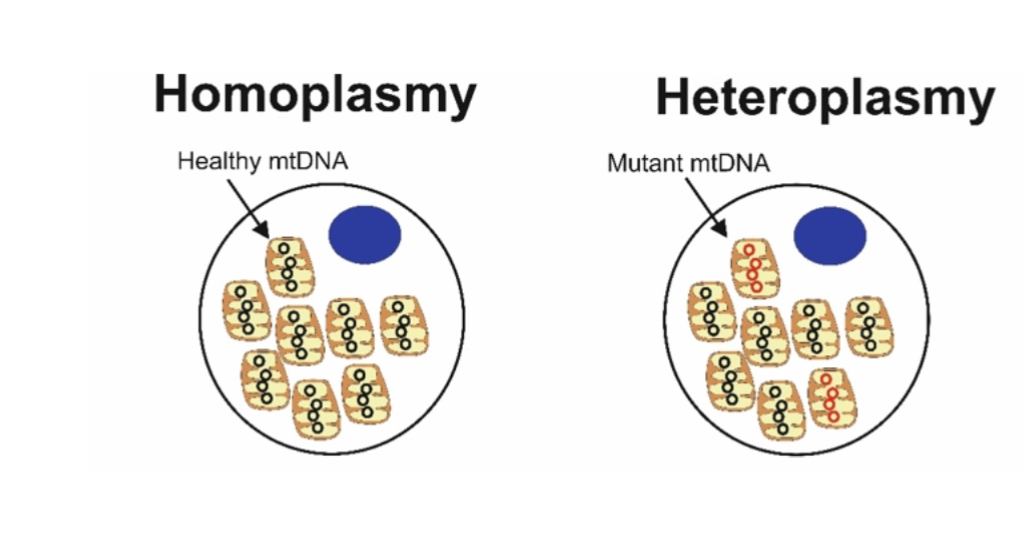
Heteroplasmy:
Coexistence of mutant and wild-type mtDNA. Disease depends on mutation load (threshold effect).
Homoplasmy (mtDNA) means all mitochondria within a cell contain identical mitochondrial DNA sequences (either all normal or all mutant).
Heteroplasmy (mtDNA) means a mixture of different mitochondrial DNA sequences exists within the same cell (some normal, some mutant).
