PSYC 304 Final
1/139
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
140 Terms
amygdala and fear
some stimuli evoke innate fear response (no learning required) - animals/humans with lesions to amygdala display fearless behaviours
amygdala plays essential role in learning to be afraid of potentially harmful things
Pavlovian (classical) conditioning
displayed by virtually all animals - helps prepare for biologically significant events in response to cues that predict them
unconditioned stimulus = biologically significant event (meat in dog’s mouth)
unconditioned response = normal response to significant event (salivation)
conditioned stimulus = previously neutral cue that reliably predicts significant event (bell predicts arrival of meat)
conditioned response = body’s response to CS alone (salivating to bell)
automatic response, neutral + unconditioned stimulus, involuntary, no reward or punishment
5 key points for classical conditioning
CS must reliably predict US
delivery of CS and US are uncontrollable by organism
CR is also uncontrollable (automatic)
in humans, CR typically occurs in absence of conscious knowledge
very long lasting (can be extinguished, but reinstated quickly)
auditory fear conditiong
CS = tone, US = shock, CR to tone = freeze (expecting shock
lesions to amygdala subnuclei in rats abolish increase in freezing and autonomic response to tone (CS)
lesions made prior to conditioning or after conditioning both disrupt CR - need amygdala to learn and recall fear
similar results in humans with amygdala damage = disrupted fear conditioning
neutral + aversive stimulus, aversive, no reward or punishment involved
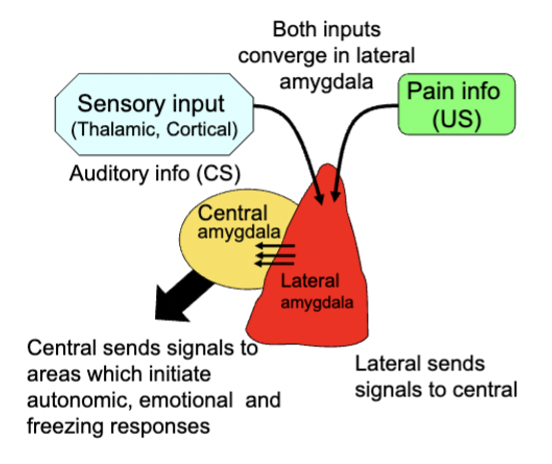
activity in lateral amygdala during painful stimulation
sensory input and pain info both converge in lateral amygdala → send signals to central amygdala → central amygdala sends signals to areas which initiate bodily and emotion responses and late
neurons in lateral amygdala show changes in firing to CR over course of learning, parallels emergence of conditioned response
increase activation in amygdala to CS after conditioning in humans
amygdala puts pain and sensory input together
amygdala an appetitive conditioning
amygdala regulates conditioning for rewarding stimuli - helps brain get to the good stuff
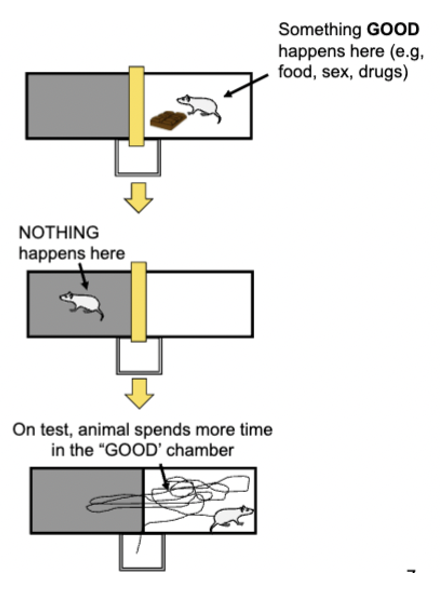
CR = typically approach behaviour (place where reward behaviour was received becomes attractive)
lesions on lateral amygdala disrupts conditioned place preference for all types of rewards (does not affect consumption of the reward) - affects how conditioned stimuli link with primary reward affect behaviour
approach/seeking response, neutral + rewarding stimulus, involuntary
testing amygdala’s appetitive control
subjects conducted a memory task of finding a red ball over a black one - finding the red ball = pleasant tone with distinct patterned background and food reward, black ball = buzzer, no reward, different pattern
healthy subjects preferred patterns associated with reward (couldn’t say why the preferred them)
amygdala lesions = no preference
reward associated cues can control behaviour sometimes without us being aware
rats will just press levers to get the conditioned stimulus, even though it never gives the reward (lesions to lateral amygdala disrupt preference for the CS level)
instrumental (operant) conditioning
association with action/motor and its consequences - organism can control what happens
any motor skill can be viewed as operant conditioning
goal-directed response, behaviour + consequence, voluntary, reward or punishment
reinforcer
something that increases or decreases likelihood of response occurring again
what parts of the brain involved in instrumental conditioning?
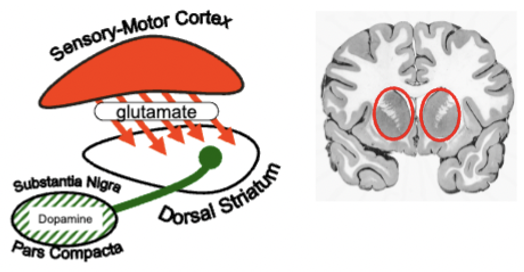
striatum (part of basal ganglia) regulates action selection + instrumental conditioning
receives converging inputs from sensory/motor cortex and DA system (both are active when action facilitate learning)
inhibits any inappropriate actions
amygdala sends input to striatum and can influence instrumental action
phases of instrumental learning
early in learning: goal-directed, responses are made to obtain goal, sensitive to levels of motivation
late in learning: responses become more automatic (habitual) - mediated by dorsal striatum
key components of memory
encoding (translating sensory info into neural code) - typically requires attention
sorting (retaining info over time) - biological memories more fragile (incorrect memories)
retrieval (active processes of using stored info) - reactivate neural networks similar to how they were when memories were coded (some retrieval more taxing than other (free recall vs. recognition)
main points about neural basis of memory
multiple forms of memory
different types of memory regulated by distinct brain regions
one type of memory regulated by interactions between multiple brain regions
short term memory
info held for short periods (seconds to hours) while physiological changes for long-term are made
