Module 2: Oral Manifestations of Hematological Disorders
1/75
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
76 Terms
anemia
-decreased volume of RBCs (hematocrit)
-decreased concentration of Hb and/or O2 binding capacity of Hb
-often a sign of an underlying disease (liver disease, chronic inflammatory conditions, malignancies, vitamin/mineral deficiencies)
anemia signs/symptoms
-mostly non-specific: weakness, fatigue, poor concentration, shortness of breath on exertion, skin and mucosal pallor
-less common: swelling of legs/arms, chronic heartburn, vague bruises, vomiting, increased sweating, blood in stool
-in severe anemia: body may compensate by increasing CO (symptoms may include palpitations)
anemia diagnosis
*Blood test:
1. RBC count
2. Hb concentration
3. MCV (normo-, micro-, macro- cytic)
4. Red cell distribution width (RDW)
→ calculate hemocrit, MCH, MCHC to compare values adjusted for age and sex
*Other tests: serum iron, serum B12, Hb electrophoresis
*Microscopic examination of a stained blood smear
*Can also do bone marrow biopsy if diagnosis still difficult
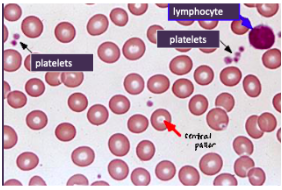
NORMAL RBCS:
*uniform in size and shape
*diameter ~ nucleus of a lymphocyte
*central pallor = 1/3 of rbc diameter
iron-deficiency anemia etiology
*Occurs in 4 different settings:
1. Excessive blood loss
2. Increased demand for rbcs (pregnancy)
3. Decreased uptake of iron (dietary restriction)
4. Decreased absorption of iron (Celiac Disease)
*Most common cause of anemia in the world
- U.S: men → GI disease; women → excessive menstrual flow
- Developing nations: parasite infection
iron-deficiency anemia clinical features
SYSTEMIC:
*Fatigue, lightheadedness, lack of energy
*Skin pallor
*Atrophy of gastric mucosa
*Alopecia
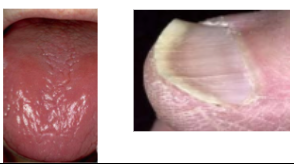
*Koilonychia (spoon-shaped nails)
*Restless Leg Syndrome
*Pica (consumption of non-food based items)
ORAL:
*Atrophic glossitis +/- burning sensation
*Generalized mucosal atrophy
*Gingival pallor
*Angular cheilitis (may predispose to candidiasis)
iron-deficiency anemia diagnosis
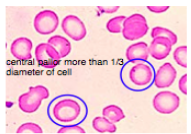
*Complete blood count: decreased MCV and MCHC
*Peripheral blood smear: hypochromic (paler) micocytic (smaller than nucleus of lymphocytes)
*Reduced blood iron levels
iron-deficiency anemia treatment
*Dietary iron supplements
*IV iron supplements
*Treatment of underlying cause
Plummer-Vinson Syndrome (Paterson-Kelly Syndrome)
*Rare
*F, 30-50 yrs, Scandanavian/N. European
*Significant increased risk for oral and esophageal carcinoma
Clinical Features:
*TRIAD:
1. Dysphagia/Esophageal Webs (🡪dysphasia)
2. Atrophic Glossitis
3. Iron Deficiency (hypochromic/microcytic) Anemia
*Angular cheilitis, koilonychia, burning sensation, GERD, esophagitis
Treatment:
*Dietary iron supplementation
*+/- esophageal dilation
pernicious anemia- etiology
*Poor absorption of cobalamin (B12, extrinsic factor)
1. Lack of intrinsic factor (necessary for B12 absorption)
- usually due to autoimmune destruction of parietal cells of stomach
- absorption of cobalamin cannot occur without IF!
