PHAR 503 midterm
1/291
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
292 Terms
how is the affinity of a drug on a target experimentally determined?
competition
pure target protein purification vs pure drug purification
it's difficult to isolate proteins from cell lysate, but pure drugs are easier—just get cDNAs and synthesize then purify that via chromatography
how does chromatography work?
hopefully the compounds have different characteristics, like electronegativity, size, IMF, etc. that you can use to separate them over beads
what is size exclusion chromatography
to separate POI from other proteins based on size
how does size exclusion chromatography work?
run solution containing POI over column containing porous beads. smaller proteins will get more stuck in the beads while larger ones will flow through, so you elute differently sized groups
what does a 205nm absorbance indicate?
peptide bond!
what does a 280nm absorbance indicate?
tryptophan/any aromatics with delocalized e- side chain
the e- delocalized makes it so high
what's the downside of looking for 205nm absorbance?
it's very narrow, so it's hard to produce = machines are expensive, but the upside is you can see any protein
affinity chromatography
to separate a protein from other proteins by its biophysical traits based on a specific binding interaction between an immobilized ligand and its binding partner
examples of biophysical traits
hydrophobicity, ligands (lectins, protein antibodies, collagen...), metals, protein tags
6-His tag
can bind nickel! makes separating this recombinant protein out easy
glutathione-sepharose (GST)
helps stop bacterial proteins from aggregating during elution
myc pro-oncogene
an epitope we have good antibodies for
how do you get the protein out of the affinity chromatrography column?
wash with salts after eluting everything else
what is the first challenge to determining drug-target affinity?
getting pure drug and pure target!
concept of compound competition
you increase the amount of normal ligand:radio ligand to see at what concentration of normal ligand 50% of the radio ligand is removed—this gives IC50.
IC50
the concentration at which the competing ligand displaces 50% of the radioligand
How do you do compound competition (steps)?
1. introduce known concentration of radioligand
2. wash with known concentration of regular ligand
3. increase concentration of regular ligand and repeat
4. less and less labelled ligand will show up on the filter as you wash
5. determine IC50
Ki from IC50
The inhibition constant for a drug; the concentration of competing ligand in a competition assay which would occupy 50% of the receptors if no radioligand were present. Whereas the IC50 value for a compound may vary between experiments depending on radioligand concentration, the Ki is an absolute value. It is calculated from the IC50 using the Cheng-Prusoff equation:
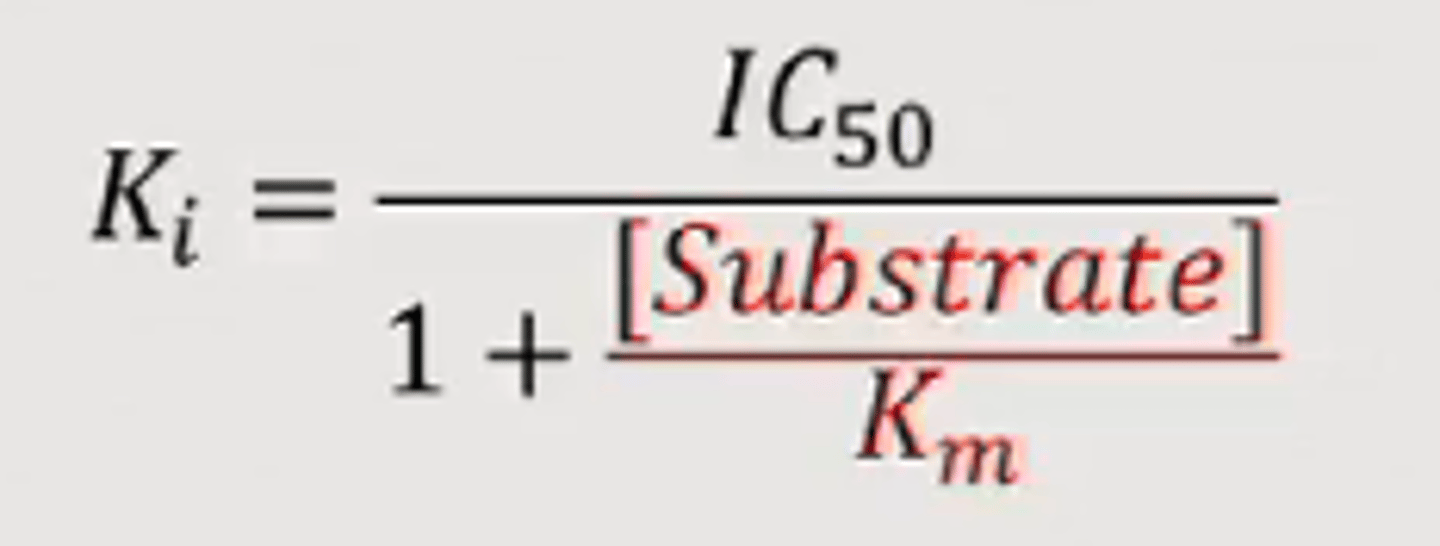
identification of same or different binding sites
if normal ligand B competes with radioligand A, they have the same binding site
if normal ligand C doesn't compete with radioligand A, they have different binding sites
imidazole
side chain of histidine
aromatic ring with two nitrogens
how is elution also a compound competition?