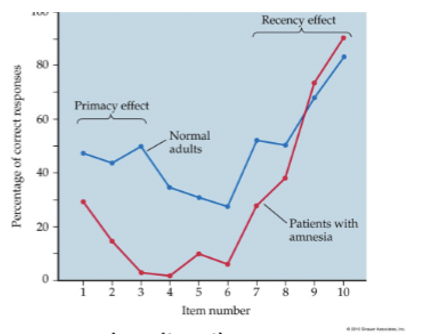
capacity of 7 ± 2 items
susceptible to distraction, requires active rehearsal
info loss occurs through displacement (something pushes it out) or decay
info gets discarded or moves to other memory stage (primacy/recency effect)
long-term memory
stable, can last lifetime of organism
potentially unlimited capacity
active or passive retention
info can be inaccurately remembered
consolidated (transferred from short-term) - involves physical changes in neuron connectivity/communication
amnesia
certain brain injuries, drugs impairing encoding, consolidation or retrieval of long-term memories
categorized by info lost relative to time of brain insult
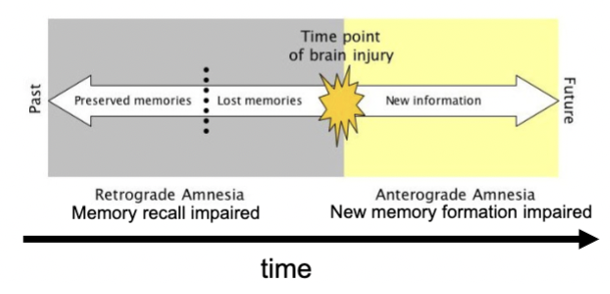
retrograde amnesia
loss of memory for events just prior to an injury (info did not get from short to long-term)
anterograde amnesia
inability to form new memories after an injury
patient HM
HM had severe epilepsy - surgeons removed both medial temporal lobes (most of hippocampus, amygdala, adjacent temporal cortex) which cause him to have severe anterograde amnesia (he couldn’t form new memories) + retrograde amnesia from ~3 years before surgery
had normal short term, forgot once his attention shifted
evidence that long-term was different than short-term
patients with amygdala damage only do not show memory loss
what HM could not do
digit span + 1 test: have subject recall numbers, keep adding digits until subject makes errors → HM could do up to 8 digits
matching to sample: shown a sample icon (verbal or nonverbal) and then asked to match the sample among a series of panels after a delay → had no problems if he could repeat the letters during the delay (up to 40 sec delay), could not do the non-verbal matching
what HM could do
learn a variety of motor skills (couldn’t remember learning them)
Mirror Drawing Task: draw object from reflection in mirror - he gradually got better at the task over 7-days
could form long-term non-declarative/procedural memories (patients with striatum damage can’t learn skills, but remember learning them)
could learn semantic facts over a period of time
conclusions from HM
two-stage model for memory formation supported - suggests memories can be stored elsewhere, but hippocampus is crucial for long-term storage
not all types of learning/memory are mediated by hippocampus
memory systems can be dissociated by type of info stored
types of long term human memory
declarative (explicit): things you know that you can tell others
procedural (implicit): things you know that you can show by doing
hippocampal damage causes deficits in declarative memory but not procedural
striatum and amygdala mediate types of procedural memories
types of declarative memory
episodic: remembering your first day of school
semantic: knowing the capital of France (don’t necessarily remember where you learnt the fact)
most memory for facts/events consists of familiarity with features of item and recollection in its original context (perirhinal cortex may be involved with sense of familiarity)
types of procedural memory
skill learning: knowing how to ride a bicycle
conditioning: salivating when you see your favourite food
priming: more likely to use a word you heard recently
hippocampus and spatial memory in rats
tested with Radial Arm Maze (rat locates food around maze using spatial cues) - rats remember where they have been if they do not re-enter arms
lesions cause rats to make more errors
and Morris Water Maze (rat swims around pool until it finds hidden escape platform) - rats use spatial cues to navigate the pool
lesions cause rats to never find hidden platform efficiently
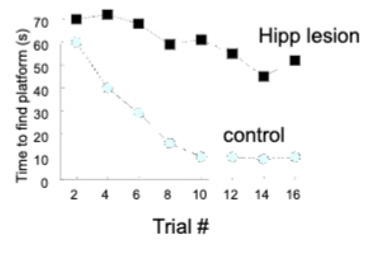
hippocampus and spatial memory
studies using virtual 3D environments: humans w hippocampal damage are impaired when navigating space and remembering routes - healthy humans show increased activity in hippocampus when learning routes
can be activated just through verbal recall of spatial memories
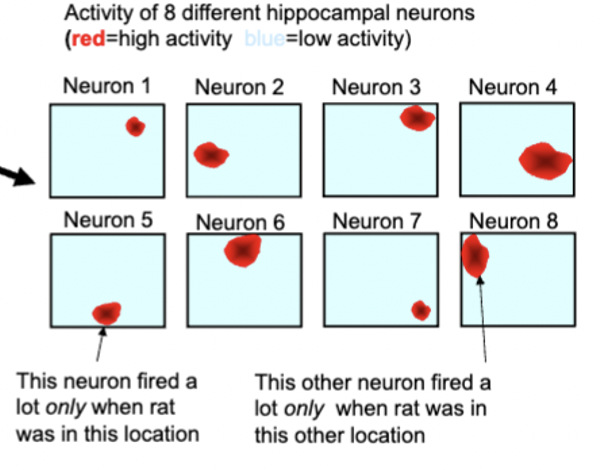
hippocampal place cells
different neurons fire preferentially when animal is one particular location (place field)
separate groups of cells encode for different environment locations
reorienting spatial cues causes place fields to reorganize (neurons fire in different locations relative to position of different cues)
hippocampus forms a map of environment
encodes relations between different stimuli in environment
grid cells fire selectively when animal crosses intersection points of abstract grid
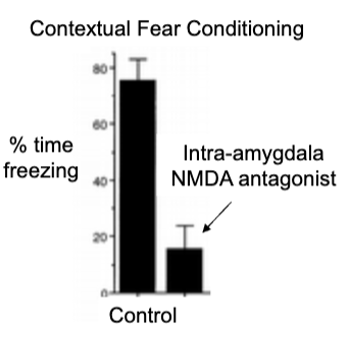
contextual fear conditioning in rats
give CS tone-shock pairings in one context (A) and then another distinct context (B) → normal rats show fear freezing and hippocampus lesion rats show normal fear to tone (implicit learning is only intact)
rat in shock context (A) with no tone = normal rats freezing, hippocampal lesioned rats not showing contextual fear conditioning
hippocampus for remembering relations between contexts (amygdala cause no fear for tone or context)
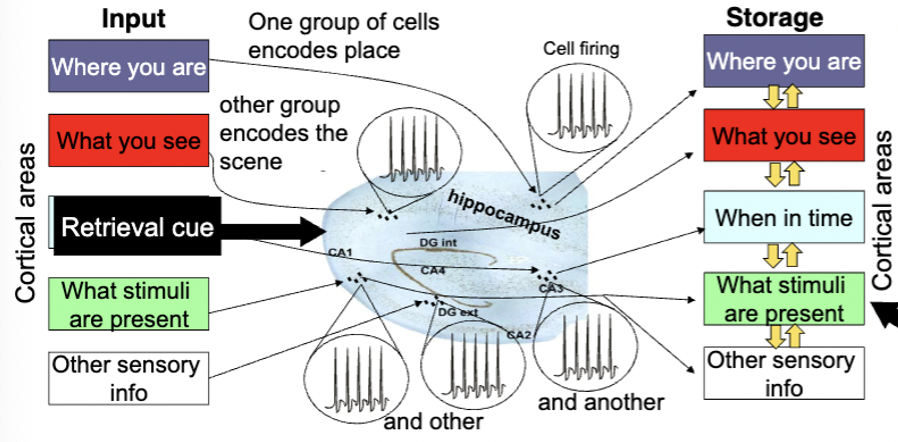