2. Poor absorption of Cobalamin
- GI bypass surgery
- Strict vegetarian diet (food sources = eggs, meat, poultry, shellfish, milk, milk products)
pernicious anemia clinical features
*More common in elderly
*Fatigue, weakness, lightheadedness
*Paresthesia, tingling, numbness of extremities
*Atrophy of gastric mucosa
*Increased risk of gastric carcinoma (1-2%)
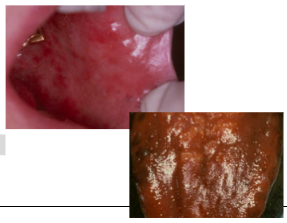
ORAL:
*Burning sensation of tongue, lips, buccal mucosa
*Oral mucosal atrophy and erythema
*Atrophic glossitis
*”Beefy red tongue”
*Yellow-tinged mucosa
pernicious anemia diagnosis
*CBC (Increased MCV)
*Peripheral blood smear: megaloblastic (seen in marrow), macrocytic (seen in peripheral blood)
*Low serum cobalamin
*Schilling Test: measures radiolabeled Cobalamin absorption & excretion rates
pernicious anemia treatment
*Monthly intramuscular injections of Cobalamin
*High-dose oral cobalamin
*Periodic evaluation by GI doctor (increased risk of gastric carcinoma)
aplastic anemia etiology
*Failure of precursor cells in bone marrow to mature into all types of blood cells
- environmental toxins (benzene)
- drugs (chloramphenicol)
- infection (non-A, non-B, non-C, non-G hepatitis)
- genetic disorders (Fanconi’s anemia, dyskeratosis congenital)
- idiopathic
*Risk of developing recurrent aplastic anemia and acute leukemia
aplastic anemia clinical features
SYSTEMIC:
1. Anemia → fatigue, lightheadedness, weakness
2. Neutropenia → bacterial & fungal infections
3. Thrombocytopenia → bleeding, bruising
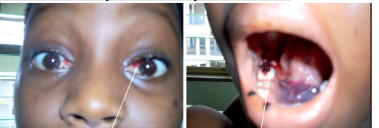
ORAL:
1. Anemia → mucosal pallor
2. Neutropenia → oral ulcerations with minimal peripheral erythema
3. Thrombocytopenia → gingival hemorrhage, mucosal petechiae, purpura
*Also see ecchymoisis of eye and mouth
aplastic anemia diagnosis
*Pancytopenia (>2 of the following):
- <10,000 rbcs /mm3
- < 500 wbcs/mm3
- < 20,000 platelets/mm3
aplastic anemia treatment
*Discontinue inciting agent (if drug or toxin)
*Supportive therapy (Abx, blood transfusions)
*Androgenic steroids
*Bone marrow transplant
*Immunosuppressive therapy (Cyclosporine)
Prognosis is guarded – risk of recurrent aplastic anemia and acute leukemia
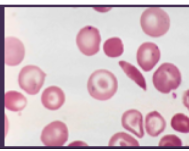
sickle cell anemia etiology
*AR genetic disorder of Hb synthesis
*Hb molecule prone to molecular aggregation in deoxygenated state → Hb forms long, inflexible chains → rbcs assume rigid, curved shape
*Reduced O2 carrying capacity of sickled rbcs
*Sickled rbcs aggregate and block capillaries
*One allele affected → 40-50% abnormal Hb, sickling only under certain conditions (low O2)
*Both alleles affected → most abnormal Hb
sickle cell anemia epidemiology
*Trait: 8% of AA
*Disease: 1:350-400 AA
*Geographic predilection – where malaria is endemic (Africa, Mediterranean, Asia)
→ sickle cell trait = resistance to malaria
clinical features of sickle cell anemia
SYSTEMIC:
*Chronic hemolytic anemia
*Ischemia, infarction, ulcers of distal extremities