- increase ion concentration to wash proteins off beads
- compete with imidazole, glutathione, etc. to bind the compound
Surface Plasmon Resonance
Technique used to characterize molecular interactions
Binding interactions are detected by monitoring the reflection of a beam of light off the interface between an aqueous solution of potential binding molecules and a biosensor surface carrying the immobilized bait protein
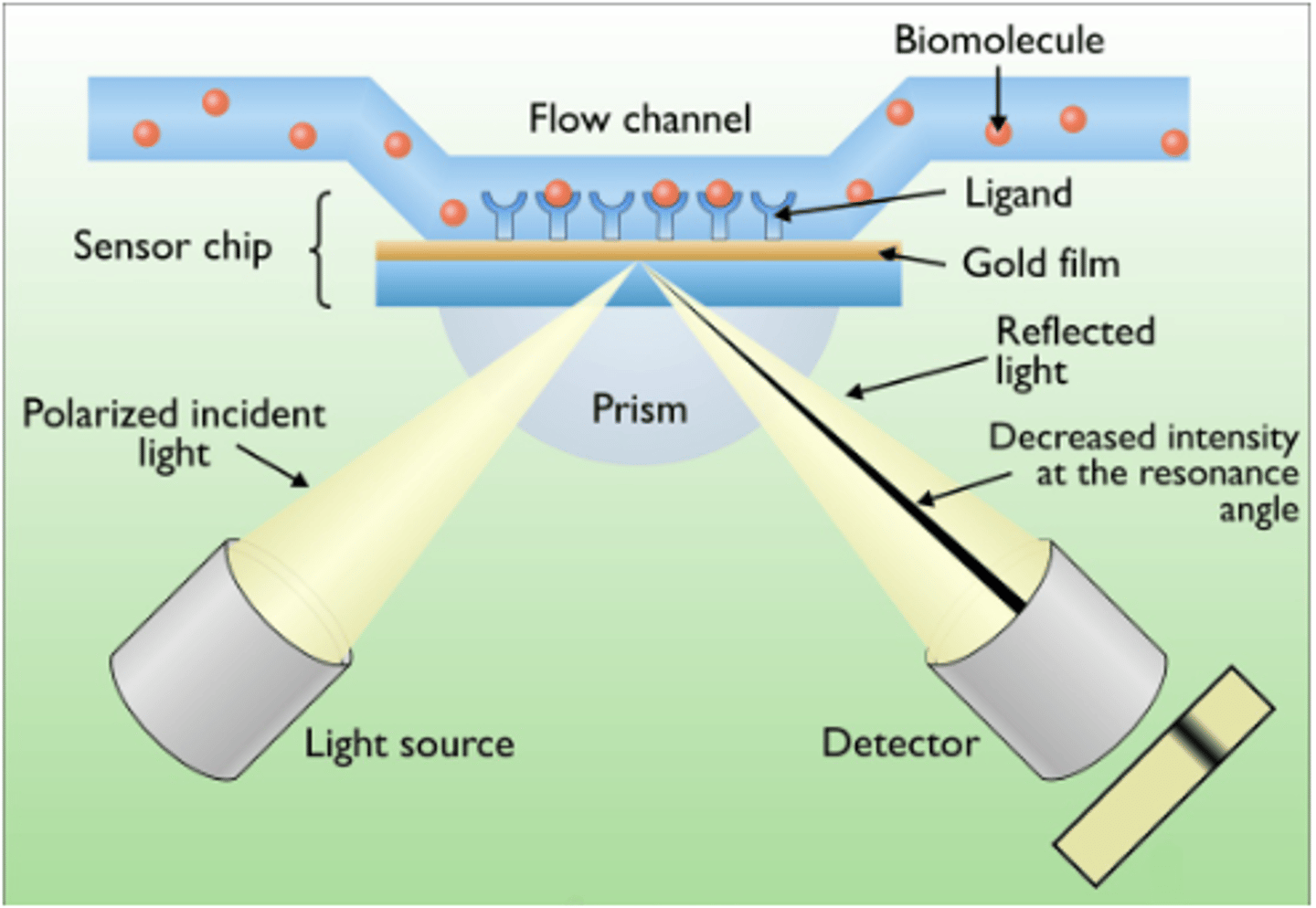
how does surface plasmon resonance work?
detects changes in reflectance properties on surface of antigen coated sensor when it binds with antibody
what is the biophysical phenomenon behind surface plasmon resonance?