hippocampus and episodic memory
records info about episode in relation to each other - encodes info as distinct patterns of activity - and helps consolidate info linked to a particular memory in same cortical regions originally processed in
when you remember the episode, similar patterns of activation in hippocampus can activate similar regions of cortex as when event memory
different groups of cells for each sensory input (where you are, what you see)
easy to remember unusual memories because they have a unique coding pattern
hippocampus
aids declarative memory (human) and spatial/relational memory (rats)
striatum
controls procedural memory/skill/habit learning (humans), instrumental conditioning (rats)
amygdala
controls pavlovian conditioning for appetitive or aversive events (humans and rats)
multiple brain regions interacting for memory
can interact to regulate a type of memory or participate in independent forms of learning/memory
when 2+ brain regions are independently involved in separate forms of learning = dissociation of memory systems
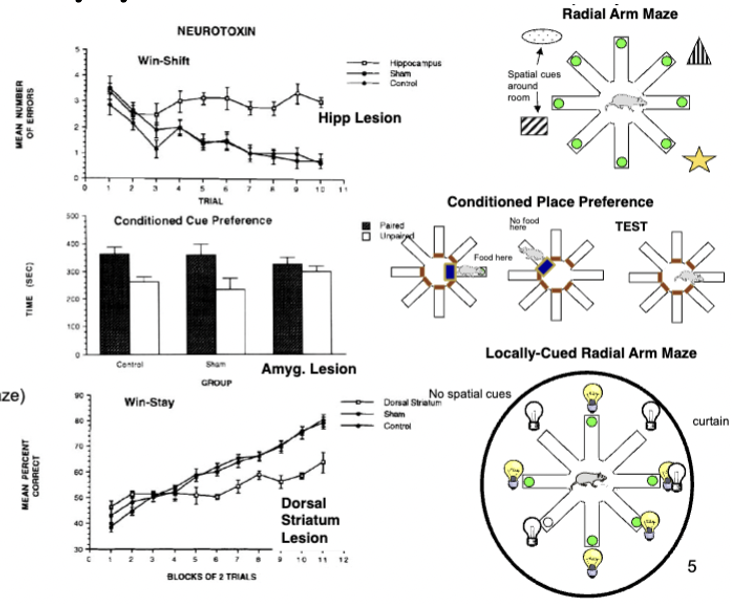
triple dissociation of memory systems in rats
three radial arm maze tasks: spatial radial maze (don’t go back to previously entered arm), conditioned place preference (go back to arm where food was), locally cued-radial arm maze (go to arms where light is on - light turns off after food is eaten)
spatial radial impairment = hippocampal lesion
conditioned place impairment = amygdala lesion
cued-radial arm impairment = dorsal striatum lesion
processing in rats during maze training
training rats to always enter one maze arm → rats can use place (hippocampal) or response (striatal) strategy
early in training = hippocampal strategy (healthy rats) → inactivate hippocampus = striatal strategy
late in training = striatal strategy (healthy rats) → inactivate striatum = hippocampal strategy
what does rat maze learning say about memory?
different systems can learn independently and in parallel to each other
suppression of one system allows behaviours driven by another to emerge
different systems learn at different rates (hippocampal = rapid, striatal = more gradual)
emotions and memory consolidation
strong emotional states can enhance memory consolidation - may be mediated by noradrenaline
NA beta-receptor antagonists = reduction of better memory for emotional part of story even though they still report emotional response to story (tone down physiological aroudal)
may explain PTSD, beta blockers to prevent PTSD
emotional enhancement of memories
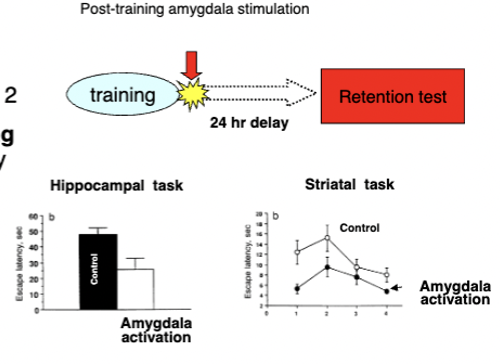
mediated by amygdala
stimulating amygdalas of rats with drugs immediately post-training on Day 1 = improved memory consolidation tested the next day on spatial (hipp.), instrumental (stri.), and aversive (amyg.) learning
can modulate consolidation of memories by other systems
synaptic plasticity
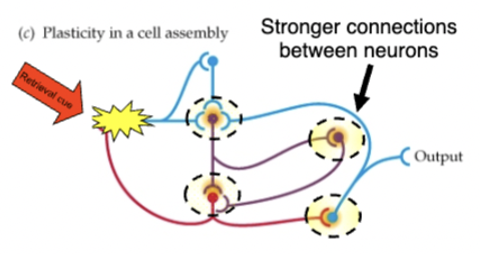
brain is plastic - every time learning/encoding a memory happens, changes occur in neurons in brain
short term memory = cell assemblies (groups of interconnected neurons) → start a chain of activation (circuit) that can continue for some time and encode a memory
if circuit is strong enough, long term memories can be formed through altering synapses (changes in synaptic strength )
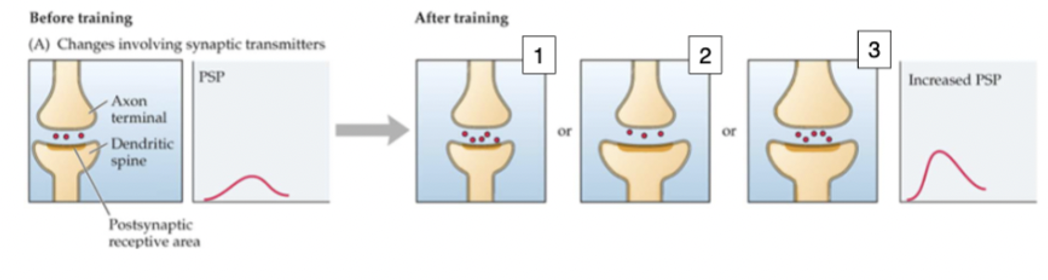
synaptic changes
changes in depolarization/hyperpolarization, nts released, size of membrane and sensitivity, interneuron modulation, new synapses formed, shift in synaptic input
measured by high frequency excitatory post synaptic potential (EPSPs) which make synapses stronger (long-term potentiation)
can also decrease synaptic strength
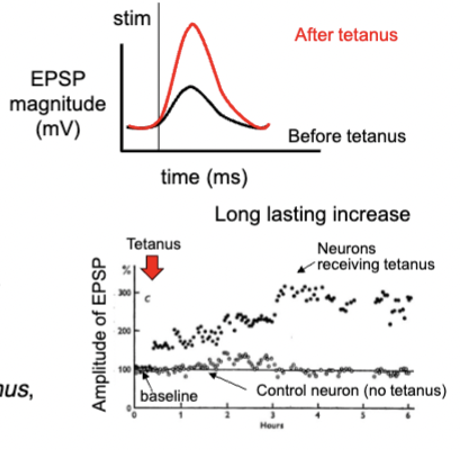
long term potentiation
stimulate presynaptic axons at low frequency to get subthreshold EPSP, establish baseline
stimulate axons at high frequency and gets lots of action potentials in postsynaptic neuron
axons at low frequency again, but EPSP is much bigger than before
input is stronger → larger EPSP → more likely to evoke AP
dependent on NMDA receptors (glutamate) - blocking prevents new LTP in hippocampus
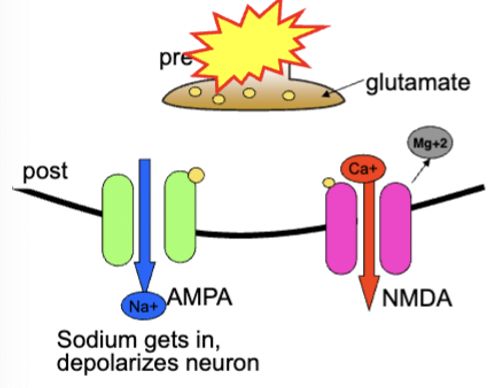
glutamate mechanisms of LTP
can occur anywhere in brain where there are glutamate synapses (hippocampus, cortex, amygdala, striatum) - 2 types of glutamate receptors
AMPA and NMDA: both allow Na+ to pass through and depolarize neuron
only NMDA: allows Ca2+ to get into neuron - if neuron is hyperpolarized, NMDA receptor is blocked by Mg2+ ions and cannot be activated by glutamate
AMPA receptors are never blocked, can always be activated by glutamate - depolarized enough, Mg block is removed and glutamate can activate NMDA receptors (long term memory activation)
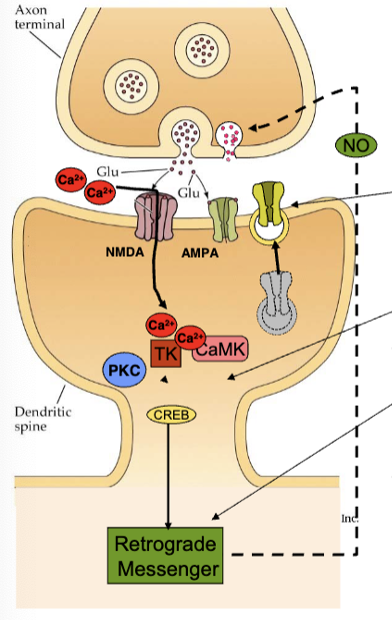
what happens when Ca2+ enters the cells?
activates multiple enzyme pathways (kinases = phosphorylate proteins, turn on other proteins and metabolic processes) → CaM first, which activates other kinases and hits latent AMPA receptors to insert it into membrane (more receptors) → PKC and TK activate CREB which forms retrograde messengers that promote more nt release