*Infections (due to destruction of spleen)
*Impaired kidney function
*Ocular damage
*CNS damage/stroke
*Sickle Cell Crisis = severe sickling of rbcs
- precipitated by hypoxia, infection, hypothermia, dehydration
- extreme pain from ischemia and infarction of tissues
- mostly affect long bones, lungs, and abdomen
- pulmonary involvement = acute chest syndrome
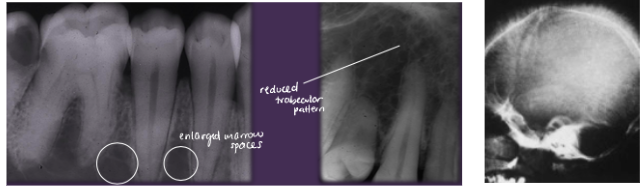
ORAL:
*Reduced trabecular pattern
*Enlarged marrow spaces
*“Hair-on-end” on skull x-ray
*Increased prevalence of osteomyelitis
*Spontaneous, asymptomatic pulpal necrosis
sickle cell anemia treatment
*Avoid precipitating events
*Supportive care (fluids, rest, anelgesics)
*Hydroxyurea (inhibits polymerization of abnormal Hb, reduces adherence of RBCs to vessel walls)
*Bone marrow transplant
*Genetic counseling
hemoglobinopathies- thalassemia etiology
*AR genetic disorder characterized by reduced synthesis of alpha-globin or beta-globin chains of Hb molecule
*Hb structure:
- tetramer of 2 alpha and 2 beta chains
- 4 genes → alpha chain; 2 genes → beta chain
*Defective production of 1 chain → reduced amounts of Hb and abnormal rbcs
*Abnormal rbcs sequestered in spleen & destroyed → hemolytic anemia
thalassemia epidemiology
*Geographic predilection: Mediterranean, Africa, India, SE Asia
*Thalassemia trait = malaria resistance
beta-thalassemia genetics
*remember: 4 genes code for alpha, 2 for beta
*Beta-Thalassemia Minor:
- 1 beta globin gene defective
- clinically insignificant disease
*Beta-Thalassemia Major (Cooley’s Anemia)
- 2 beta globin genes defective
beta-thalassemia clinical features
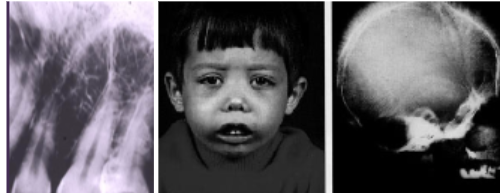
Thalassemia Major
*Manifestations at ~1 yr
*Hemolytic anemia
*Increased hematopoiesis with bone marrow hyperplasia
and enlarged marrow spaces
*”Hair-on-end” appearance on skull xray
*Enlargement of liver & spleen, lymphadenopathy
*Enlargement of maxilla and mandible (“chipmunk faces”)
*Altered trabecular pattern
beta-thalassemia treatment
*Blood transfusions every 2-3 weeks
- complicated by iron overload
*Bone marrow transplant
*Surgical Recontouring for abnormal faces
beta-thalassemia prognosis
*Without therapy: tissue hypoxia, bacterial infections, cardiac failure by 1 yo
*With therapy: relatively normal lifespan
alpha-thalassemia genetics
*remember: 4 genes code for alpha, 2 for beta
*No disease – 1 gene defective
*Alpha Thalassemia Trait – 2 genes defective
*HbH Disease – 3 genes defective
*Hydrops Fetalis – 4 genes defective (fatal within a few hours of birth)
alpha-thalassemia clinical features
*Alpha Thalassemia Trait
- mild anemia, may be clinically insignificant
*HbH Disease
- more severe hemolytic anemia, splenomegaly
alpha-thalassemia treatment
*Iron and Folic Acid supplementation
- caution: iron overload (hemochromatosis)
*General Supportive care
- transfusions, may be needed periodically
*Splenectomy – risk of infection
*Severe cases: allogeneic hematopoetic stem cell transplant (curative)