buffer flows over proteins bound to chip. drug binds protein. change in mass on chip surface. change in diffractive index between media. evanescent wave changes, causing a measurable change in its 'shadow' angle in diffracted light
SPR resonance signal
the change in the angle of the 'shadow' in the diffracted light
SPR sensorgram
senses change in resonance signal
SPR sensorgram graph
resonance signal vs. time
shows buffer flow, then association of drug with protein til equilibrium, then dissociation of drug as injection of drug stops
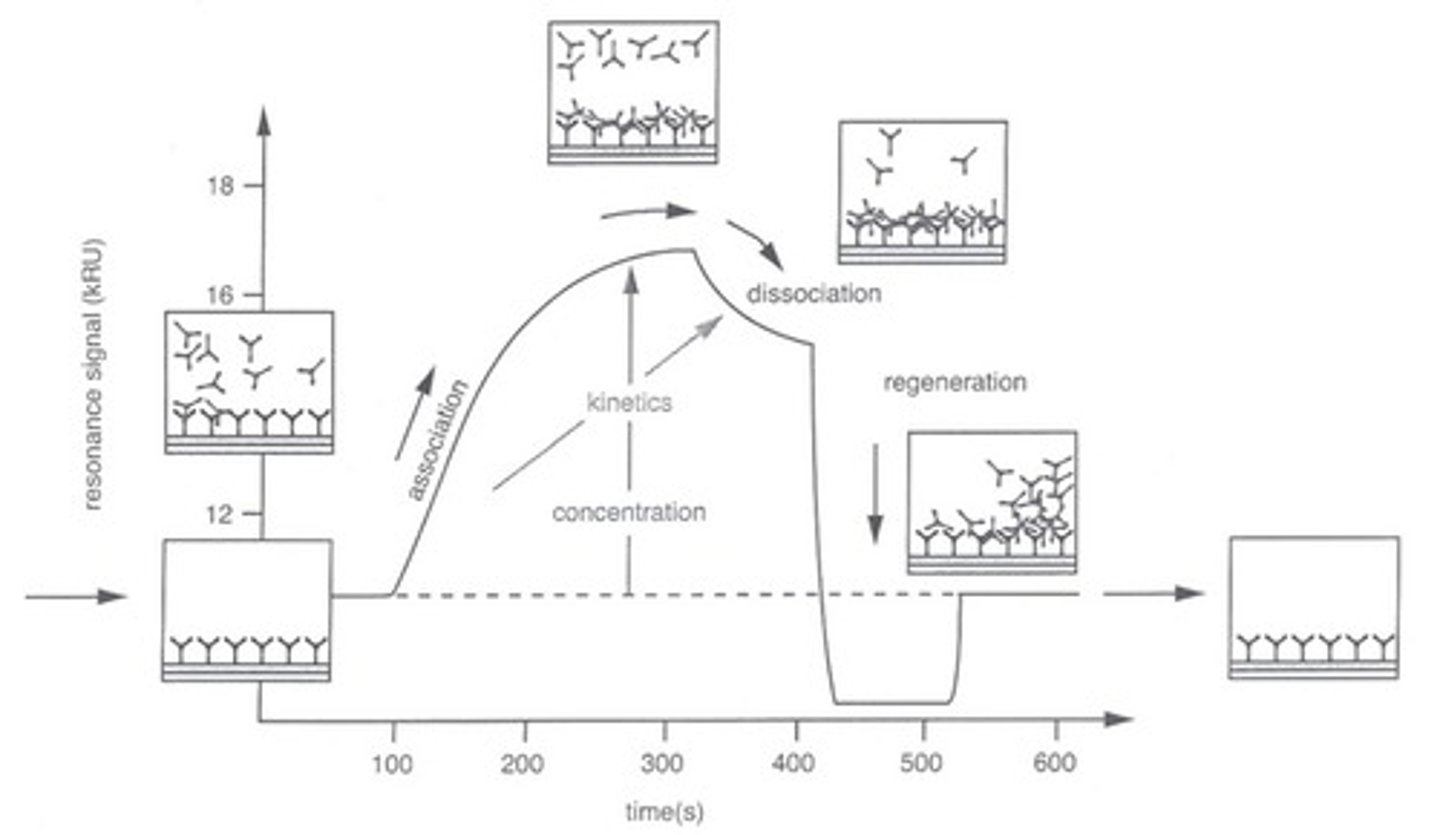
what does SPR measure?
change in resonance signal, which shows online kinetics i.e. association dissociation rates over time + equilibrium constants
low Kd means what?
high affinity (it's not dissociating!)
high Ka means what?
high affinity; Ka describes binding affinity
Ka =
= [target + drug]/[target]*[drug]
Kd =
= [target]*[drug]/[target+drug]
when a drug is given at a concentration equal to its Kd, how many receptors will be occupied?