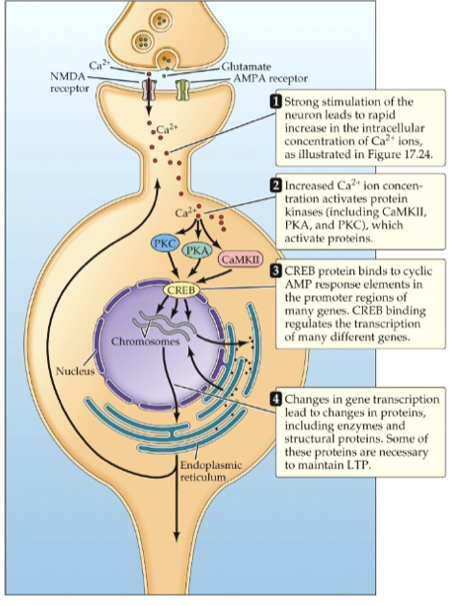
LTP phases
increase in receptors and glutamate release occur quickly - changes blocked by NMDA receptors
CREB activates protein synthesis - dendrite shape and size changes, more ion channels and dendrites
longer lasting changes
also blocked by protein synthesis inhibitors
both phases blocked by prevention of Ca entry (only certain synapses on neurons will change synaptic strength, entry is localized
LTP as a mechanism for memory
drugs that block LTP formation also disrupt learning (in hippocampus = spatial, amygdala = fear, striatum = instrumental)
treatments immediately after training do not disrupt memory formation
blocking protein synthesis also disrupts different forms of learning (treatments after training do disrupt memory formation)
behaviour electrophysiology: changes in neural activity resembling LTP that occur after learning
prefrontal cortex anatomy
moving up evolutionary scale, relative % size of PFC increases - largest in humans
medial/orbital regions = emotion regulation
dorsolateral regions in primates = working memory, planning
PFC functions develop late in humans (~2-3 years)
rat shares some functions of dorsolateral PFC in primates/humans
PFC and working memory
PFC regulates executive functions (command and control function), viewed as conductor of various cognitive skills
working memory part of EF - short term manipulation and retrieval of trial unique info (temporary, manipulated, discarded)
info encoded in one form, used to guide other behaviour in other form
recalling numbers in sequence = short term memory, recalling them backwards = working memory
using information to guide a response after a delay (using storage and manipulation of short term info to complete tasks) (PFC lesions can not do it)
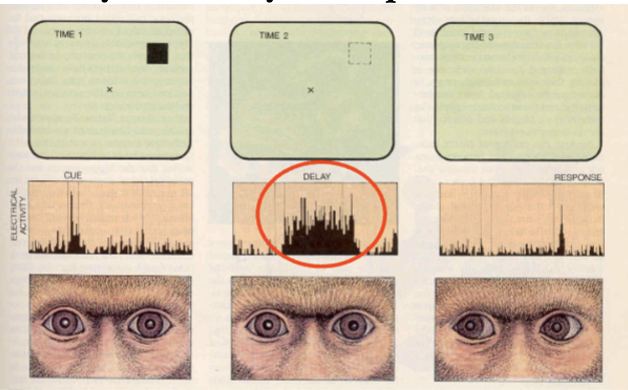
PFC neural activity and delayed response
record from different neurons, fire at different parts of task - neurons fire the most when the monkeys need to remember where the cue was during the delay
multiple PFC neurons may encode different bits of info - possibly activity of different PFC cell groups hold different bits of info
PFC and behavioural flexibility
wisconsin card sorting task (test ability to change strategies)
ps sort card according to one category, then they have to switch and figure out new sorting strategy (experimenter doesn’t tell them until they are right)
patients with dorsolateral PFC damage can learn first discrimination but cannot switch
seen rats too - inactivating PFC during initial learning has no effect, but during the shift causes a major impairment
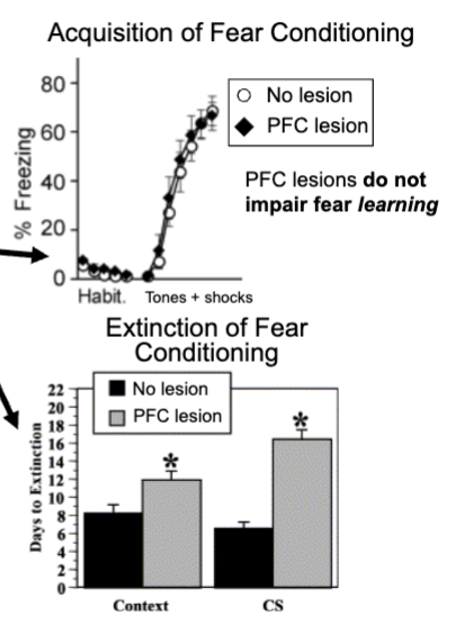
prefrontal cortex and extinction learning
keep giving tones with no shock to rats causes them to eventually stop freezing (extinction) - new learning that suppresses old response
PFC damage in rata does not disrupt learning of fear conditioning, rats with PFC damage take longer to extinguish fear response during extinction
PFC is connected to amygdala and PFC inputs can inhibit neural activity in amygdala
medial diencephalon damage also causing amnesia
patient NA had profound anterograde amnesia due to damage to thalamus, hypothalamus, mammillary bodies - could not form declarative long-term memories (all needed to form them)
Korsakoff’s syndrome shows similar effects (fill memory gaps with false info, don’t think anything is wrong with them) - caused by thiamine deficiency due to alcoholism
how is the cortex essential for long-term storage of memories?
patient KC could not access episodic memories after a motorcycle accident caused damage to cerebral cortex = severe shrinkage of hippocampus and parahippocampal cortex
semantic memories could still be accessed
sensory buffer memories
stores sensory impression of a scene - lasts for a few seconds
working memory
where we manipulate and process short term memories - last for around 30 secs, 7 chunks of info
deficits associated with PFC damage
3 components: phonological loop of auditory info, visuospatial sketch pad of visual impressions, episodic buffer of more integrated info
system for creating memories
encoding of raw info from sensory channels into short-term memory, involves encoding, flow of info in and out of working memory supervised by central executive
consolidation of STM into long-term memory - medial temporal lobe crucial for consolidation, permanent storage tends to be in regions of cortex where info was first processed
retireval of stored info for future use, makes memories temporarily plastic again and susceptible to updating before reconsolidation
adult neurogenesis
occurs primarily in dentate gyrus of hippocampal formation, also observed in olfactory bulbs and some cortical location
enhanced with exercise, enriched environments, memory tasks
learning and memory with age
healthy elderly people show some memory impairment in tasks of conscious recollection, internal generation of memory