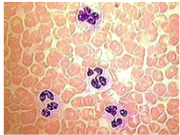
polycythemia vera etiology
*Idiopathic disorder characterized by increased mass of rbcs
and abnormal proliferation of marrow stem cells
*Myeloproliferative Disease
*Increase in wbcs and platelets
*Cells function normally, just in excess (very crowded smear)
→ % of rbcs in blood may be so high
→ blood ceases to flow in some smaller vessels and capillaries
polycythemia vera clinical features
*Older adults ~ 60 yrs
*Symptoms: nonspecific – HA, weakness, dizziness, visual disturbances, sweating, weight loss
*Ruddy complexion
*Pruritis – 40%
*Transient ischemic attacks, stroke, heart attacks – related to thrombosis
*Erythema and burning of hands and feet, gangrene and necrosis (related to thrombosis)
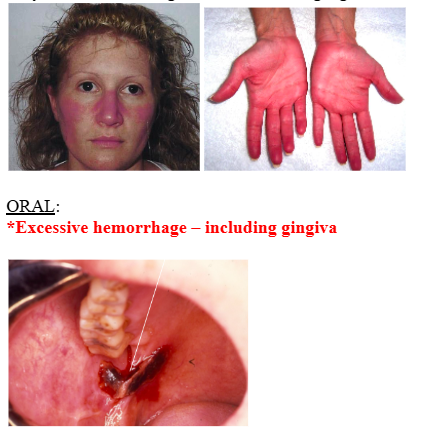
ORAL:
*Excessive hemorrhage – including gingiva
polycythemia vera treatment
*Phlebotomy – removal of blood (risk of thrombosis)
*Aspirin therapy – to reduce thrombotic events
*Myelosuppressive Therapy/Chemotherpeutic Drugs (risk of leukemia)
polycythemia vera prognosis
*Fair: average survival 10-12 years after diagnosis
*2-10% develop acute leukemia
leukemia etiology
*Malignant proliferation of hematopoetic stem cell derivatives (may be component of a syndrome – Down, Bloom, Klinefelter)
*Increased risk associated with exposure to certain environmental agents (pesticides, benzene, ionizing radiation)
*Generally bone marrow involvement
Classification:
Acute Myeloid – broad age range
Chronic Myeloid – primarily affects adults
- Philadelphia Chromosome – translocation of long arms of chromosomes
Acute Lymphoblastic – primarily affects children
Chronic Lymphoblastic – primarily affects adults
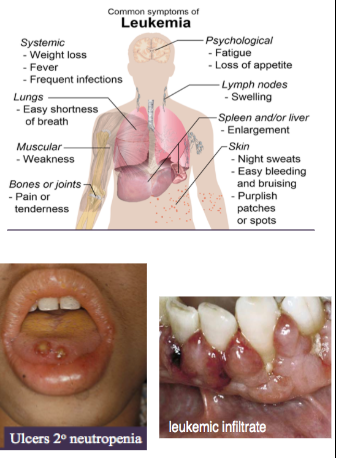
clinical features of leukemia
*Due to reduction of rbcs and wbcs caused by crowding out of normal marrow cells by malignant leukemic cells → myelophthisic anemia
SYSTEMIC:
*Anemia → fatigue
*Neutropenia → ulcerations, bacterial, fungal, viral infections
*Thrombocytopenia → bleeding, bruising
ORAL:
*Mucosal pallor
*Mucosal petechiae, purpura
*Ulcerations
*Fungal infections (eg candidiasis)
*Viral Infections (eg herpetic lesions)
*Gingival Leukemic Infiltrate = tumor-like collection of leukemic cells (granulocytic sarcoma or extramedullary myeloid leukemia, old term = chloroma), usually myeloid, type, spontaneous bleeding (no spontaneous bleeding in benign hyperplasia)
treatment of leukemia
*Chemotherapy + radiation
*Bone marrow transplant
*Supportive care (blood transfusions, Abx)
*Encourage optimal oral hygiene
leukemia prognosis
*Depend on type, age, other factors
*5 year survival rates:
- older patients with AML = 10-20%
- children with ALL = 70%
lymphoma etiology
*Group of malignant solid tumors involving cells of the lymphoreticular or immune system (T/B Cells)
*Most arise within lymph node (vs leukemias which begin in bone marrow and are characterized by malignant cells circulating in peripheral blood)
*Unknown etiology – viruses?