50%
how can competition be used to show specificity on a sensorgram?
flow a soluble target protein over the chip to compete as a control, because this will eat up any unbound proteins = you make sure measured signals are only of bound proteins
IC50 stands for?
inhibitory concentration 50
mass spectrometry
a technique that separates particles according to their mass
how do you 'fish' for side-effects you like?
modify the drug at random sites and see if those sites cause GOF/LOF of your target side effect
trifunctional capture compound
1. reactive azide
2. biotin for purification
3. drug you're interested in
reactive azide
forms a covalent bond with whatever it's close to upon UV light exposure
biotin for purification
binds with 10^-15 affinity for avidin so makes purification easy
biotin avidin affinity
10^-15 = very low Kd = very high affinity
what do you do once you purify your trifunctional compound?
mass spectrometry to identify proteins that were co-purified and thus potentially drug-binding partners
trifunctional compound workflow
1. modify
2. culture cells, wash with trifunctional drug
3. shine UV 5-10min
3.5 azide binds interaction protein
4. pull out drug-protein complex with biotin-streptavidin
5. wash hard to get the complex off the streptavidin
6. anything unspecific is gone! mass-spec to find the drug + protein!
why protein digestion?
to remove protein from avidin beads via proteases
to make the protein bite-sized so you can run mass spec (too big otherwise)
importance of trypsin in mass spec
this protease only cleaves after K (lysine) and R (arginine), so you can piece the mass spec fragments together based on R and K
how does mass spec work?
negatively ionize your peptides, then accelerate by adding a positive voltage