working memory, ability to form new memories also declines
autobiographical and semantic knowledge remain stable
happens through impairment of encoding/retrieval, loss of neurons or connections, problems with neurotransmission
physical activity, mental activity, adequate sleep, effortful learning can postpone cognitive decline
prefrontal cortex and planning
keeps track of/plans sequences of action - their temporal organization
patients with damage can remember all the the ingredients and actions when cooking a meal but cannot put them in the correct sequence (long term memory intact)
impairs recall of temporal order of events in memory
tower of london task: plan movement of balls to get them from one position to the next in least amount of movements - patients with damage need many more moves
patients with damage could not form a plan and follow it when attempting to multi-task for a dinner party although they remembered/understood what they had to do
schizophrenia
early dementia, split mind (thought disorder) - family of disorders with at least a few distinct symptoms
positive and negative symptoms
schizophrenia positive symptoms
abnormal behaviours that have been gained - hallucinations (typically auditory), delusions (paranoia), psychosis
schizophrenia negative symtoms
normal functions that have been lost - blunted emotional responses, poverty of speech, social withdrawal, anhedonia, lack of insight, cognitive deficits
diversity of symptoms in schizophrenia
at least 1 of 3 core symptoms is required for diagnosis (presentation varies greatly): hallucinations, delusions, disorganized speech
common symptoms: lack of insight into illness = 97%, auditory and verbal hallucinations = 72%, delusions of reference = 70%, suspiciousness = 65%, flatness of affect = 65%, paranoid = 64%
~1% of population - onset after puberty (18 years), some women after menopause (+45 years)
cognitive abnormalities in schizophrenia
exhibit some cognitive deficits - level of cognitive functioning is #1 predictor of long term outcome (better function = better prognosis)
severity of psychotic symptoms not related to severity of deficits
many impaired functions mediated by prefrontal cortex/hippocampus
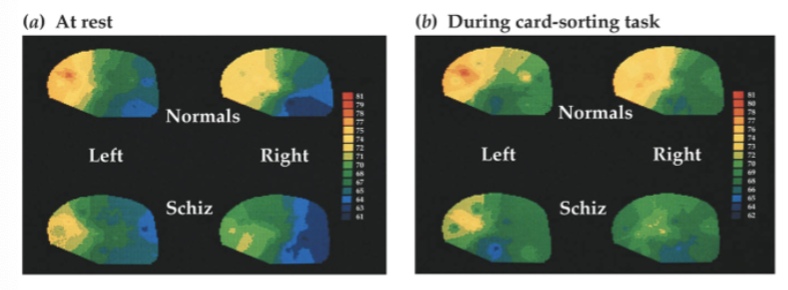
schizophrenia patients showed minimal PFC activity increase when performing Wisconsin Card Sort task, unlike controls - other brain regions similarly activated
current medication mutes cognition, which makes it hard to function
genetics of schizophrenia
odds of developing it increase if one has a relative with it - odds go up as relative gets closer (highest concordance in identical twin and both parents having it [~50%])
strong genetic component but altered genes are not the only cause
various genes associated with schizophrenia
![<p>odds of developing it increase if one has a relative with it - odds go up as relative gets closer (highest concordance in identical twin and both parents having it [~50%])</p><ul><li><p>strong genetic component but altered genes are not the only cause</p></li><li><p>various genes associated with schizophrenia</p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/2c55ca94-dca6-4afa-9f15-7add4d154e29.png)
neural development and schizophrenia
alterations during development thought to be critical to disorder
in utero: poor nutrition during pregnancy, premature birth/low weight, physical/immune stressors during pregnancy
early developmental insults = brain abnormalities
stressors later in life (after puberty) can trigger onset
genetics increase sensitivity to stressors (some people have susceptibility to acquiring disease)
living in a city also raises risk (stress, pollutants, disease exposure)
hippocampus abnormalities in schizophrenia
enlarged ventricles (transport cerebrospinal fluid) due to smaller hippocampus/temporal lobe regions with schizophrenia
altered organization of hippocampal neurons - during development can disrupt how brain processes info
not lesioned, just a size thing
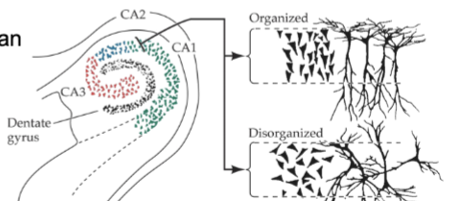
prefrontal cortex abnormalities in schizophrenia
pyramidal neurons have reduced # of dendrites (reduces processing power of these cells)
causes hypofrontality (reduced PFC function)
GABA interneurons serve as major info filter for PFC (and hippocampus) - people with schizophrenia don’t have as many in these regions → noisy cortex (reduces info filtering and allows for more external distractors and impairs function of these regions)
dopamine hypothesis of schizophrenia
most of DA in brain is produced in midbrain
1960s: found that drugs that increase DA release could induce psychotic symptoms
1970s: DA receptor subtypes discovered - antipsychotic potency of a drug correlated with binding to D1 receptors specifically
excessive amphetamine use = similar symptoms as schizophrenia (they promote release of DA and halt reuptake)
hypothesis: schizophrenia is caused by an abnormal increase in DA transmission → overstimulation of D2 receptors
support for DA hypothesis
all drugs that are effective in treating psychosis block D2 receptors to some degree - how?