*2 major categories:
- Hodgkin’s Disease
- Non-Hodgkin’s Lymphoma
Non-Hodgkin’s lymphoma epidemiology
*Lymphoma of oral cavity is rare (more often Non-Hodgkin’s type: 3-5%)
- oral lesions rarely initial presentation
*Primarily affects adults (66 yrs)
*Male
*Increased incidence in AIDS
Non-Hodgkin’s Lymphoma clinical features
*Most common presentation: painless, persistent enlargement of lymph nodes
*Extra nodal disease can occur → mainly presents in soft tissue
*Usually in patients >40 y/o
*Increased incidence in AIDS patients
ORAL:
*Enlarged tonsil, painless palatal swelling, or gingival mass (may mimic inflammatory disease)
*Tumor isnontender, diffuse, boggy, discolored swelling (+/- ulcerated)
*Rarely can occur in jaw bones
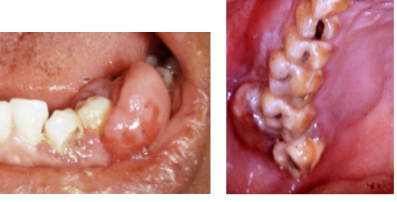
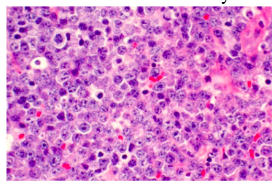
Non-Hodgkin’s Lymphoma pathology
-diffuse sheet of inflammatory cells
Non-Hodgkin’s Lymphoma treatment
-radiation and/or chemotherapy
Non-Hodgkin’s Lymphoma prognosis
-5 year survival ~50%
Burkitt’s lymphoma epidemiology
*Unique high grade non-Hodgkin’s Lymphoma, very rapid growth
*t(8:14)
*2 types:
- African – 6 years, 2x M, EBV found in 90%
- American – 11 years, no gender predeliction, EBV found in 10% of American
Burkitt’s lymphoma clinical features
African Type:
*Primarily involves jaw bones (most often maxilla) with secondary visceral involvement
American Type:
*Primarily presents as abdominal swelling - only 15% jaw involvement → Jaw involvement in both types = rapidly growing mass, teeth become mobile and can exfoliate
Radiograph:
*Destructive, ragged, “moth-eaten” radiolucency
*Teeth “floating in air”
Histology:
*Diffuse small lymphocytes with “starry sky”
- sky = lymphocytes
- stars – macrophages
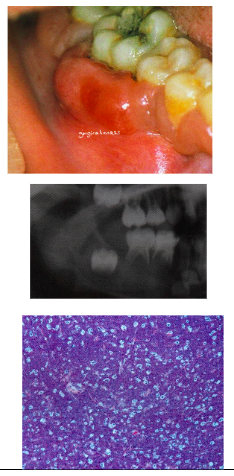
Burkitt’s lymphoma treatment
-chemotherapy
Burkitt’s lymphoma prognosis
-depends on volume of tumor at initiation of chemo → tumor often surgically debulked first
-survival rate ~70%
extranodal NK/T-cell lymphoma (angiocentric T cell lymphoma, midline lethal granuloma) clinical features
*Rare, affects mainly adults
*Most frequent in Asian, Guatemalan, & Peruvian
*Destruction of midline structures of palate and nasal fossa
*Pain, nasal stuffiness, epistaxis, swelling of palate,
ulceration, oral-antral communication
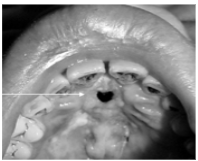
extranodal NK/T-cell lymphoma treatment and prognosis
*Tumor responds well to radiation
*Good prognosis with early treatment
*Untreated → secondary infection, hemorrhage, involvement of vital structures → death
neutropenia etiology
*Decreased number of circulating neutrophils (<1500/mm3)
*Decreased production or increased destructed of neutrophils
*Congenital or acquired
*Benign Ethnic Neutropenia: several ethnic groups (African, Middle eastern) will consistently have neutrophil counts that would qualify as neutropenia, yet are otherwise healthy
causes of acquired neutropenia
*Destruction of bone marrow by malignancy or metabolic disease
*Drugs (chemotherapy, penicillins, sulfonamides, diuretics, tranquilizers, phenothiazines)
*Nutritional deficiencies (B12, folate)
*Viral Infections (hepatitis, rubella, measles, HIV)
*Bacterial Infections (TB)
*Autoimmune Diseases (SLE)
neutropenia clinical features
*Bacterial > Viral/Fungal infections
- may demonstrate reduced suppuration/abscess formation
- Common infection site = middle ear, oral cavity, perirectal area
ORAL:
*Infection of oral mucosa may be initial sign!