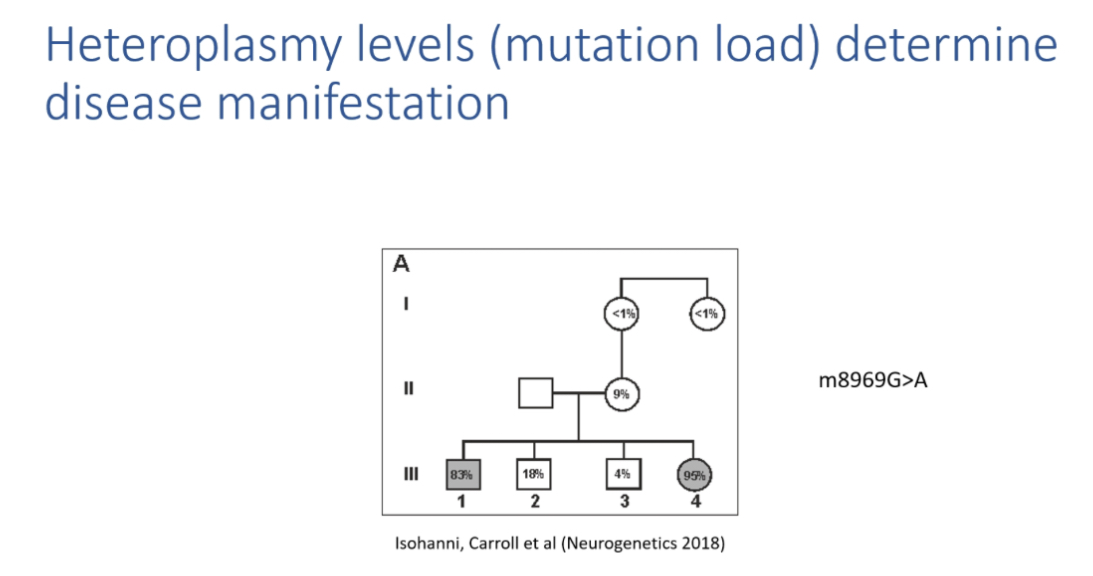
Examples of heteroplasmy thresholds
m.8993T>G:
90 percent mutant mtDNA= Leigh syndrome
60–75 percent = NARP
<60 percent = asymptomatic
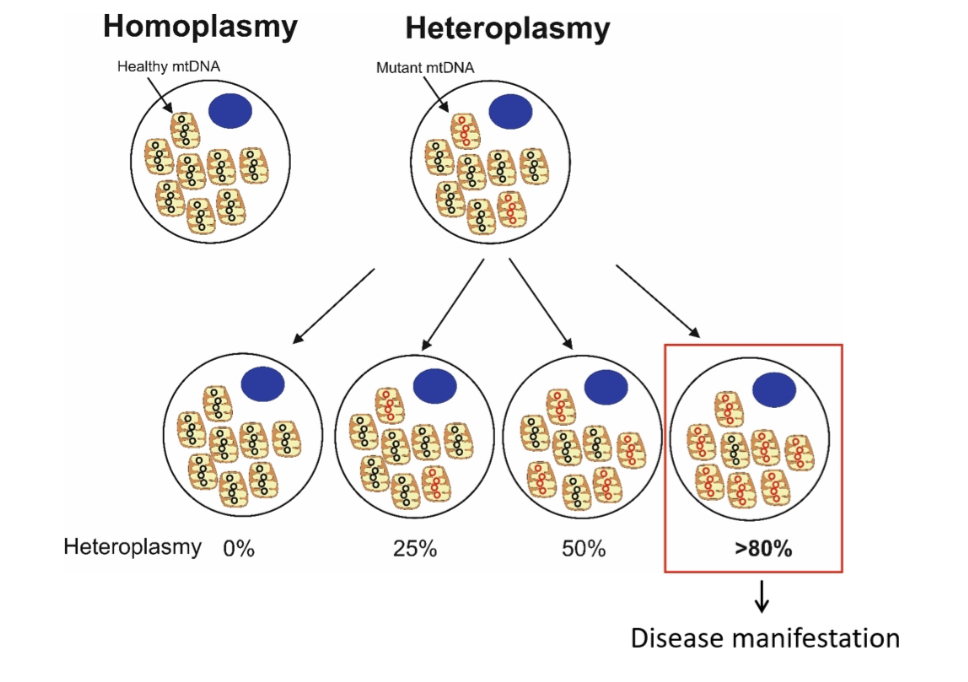
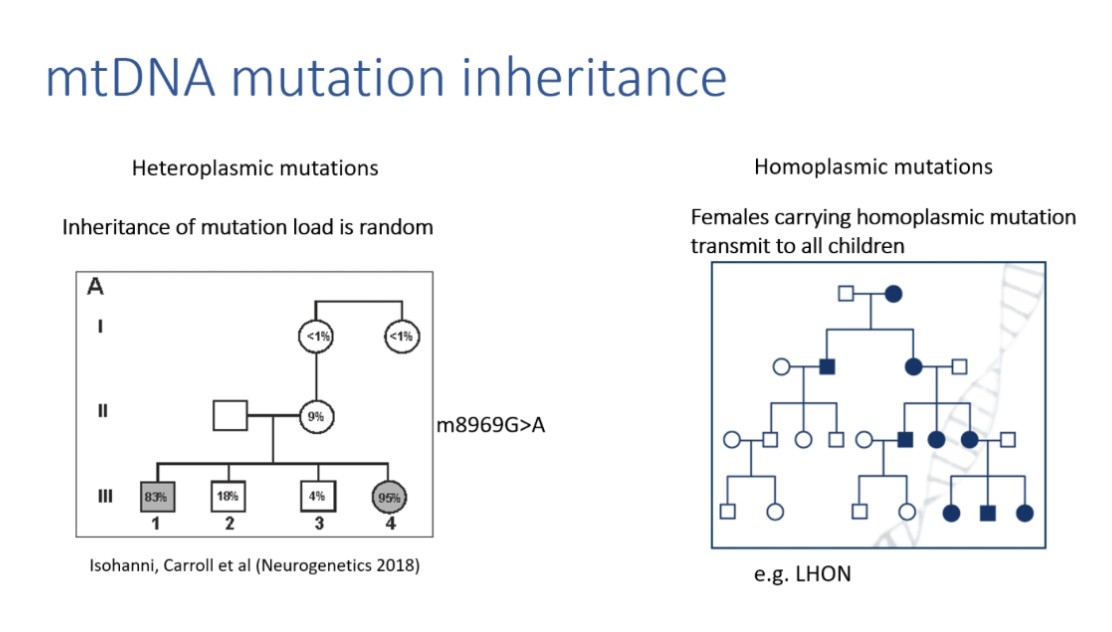
Penetrance variation in homoplasmy
LHON shows sex-biased manifestation: ~50 percent males, ~10 percent females affected.
Estrogen neuroprotection may reduce female penetrance.