more D2 receptors with schizophrenia? Unlikely (changes may be due to chronic antipsychotic medication upregulating DA receptors
more DA being released? giving amphetamine to a schizophrenia and a control sample resulted in greater DA release in striatum in those w schizophrenia
DA release may be hypersensitive in schizophrenia
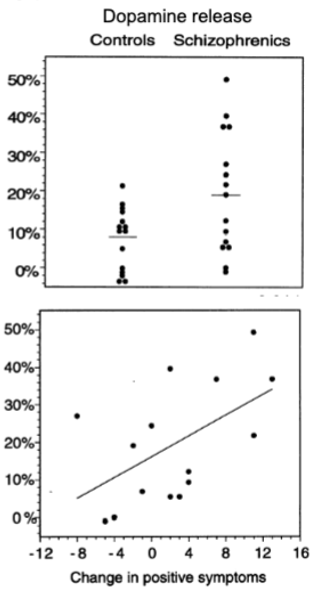
what is DA doing in schizophrenia
DA neurons show increased activity to salient/novel stimuli (may serve as signal brain uses to determine what’s important) → hyperactive DA system may tag normally irrelevant stimuli as important (leading to aberrant salience attribution that could contribute to delusions)
antipsychotics thought to reduce this salience by reducing DA activity
treatment issues with schizophrenia
dopamine is involved in motor functions so long term antipsychotic use can cause movement side effects (tardive dyskinesia: grimacing, tongue protrusion, lip smacking, rapid limb movements)
drugs that are more selective for DA vs other receptors usually cause worst effects
negative symptoms rarely improve with typical antipsychotics
limitations of DA hypothesis
antipsychotics block DA receptors right away, but drug treatment takes ~2 weeks to reach full effect (if schizophrenia was just an increase in DA, drugs would work right away)
not all with schizophrenia respond to dopamine blockers (about 1/3)
DA blockers can alleviate psychosis but not negative symptoms
some drugs that reduce symptoms do not block D2 receptors well (clozapine: has higher affinity to other receptors like 5-HT which can sometimes improve negative symptoms) - atypical antipsychotics shouldn’t work well
drugs that increase DA can improve negative symptoms (amphetamine increases DA and improves memory performance)
there are other processes at play
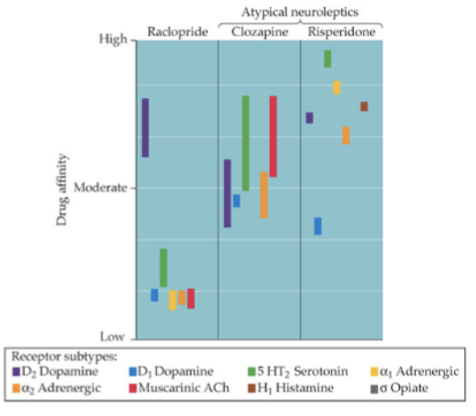
glutamate and schizophrenia
when people abuse PCP or ketamine, they can have psychotic symptoms and cognitive deficits that resemble schizophrenia - these drugs block NMDA glutamate receptors by blocking the ion channel so glutamate can’t activate receptors
glutamate hypothesis of schizophrenia
sz is caused by decreased glutamate transmission
PFC and hippocmpus use glutamate as a transmitter
degeneration of these neurons in schizophrenia disrupt their function, less glutamate released in these areas
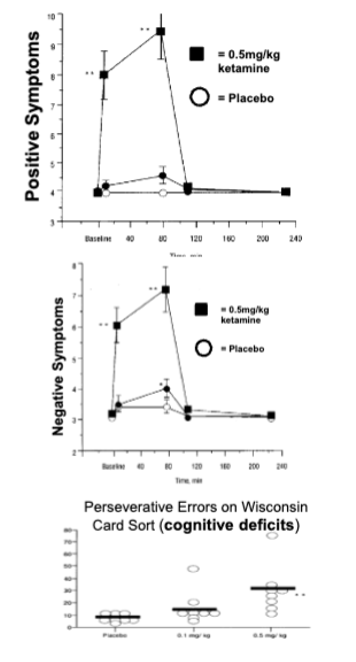
insight from animal models of schizophrenia
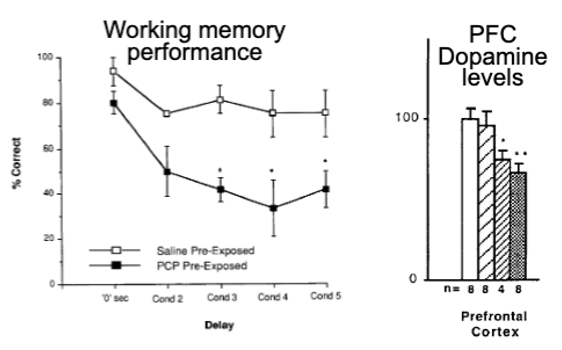
repeated PCP in rats causes:
cognitive deficits on PFC-dependent tasks
decreases in PFC DA levels
sz also associated with reduced PFC DA activity in rat
different symptoms may be drive by imbalance of DA transmission in PFC (too little) and striatum (too much)
may explain why drugs that increase DA improve cognition - DA imbalance hypothesis (increase of subcortical DA and decrease of cortical)
salience attribution in schizophrenia
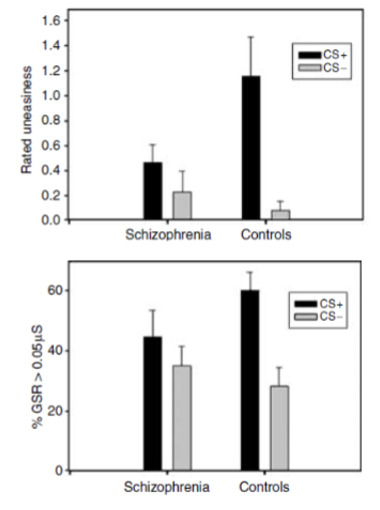
people with sz show abnormal fear learning
high comorbidity with anxiety disorders and inappropriate discriminative fear conditioning
decrease in fear to CS that was paired with shock
increase in fear to CS that was presented alone (increased PFC activity)
identify with stimuli that are not of emotional importance - may contribute to delusions
PFC GABA and positive symptoms - salience attribution
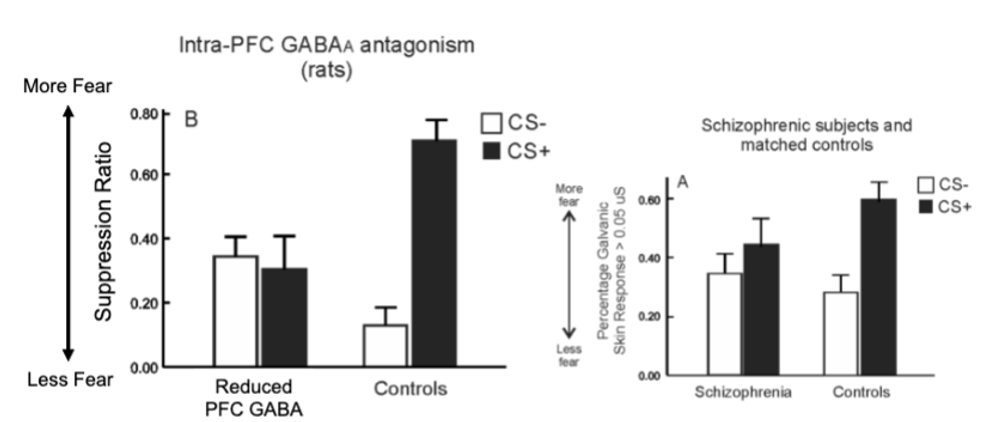
discriminative fear conditioning with rats - pairing a tone with a foot shock, pairing ANOTHER tone with no consequence
neutral tone = rats displaying little/no fear (continue to press lever)