ions fly through mass analyzer and hit the detector at different times depending on their mass
Only particles with a certain mass/charge (m/Z) ratio will NOT hit the walls and exit onto the detector at the end of the flight tube.
how to read a mass spectrum
y axis = relative intensity
x axis = mass per charge
fingerprint analysis of mass spectrum
list with exact peptide masses measured in a spectrum is compared to 'in silico' cleavage of known proteins in a database
Mascot search
can do the fingerprint analysis for you!
parts 1 and 2 of mass spec
1. generate mass spec, fingerprint analysis
2. validate
how to validate mass spec
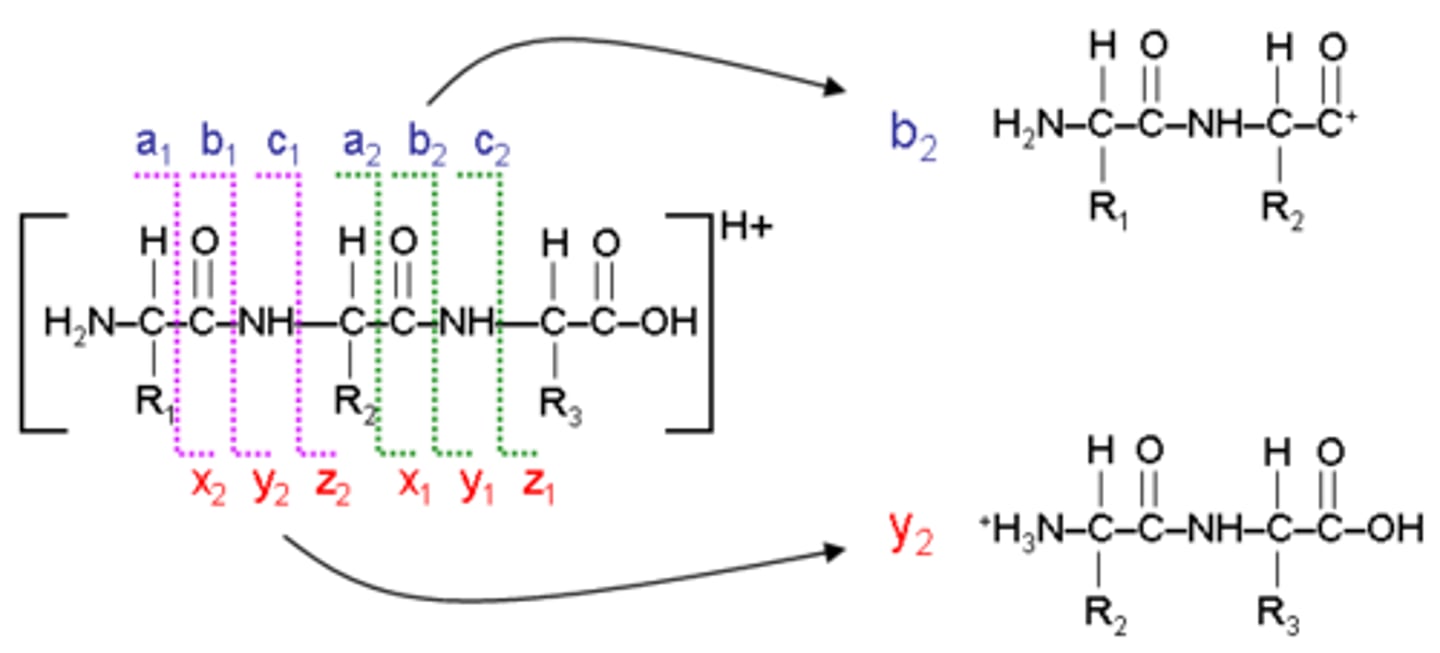
crush peptide with air before secondary analysis to figure out where Y and B ions line up
Y and B peptide ions
most common cleavage occurs between C=O and NH group of peptide bond
MALDI-TOF
matrix assisted laser desorption ionization time of flight
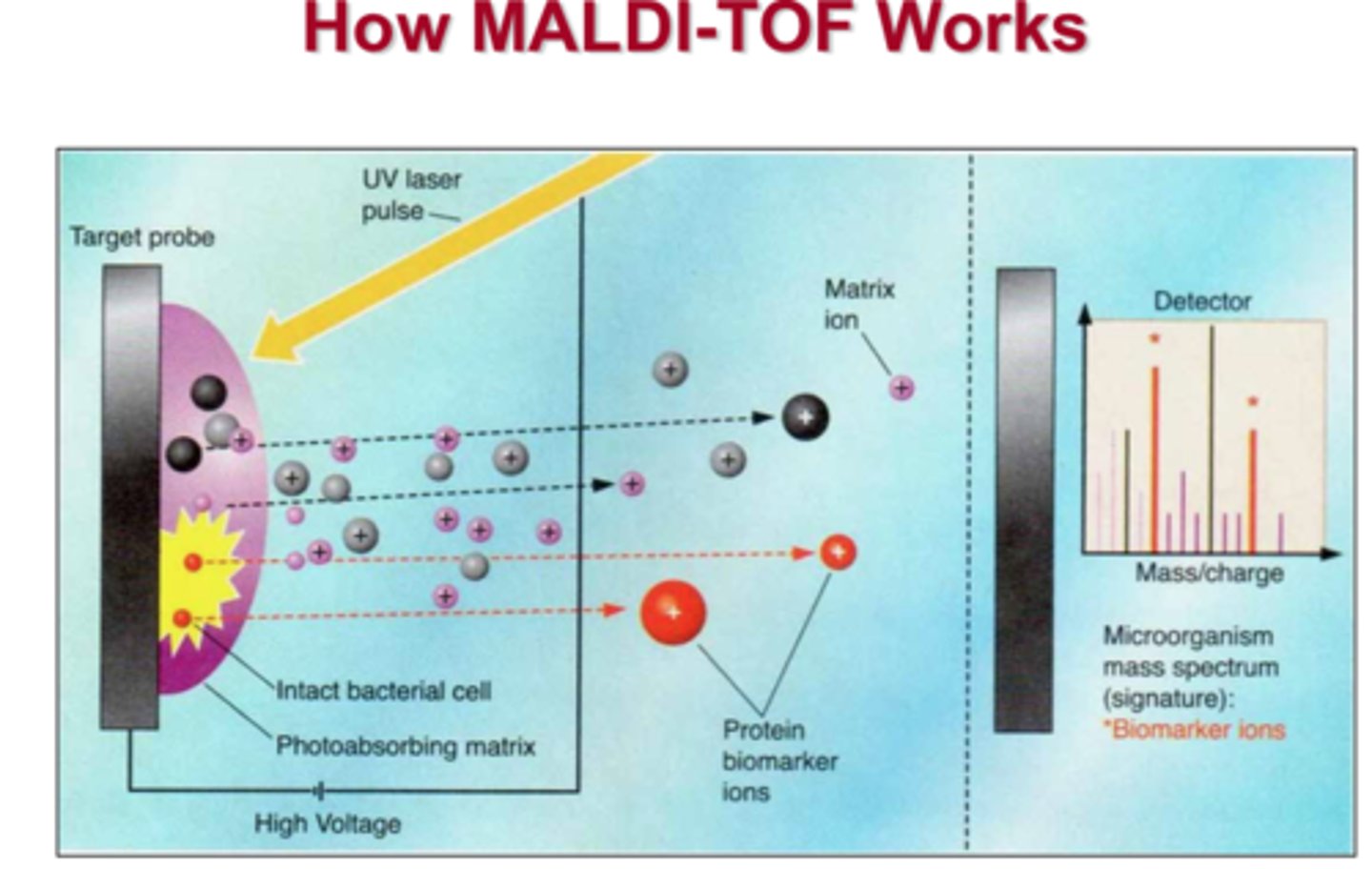
what is the distance between two peaks in MALDI-TOF sequencing?
the mass of one amino acid—so you can read the sequence out like a book!
Fluorescence Anisotropy
Depolarized emission for free molecules (low anisotropy)
Anisotropy
having a different value when measured in different directions
fluorophore
molecules that absorb light and reemit it at longer wavelengths
why is emission always at higher wavelengths?
red shift due to energy being lost = higher wavelength = lower energy
this is Stoke's shift!
why are emission/excitation peaks mirror images?
The same vibrational levels being involved in absorption and emission.
Franck-Condon principle: the best overlap between discrete excited and ground state wave functions just happens to be a mirror image of the excitation
Jablonski Diagram
Shows the probabilities of going from one state to another.
