General and specific transcription factors
1/17
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
18 Terms
How do cells become defined by their proteins during development?
Normal development begins at fertilisation – the zygote is totipotent. During development, stem cell lineage becomes restricted and they acquire: pluripotent, then multipotent and eventually unipotent capabilities. This process correlates with their ability to express proteins of a specific cell lineage. The human body has around 250 distinct cell lineages.
Differentiated cells are defined by the functions they perform – which in turn are determined by the specialised proteins that they are programmed to express.
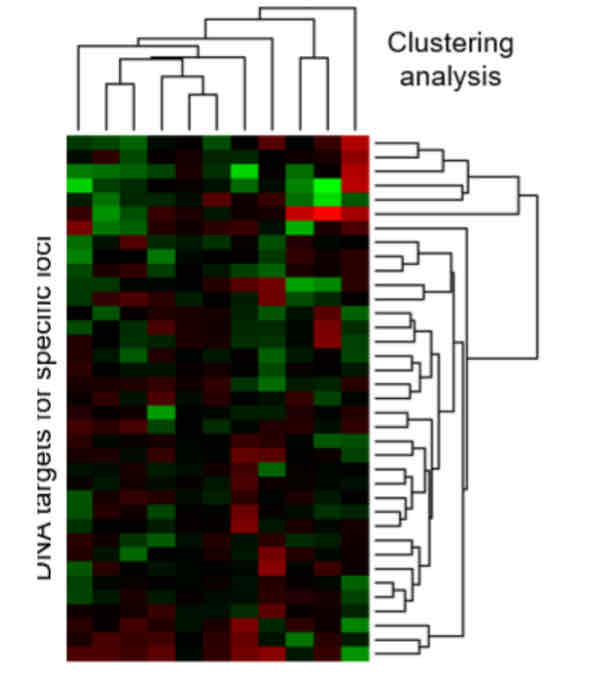
What is microarray analysis and what is it used for?
One technique to look at differentiation expressions is microarray analysis. RNA (usually mRNA) is extracted from comparator samples: 2 different cell types, or 1 cell type in different conditions. They are then prepared and labelled with fluorescent tags. Probes are hybridised to DNA target sequences bound in a known pattern on the microarray surface. After hybridisation, samples are washed and fluorescence indicates bound probe quantity. Expression of specific loci is defined and loci clustered to monitor cell-type specific (a) or condition specific (b) patterns of expression. RNA seq (Illumina HiSeq technology) provides a state-of-the-art alternative which delivers improved quantification and can monitor all classes of RNA.
The brighter the colour, the more of the protein expressed. There can be a mix of probes to show equal amounts in both cells.
What is the general outline for eukaryotic gene expression?
During eukaryotic gene expression, the chromatin relaxes, then the DNA gets transcribed and processed to form mRNA. It is then spliced before transporting to a ribosome for translation.
What is the structure for a class II gene regulatory region?
The class II gene regulatory region has a promoter and an enhancer.
The promoter has a TATA region (~-25) which binds a TATA protein which recruits sequence-specific general TFs. It is the location of assembly of RNA polymerase complex. Most contain an Initiator (Inr) element. It is position and orientation sensitive. General transcription factors are the same in all genes.
The enhancer has multiple binding sites for sequence-specific specific TFs. They bend the DNA to interact with RNA polymerase II and general TFs to allow the start of transcription. It is position and orientation independent. Specific transcription factors are different depending on the cell and its protein needs.
How do general and specific transcription factors work?
General transcription factors will accumulate at the TATA region, but work very slowly on their own. Specific factors will bind to the enhancer region, which will bend the DNA to interact with proteins bound at the core promoter which will activate the accumulated general TFs to start transcription.
How does phosphorylation allow for elongation?
Phosphorylation of Pol II C-terminal domain causes TFII DAB to become dispensable for elongation, and TFII E and H become dispensable for elongation. Other proteins associate with TFIIH to form the repairosome. Defects in this function cause Xeroderma Pigmentosum.
The CTD has many repeats of 7 amino acids (YSPTSPS), and the 5th serine is phosphorylated each time. Therefore, serine 5, 12, 19… are phosphorylated.
How does elongation occur?
Initiation of transcription occurs when RNA pol II is phosphorylated at ser5. After transcribing a few hundred bases, the polymerase stalls to prevent the mRNA from being degraded. At this point, elongation may abort or it will continue once the capping complex is recruited to add a 5’ cap to the mRNA which is then committed to complete synthesis. During this step, the CTD is additionally phosphorylated at ser2 by CDK9 (serine 2, 9, 16 etc are phosphorylated) which provides a signal for continuation.
During elongation, nucleosomes are continually displaced by RNA pol II and reformed after it passes, and enzymes associated with the synthetic complex continually remodel the post-translational histone states. Sometimes, splicing can happen at the same time as elongation.
How does termination occur?
Termination occurs when RNA pol II arrives at the poly-A-signal (3’-TTATTT-5’) where it then adds 5’-AAUAAA-3’ to the mRNA. The cleavage site (CA) is located 15-30 bases downstream of the poly-A-signal. When the polymerase reaches here, it will start adding A bases to the 3’ end to protect it.
This process is more efficient if there is also a U-rich upstream signal element and GU-rich downstream signal element.