*Ulcerations (minimal peripheral erythema, usually gingiva,
biopsies show neutrophilic infiltrate)
*Premature periodontal disease
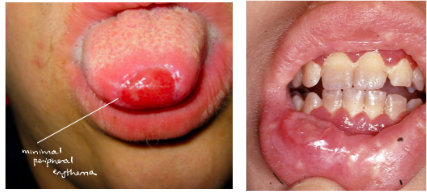
neutropenia treatment
*Abx (for infections)
*Encourage optimal oral hygiene
*G-CSF – promotes growth and differentiation of neutrophils
cyclic neutropenia etiology
*Idiopathic periodic reductions in neutrophil counts
*May be related to defect in bone marrow stem cells
*Most sporadic, some inherited (AD)
*Neutrophil count lowest (nadir) for 3-5 days in ~21 day cycles
*Incidence 1/1,000,000
*Diagnosed in childhood
- diagnosed with sequential complete blood counts (2-3x/week for 8 weeks)
- neutrophil count <500/mm3 for 3-5 days for at least 3 successive 21 day cycles
cyclic neutropenia clinical features
*Related to neutrophil count at nadir
- recurrent fever, cervical lymphadenopathy, pharyngitis, malaise, ulcerations of GI tract
ORAL:
*Ulceration of mucosa with minor trauma
- gingiva, lips, tongue, buccal mucosa
- with reduced neutrophilic infiltrate
*Severe periodontal bone loss
*Tooth mobility
*Marked gingival recession
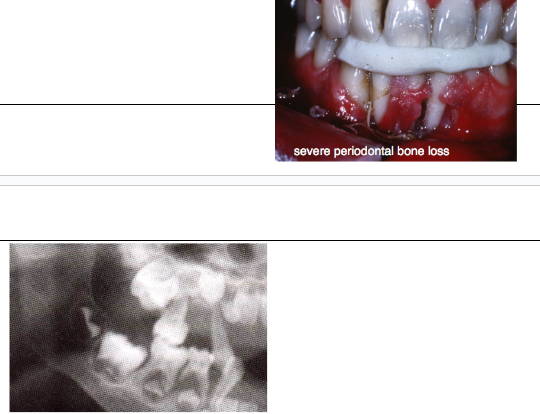
cyclic neutropenia treatment
*ABx (for infections)
*Encourage optimal oral hygiene
*G-CSF several times weekly → reduced duration of nadir and severity of infections/ulcerations
agranulocytosis etiology
*Absence of cells of granulocytic series – especially neutrophils
*Decreased production or increased destruction
*Most drug-induced, some idiopathic
agranulocytosis clinical features
*Bacterial infections, malaise, sore throat, fever (a few days after drug is taken)
ORAL:
*Necrotizing, punched-out, deep mucosal ulcerations
- buccal mucosa, tongue, palate
- sparse inflammation and abundant bacterial colonies
*Gingivitis resembling necrotizing ulcerative gingivitis (NUG)
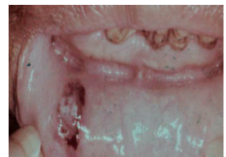
agranulocytosis treatment
*Discontinue offending drug (count returns to normal in 10-14 days, longer for chemotherapeutic drugs)
*Abx (for infections)
*Encourage optimal oral hygiene
*Chlorhexidine mouth rinses
*G-CSF when counts don’t return to normal
multiple myeloma etiology
*Malignant neoplasm of plasma cells
*Involves many bones in the body: vertebrae, ribs, skull, jaw (5-30%)
*40+ years
multiple myeloma clinical features
SYSTEMIC:
*Most common: skeletal pain – due to bone lysis by accumulation of tumor cells → multiple punched out radiolucencies with ill-defined margins/lack sclerotic border → pathological fractures
*Renal failure
*Severe bacterial infections
*Clotting defects → dental management considerations (bisphosphonate exposure?)