An energy diagram that illustrates the electronic states of a molecule and the transitions between them; the states are arranged vertically by increasing energy and grouped horizontally by spin multiplicity.
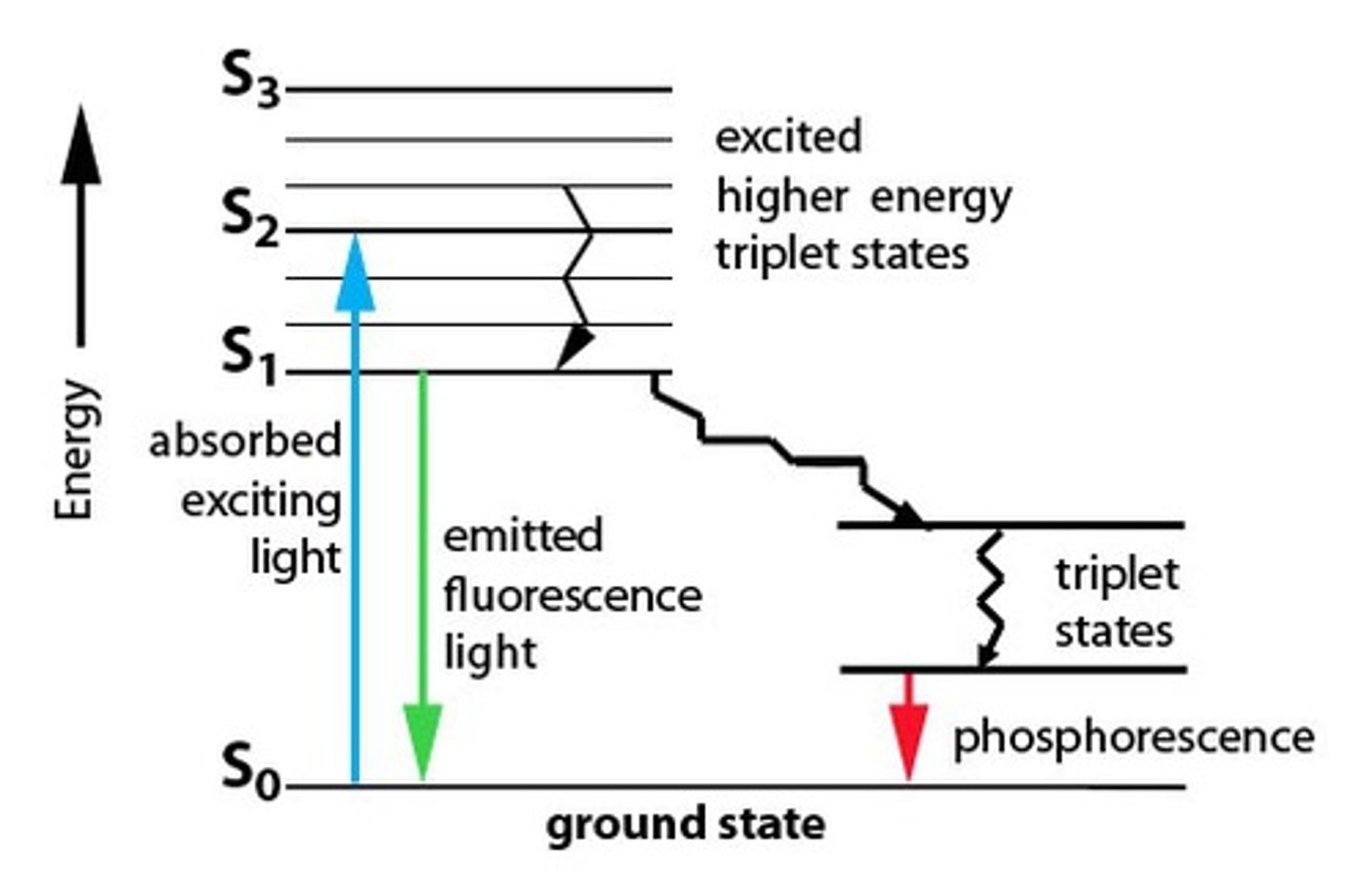
Franck-Condon Principle
best highest overlap between discrete positions will occur, meaning it is most likely that transfer happens between the best overlapping vibrational wave functions
vibrational levels are DISCRETE
the best overlapping vibrational waves just happen to be mirror images of each other
Stoke's shift
difference between excitation and emission wavelengths
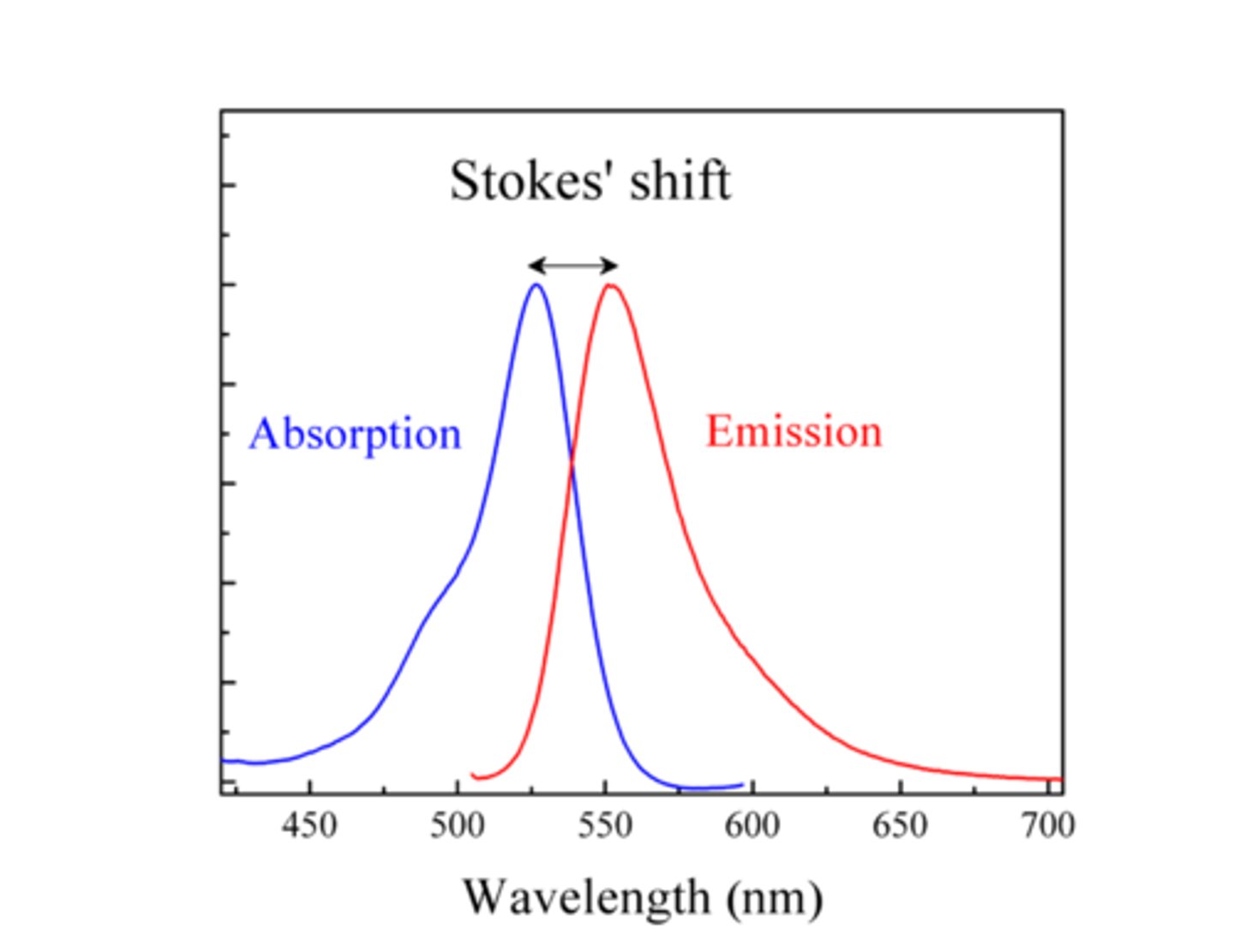
how does Stoke's shift and Jablonski diagrams explain the mirror image of emission spectra?
if you go up to v'=1, you'll go back down to v=1—due to the same vibrational levels being involved in absorption and emission.
monochromator
A device for isolating individual wavelengths or frequencies of light. the ideal fluorescence measurer because they don't need filters
why are 96-well fluorescence plate reader plates opaque black?
so fluorescence doesnt leak between samples, contaminating them
FRET
fluorescence resonance energy transfer
BRET
bioluminescence resonance energy transfer
quenching
gain fluorescence after enzymatic activity
FRAP
fluorescence recovery after photobleaching
TIRF
total internal reflection processes at the plasma membrane
when does FRET occur?
when fluorophores are close together, so it's a great way to see if your drug is binding a particular protein
how does FRET occur?