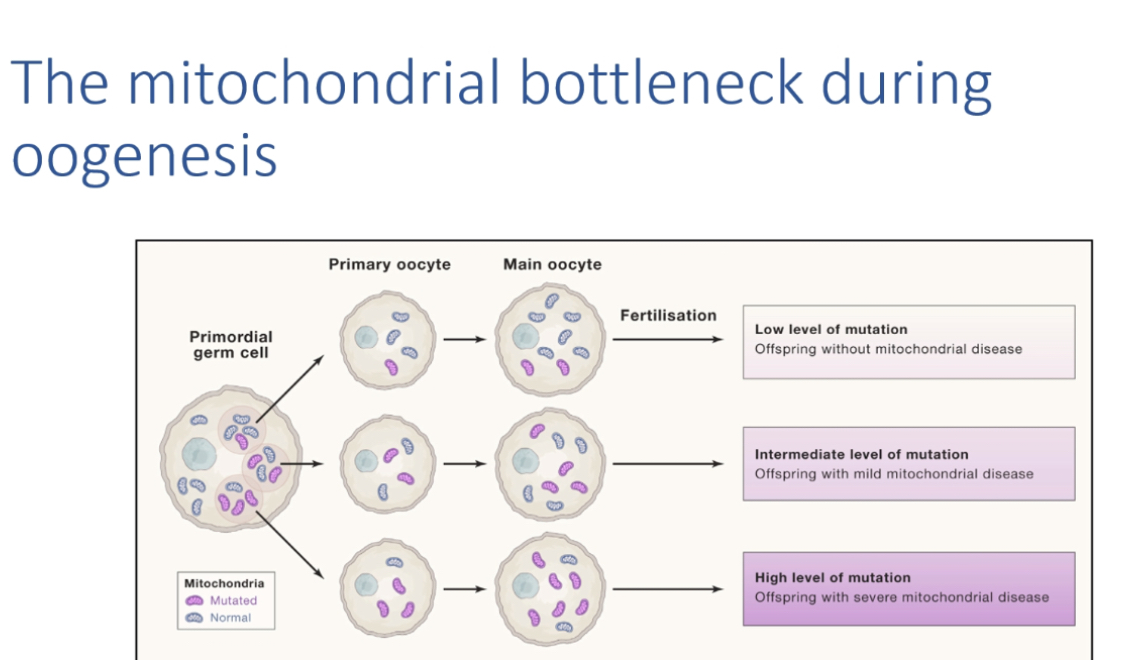
Mitochondrial bottleneck
During oogenesis, rapid change in heteroplasmy distribution creates unpredictable recurrence risk.
Nuclear gene causes
350 nuclear genes impact mitochondrial function including mtDNA maintenance genes (e.g. TWINKLE).
mtDNA deletions and depletion
Secondary consequences of nuclear gene defects, often affecting post-mitotic tissues (muscle, brain, heart, liver).
TWINKLE-associated disease
Dominant TWINKLE mutations cause multiple mtDNA deletions → PEO, progressive weakness.
Gene panels
Target a few known mitochondrial disease genes: high coverage but may miss novel genes.
Whole exome sequencing (WES)
Covers coding regions; can detect off-target mtDNA reads.
Mitochondrial DNA (mtDNA) genes are not intentionally targeted in WES.
What “off-target mtDNA” means:
During WES library prep and capture, some mtDNA fragments are accidentally sequenced even though the probes were designed for nuclear exons. These unintended reads are called off-target mtDNA reads.
Why this happens:
mtDNA exists in many copies per cell, so it’s abundant.
Capture probes and sequencing are not perfectly specific, so mtDNA can slip through.
What you can (and can’t) do with off-target mtDNA reads:
You may detect common mtDNA variants or estimate heteroplasmy roughly.
Coverage is uneven and incomplete, so it’s not reliable for full mtDNA diagnosis.
Whole genome sequencing (WGS)
Covers all DNA including mtDNA fully, but higher cost and complexity.
Counselling complexity
Prognosis depends on mutation type, heteroplasmy level, and penetrance → highly unpredictable.
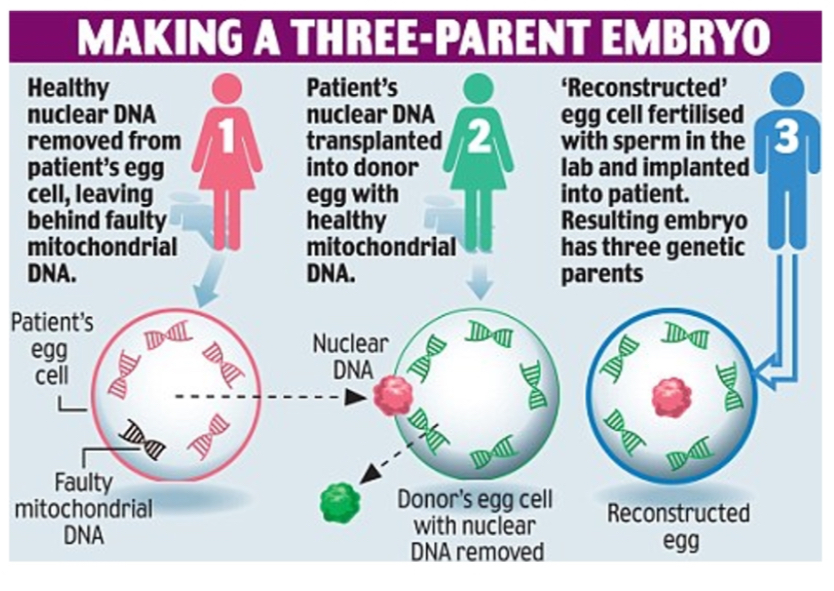
Prevention of transmission
Oocyte donation, prenatal diagnosis, PGD, mitochondrial replacement therapy (MRT/“three-parent babies”).