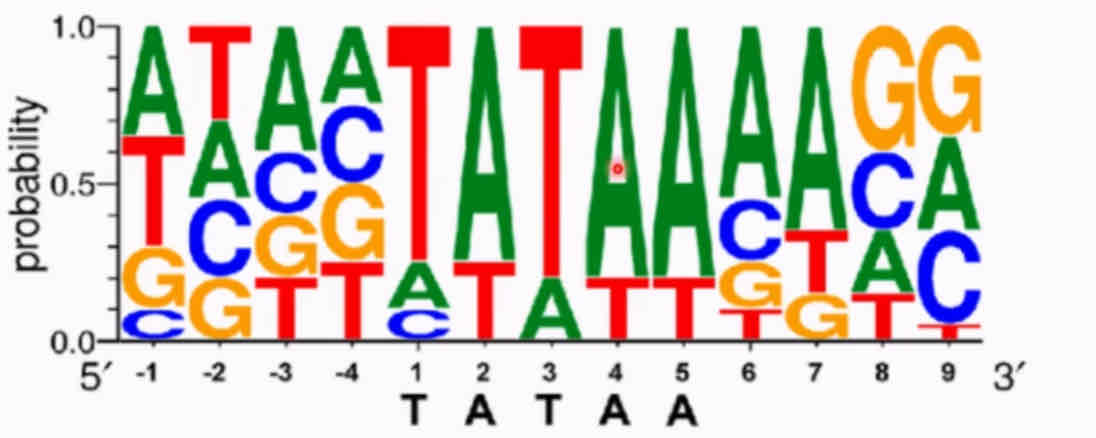
What is consensus sequence analysis and how does it work?
Consensus sequence analysis is how you find the DNA sequence that binds to a protein. This is when you generate a random sequence of oligonucleotides, anneal them in a test tube to generate all DNA combinations. The protein of interest is stuck to a column and the DNA is run over it. Separate the bound DNA from the rest via washing, then separate the bound DNA from the protein. Use a computer to align the sequence to determine the consensus sequence (often via logo plot).
A logo plot shows the probability of each base appearing at each number in the sequence. The highest probability sequence is the consensus one. You can get proteins which bind TATTA etc, because they are similar but there is less affinity therefore less probability of appearing.
What is an electrophoretic shift assay and how does it work?
Electrophoretic mobility shift assay is used if you already know the DNA sequence and want to know whether it can bind to a protein. Firstly, purify the protein and add it to a known DNA sequence that has been 5’-end radiolabelled. The protein-DNA mix is run along a native gel, and the DNA bound protein will have a higher molecular weight and run slower in the gel.
What is DNAse I footprinting and how does it work?
To find the exact sequence a protein is binding (if you know the region), DNAse I footprinting is used. Firstly, a known DNA sequence is made and radiolabelled at 5’. Purified protein and DNAse is added at a concentration where it will randomly cut once per strand. The DNA is isolated and run on a gel. Regions where the DNA was bound to a protein will have been protected, so you get a footprint on the gel - this indicates which bases were bound to a protein.
What is the structure of a transcription factor?
TFs have a modular structure. They have DNA binding domains and transcriptional regulation domains. Some of these may activate and some repress. Some may even have domains which interact with other TFs.
What different transcription factor superfamilies are there?
They are grouped into superfamilies based on their DNA binding domains; helix-turn-helix (HTH) and zinc fingers (ZF) act as monomers. Basic-Leucine-Zippers (bZIP) and basic helix-loop-helix (bHLH) act as dimers.
What is the structure of a helix-turn-helix TF?
In the HTH motif, helices 1 and 2 lie above the DNA and stabilise the structure, helix 3 binds to the major groove, and the N-terminal binds to the minor groove.
What is the structure of a zinc finger TF?
The ZFs bind to Zn molecules, causing one side to form a beta sheet and another side to form an alpha helix - these fingers interact with DNA as a monomer, or can interact with each other to form a dimer, which changes the DNA binding region (not finger). There are 3 different classes; 1 which is C2-H2 (cysteine and histidine bind to Zn), 2 which is C2-C2, and 3 which is C6 that binds to 2 Zn. The amino acids within fingers generate binding specificity.
C4 ZFs are a nuclear hormone receptor family that act as dimers. Large superfamily of ligand-dependent transcription factors/receptors. Nuclear Oestrogen Receptors are ERα and ERβ that can bind DNA as homodimer (αα or ββ) or heterodimer (αβ).
What is the structure of a basic-leucine-zipper TF?
The bZIP is an amphipathic helix with basic regions. Leucines at every 7th position form the hydrophobic side of the helix. bZip proteins form dimers because their hydrophobic helix preferentially binds to another hydrophobic helix (leucine sides interact). The foot of each helix is rich in basic residues. These basic residues then bind to the DNA, and DNA-specificity depends on the bZip dimer.
What is the structure of a basic-helix-loop-helix TF?
The bHLH have a similar structure to bZip whereby the Helix-Loop-Helix domains interact with each other. The basic-regions can then bind to DNA. They target the DNA-sequence CAnnTG. Many are heterodimers that use a common Type-1 partner but DNA-specificity is due to the Type-2 variable partner.
Muscle MyoD is a bHLH factor that binds to E47 that activates genes to make muscle cells. Atoh1 is a factor that binds to E47 that activates genes to make neuronal cells. These sequences they recognise are slightly different.
How do TF activation domains differ?
Activation domains share loose similarity - they fall into a number of classes; ACIDIC (Asp [D] and Glu [E] rich), GLUTAMINE RICH (Gln [Q]), and PROLINE RICH (Pro [P]). They interact with GTFs/TAFs.