shock-associated tone = fear response (stop behaviour
after reducing PFC GABA
decreased fear to shock tone and increased fear to no shock tone
people with schizophrenia show similar fear responses
dysfunctional PFC GABA may also contribute to positive symptoms
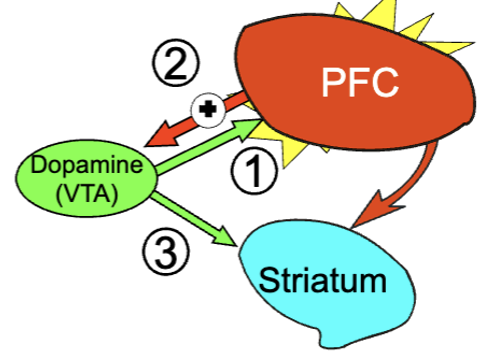
integrative hypotheses for schizophrenia
decreased PFC DA/glutamate reduces activity (negative symptoms)
noisy PFC (reduced GABA inhibition) may also contribute to cognitive dysfunction
increased subcortical DA transmission contributes to positive symptoms
appears to be a disruption in balance between general excitatory/inhibitory transmission → subtle changes in various regions can alter entire transmission systems
depression characteristics
unhappy mood (absence of happiness), worthlessness, guilt, desperation
loss of interest, motivation, appetite, blunted ability to experience pleasure (anhedonia
difficulty in concentration, restless agitation
episodes that occur with no apparent cause or triggered by external events
increased risk of suicide
DSM diagnostic criteria for MDD
>/= 5 symptoms during same 2 week period - depressed mood and/or loss of interest must be present, plus
weight loss or gain
insomnia or hypersomnia
psychomotor agitation or retardation
fatigue
feeling worthless or excessive guilt
decreased concentration
thoughts of death/suicide
symptom cluster vary with individuals - over 200 combinations of symptoms (single treatment method is difficult
epidemiology of depression
unipolar typically alternates with normal emotional states, episodes can last 6-9 months throughout life
10% of population in North American afflicted at any one time
women more likely - incidence often coincides with major hormonal changes (post partum, menopause)
incidence has increased over past 50 years
age of onset decreased (~27 years)
some types have genetic influence (strong hereditary contributions)
stress and depression
depression viewed as stress-related disorder (emerges during stressful life periods)
some forms of depression linked to alterations in HPA-axis and higher CORT levels (people with Cushing’s syndrome have high levels of glucocorticoids and are prone to depression)
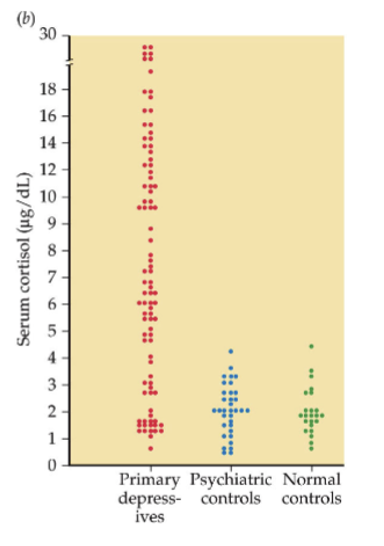
circulating CORT levels tend to be higher in depressed people
diathesis-stress model: predisposition for depression - stress triggers it
dexamethasone (synthetic glucocorticoid) can suppress CORT in normal people, but not depressed people (fools HPA axis into thinking their are higher levels)
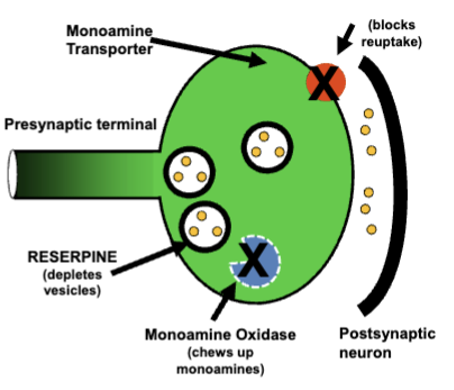
monoamine hypothesis of depression
5-HT, noradrenaline, DA - monoamine oxidase inhibitors (block monoamine metabolism) alleviated depression
selective serotonin reuptake inhibitors (SSRIs) like prozac also alleviate depression (block reuptake = more nts)
depression appears to be result of abnormal reductions in brain monoamine levels
limitations of monoamine hypothesis
antidepressants increase monoamine levels quickly, but there is a lag in reduction of symptoms (~4-6 weeks)
not all depressed people respond to these drugs (large placebo effect, SSRIs are not more effective than classic tricyclics) - appear to only work for severe depression
SSRIs associated with increased risk of suicide in children and adolescents (give enough energy for the act)
in animals, depletion of 5-HT does not cause depressive-phenotype (more than just overall 5-HT decrease)
brain changes with depression
increased blood flow to amygdala and ventral parts of medial prefrontal cortex (depression = disrupted regulation of amygdala emotion processing by PFC?)