*Histology: sheets of plasma cells in various degrees of differentiation
ORAL:
*Jaw lesions → pain, swelling, numbness, tooth mobility
*Amyloid deposits throughout body: tongue, vestibule
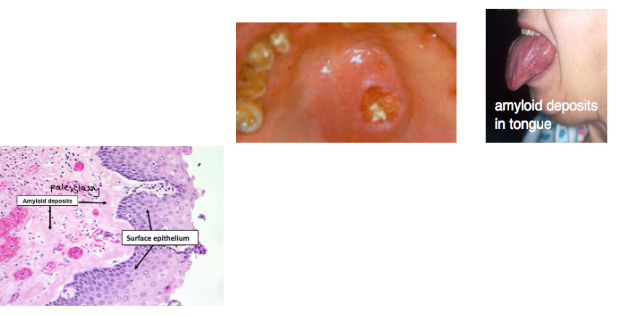
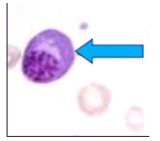
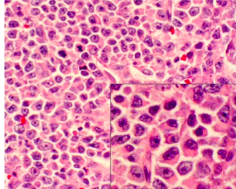
multiple myeloma histology
*sheets of plasma cells in various degrees of differentiation
*plasma cells have eccentric nucleus, perinuclear halo, clockface nucleus
multiple myeloma treatment
*Chemotherapy
*Radiation for local symptomatic lesions
*Stem cell transplant
multiple myeloma prognosis
*Poor prognosis
*Average survival: 2-3 yrs with chemo/radiation
*Younger patients have better prognosis
*Better with stem cell transplant (~25% remission)
Note: dental management considerations – increased risk of hemorrhage nad increased susceptibility to infection, bisphosphonate exposure?
Langerhans Cell Histiocytosis (LCH)
*Clonal proliferation of Langerhans Cells = type of APC usually seen in suprabasal layer of epidermis/mucosa
*Pulmonary Langerhans Cell Histiocytosis: unique form that occurs almost exclusively in cigarette smokers
- now considered a form of smoking-related interstitial lung disease
- some recover after quitting
- other develop pulmonary fibrosis, pulmonary HTN
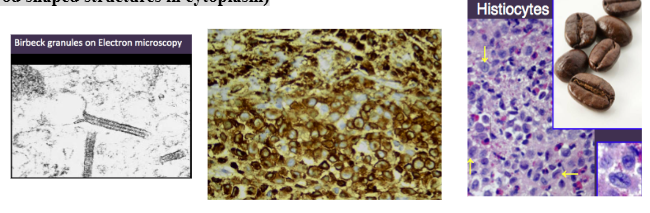
LCH diagnosis
*Biopsy: diffuse infiltration of lymphocytes, plasma cells, eosinophils,
- large pale staining “histiocytes”/Langerhans Cells indented kidney bean shaped nuclei
*Immunostaining to identify
Langerhans Cells: S-100+, CD1-a+
*Electron Microscopy: former gold standard
- Birbeck Granules (rod shaped structures in cytoplasm)
Letter-Siwe disease (multifocal multisystem) clinical features
*Occurs before age 3
*Most acute form
*Skin rash, enlarged spleen, liver, lymph nodes
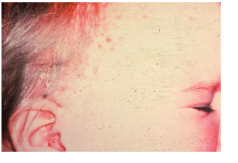
Letter-Siwe disease oral manifestations
*Ulcerated gingival lesions
*Diffuse bone destruction with loss of teeth
*Significant oral involvement may not occur due to rapid course → death
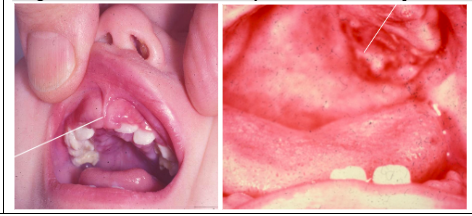
Hand-Schuller-Christian disease (multifocal unisystem) clinical features
*Children: 5yrs – adolescence
*Chronic
*50% spontaneous remission
*Skeletal and Extraskeletal lesions
*Classic Triad (25%)
1. Single/multiple punched out lesions of skull and/or jaws
2. Uni or Bilateral exopthalmus
3. Diabetes Incipitus
ORAL:
*Lesions: sore mouth, gingival ulcers, halitosis, gingivitis, loose teeth and premature loss, loss of alveolar bone - Mimics advanced periodontal disease
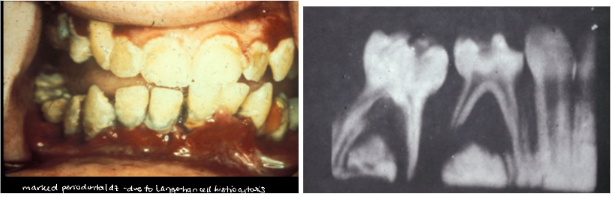
Hand-Schuller-Christian disease treatment
*Curettage, Excision, chemo, radiation
eosinophilic granuloma (unifocal) clinical features
*Older children & Young adults
*Some cases of spontaneous regression
*Solitary or multiple lesions in bone
- skull & mandible most common sites
*Radiograph: irregular radiolucent areas
- can mimic periodontal disease or periapical cysts/granulomas
*Some cases with skin involvement but NO visceral involvement
*Looks like periodontal disease but unresponsive to scaling
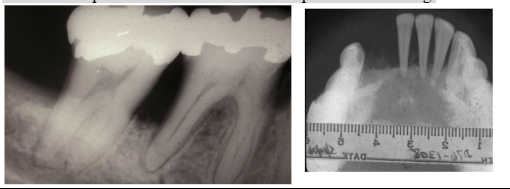
eosinophilic granuloma treatment
*Curettage, steroid injections
*Very good prognosis
thrombocytopenia etiology
*Deficiency of circulating platelets (<100,000/mm3)
*Platelets: necessary for normal hemostasis & clot formation
*Decreased production of platelets
- infiltration of bone marrow by malignant cells (leukemia)
- chemotherapeutic drugs
*Increased destruction of platelets
- immunologic drug reaction
- viral infection, vaccination → autoantibodies against plt
*Sequestration of platelets in spleen
- some conditions that cause splenomegaly can cause platelets to be sequestered in spleen (portal HTN)
thrombotic thrombocytopenia purpura (TTP) and idiopathic (immune) thrombocytopenic purpura (ITP)
A. Thrombotic Thrombocytopenia Purpura (TTP)
- Increased destruction of platelets
- Consumption of platelets by abnormal clot formation
B. Idiopathic (Immune) Thrombocytopenic Purpura (ITP)
- Usually occurs after non-specific viral infection
- Most cases resolve spontaneously in 4-6 weeks
clinical features of thrombocytopenia
*Increased bleeding, even with minor trauma
*Platelets <10,000/mm3 → risk of GI, GU, pulmonary, intracranial hemorrhage
ORAL:
*Bleeding of oral mucosa, even with minor trauma
*Spontaneous gingival hemorrhage
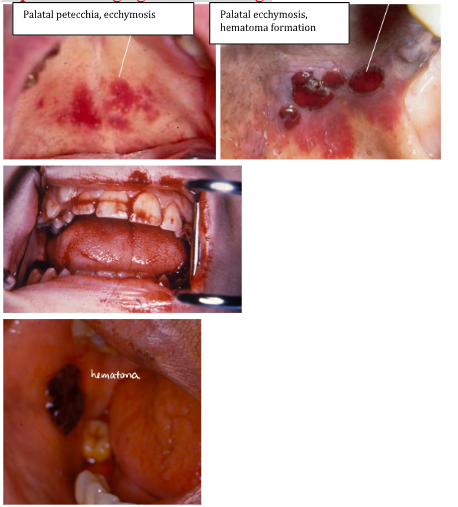
thrombocytopenia treatment
*Discontinue offending drug if drug-related
*Platelet transfusions