alterations in brain activation can be normalized with antidepressants
reduced hippocampal volumes (can’t remember when happy?)
disruptions to PFC may underly negative appraisals of life events (makes things seem worse, persistent rumination)
electroconvulsive therapy for depression
electrical stimulation to the brain - highly effective for severe depression when other treatments have not worked
transcranial magnetic stimulation
using magnetic fields to alter cortical electrical activity and improve neural connectivity in under active brain areas
deep brain stimulation for depression
electrode surgically implanted in brain, very high frequency given continuously
in ventromedial PFC or other subcortical areas shown to be effective in alleviating depression in treatment resistant patients
ketamine for depression
sub-anesthetic doses induce rapid reduction in symptoms for ~70% of treatment-resistant patients (after psychosis dissipates)
effects last 1-3 weeks, takes a day to see improvements
nasal formulation
may be linked to increased cortical activity
CBT for depression
takes a longer time but just as effective as other treatments - used in combination with other treatments is most optimal care plan
animal models of depression
learned helplessness common simulation of depression - animal is exposed to repetitive, inescapable stressful stimulus and after repeated exposure, more sensitive animals will give up instead of escape when given opportunity
tend to have cellular/neurochemical alterations in brain (decrease in 5-HT)
antidepressants can reduce the despair
bipolar disorder (mania/depression)
characterized by periods of depression alternating with expansive mood or mania
rate of cycling varied - rapid = 4+ cycles in one year
also a milder version: less extreme moods, cycling between mild depression and hypomania (increased energy and positive mood less intense than mania)
neural basis not fully understood, involves abnormalities in hippocampus and amygdala
Lithium (stabilizing drug) to treat - modulates 5-HT and DA transmission, reduces cell death associated with BPD
men and women equally affected
intracranial self-stimulation in rats
form of instrumental learning, electrode implanted in brain - acquired quickly, rats respond at very high rates
self-stimulate until exhaustion - ignore food/water to near death
all brain regions that support self-stimulation are connected to mesolimbic (midline) DA system
mesolimbic DA system
all DA cell bodies located in midbrain, send axons to many brain regions (DA neurons = projection neurons)
ventral tegmental area = heart of mesolimbic DA (send DA via medial forebrain bundle to limbic regions (PFC, amygdala, nucleus accumbens)
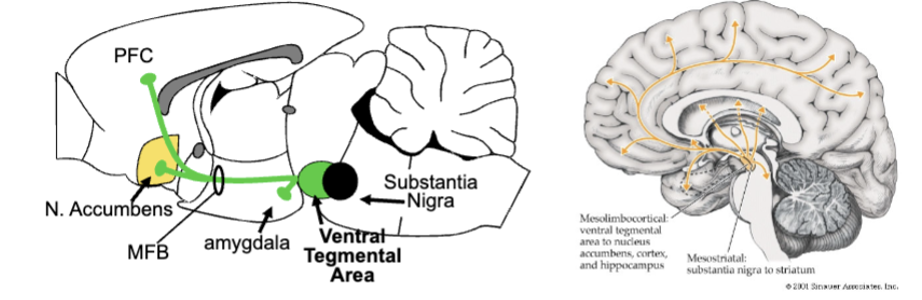
mesolimbic DA and reward
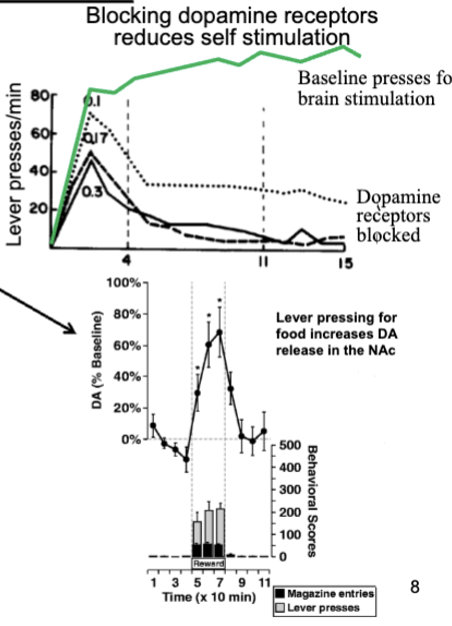
self-stimulation increases DA release in limbic system, particularly accumbens - DA plays important role in reward-related approach
reducing DA transmission reduce self-stimulation
animals work to have DA agonists infused directly into accumbens
rewards or conditioned stimuli with rewards increase DA in accumbens
all drugs of abuse with high addiction potential increase DA release in nucleus accumbens
ligand
any substance that binds to receptors (hormones and nts)
one of these effects: agonists (initiates transmitters), antagonist (blocks transmitters), inverse agonist (initiates transmitters but effect is reverse of normal receptor function)
noncompetitive ligand: bind to receptor that does not normally bind the transmitters
binding of drug molecules
do not seek out particular receptors - spread widely through body, bind briefly when come in contact with receptor that is correct shape
degree of chemical attraction = binding affinity
propensity of ligand to activate receptor = efficacy (agonists are high, antagonists are low)
larger doses = more receptors bound by drug
smoking/intravenous injection rapidly increase drug concentration
blood-brain barrier blocks many drugs from passing into brain due to molecules being too large
drug tolerance
decreased sensitivity to effects of drug after repeated use
metabolic: body (liver) becomes more efficient at metabolizing drug
functional: occurs at site in brain/body where drug exerts effect (ex. receptor # decreases)
develops to some effect of drugs but not others, can develop at different rates (tolerance to pleasurable effects develops faster than respiratory effects)
cross-tolerance can happen to similar drugs
withdrawal
rebound reaction to elimination of drug from system after repeated exposure
typically opposite effects of drugs action
may be body’s attempt to maintain homeostasis (initiates compensatory changes to counteract effects of drug → when drugs have been eliminated from system, changes can linger causing withdrawal symptoms)
additional drug taking can alleviate withdrawal
seen drug effects as conditioned stimuli - brain makes associated between drug effects and context of taking them (environment is crucial)