when emission spectrum of the donor overlaps with the excitation spectrum of the acceptor, the emissions of the donor will excite the acceptor
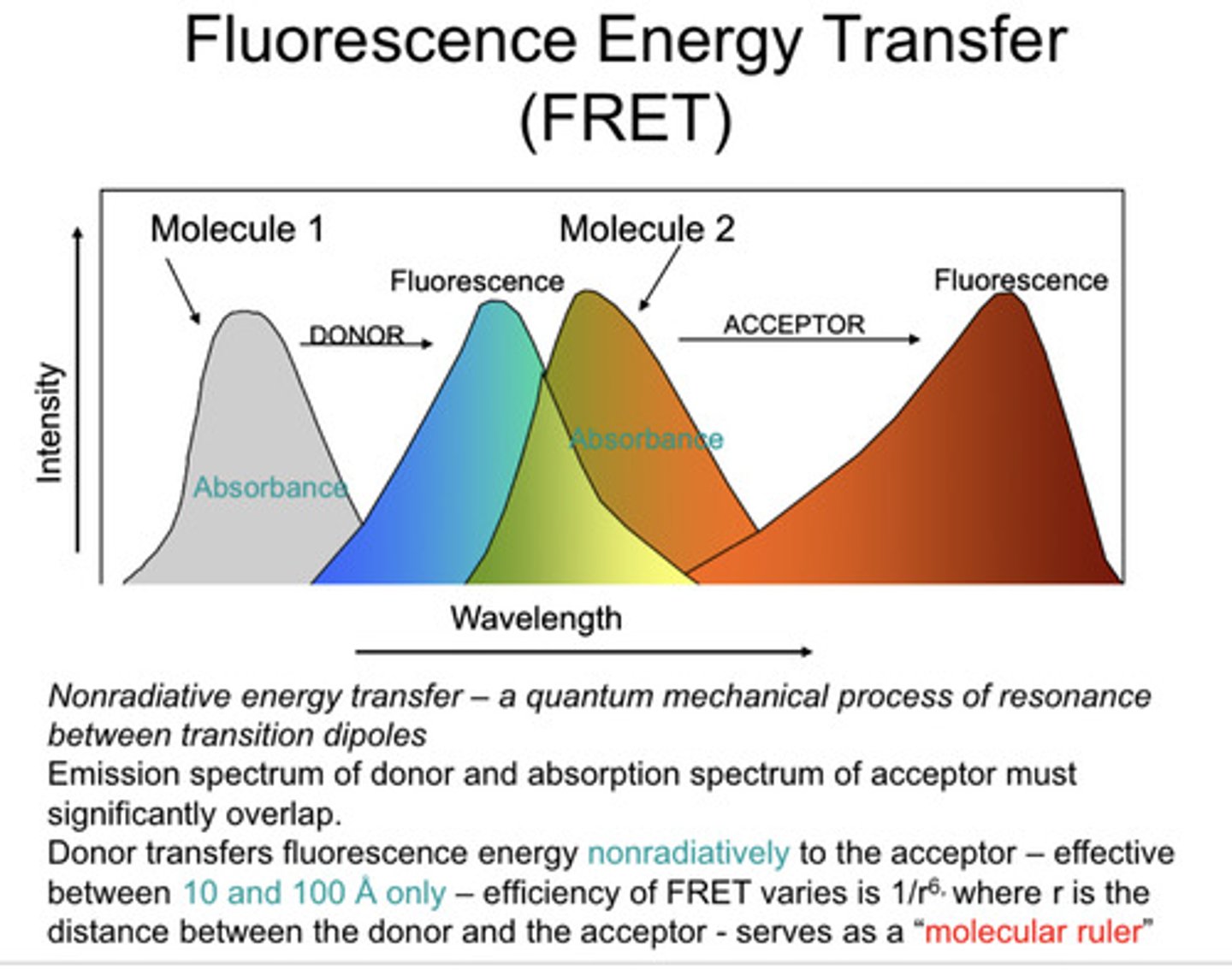
how close do fluorophroes have to be to give FRET?
10nm
how is BRET different from FRET?
the donor "fluorophore" is a bioluminescent reaction
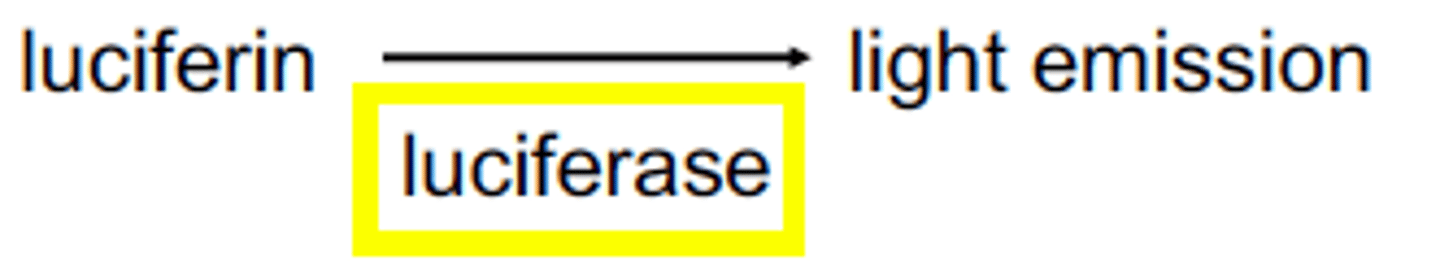
luciferase reaction
substrate: luminol
luciferase: protease
reaction with oxygen releases light
how does quenching with proteases work?
you have a peptide that includes a fluorophore and a quencher. the quencher steals energy from the fluorophore in a FRET-like manner, cancelling out its fluorescence and keeping it dark. When a protease comes along and cleaves the peptide, the fluorophore is released from the quencher and it lights up.
how is quenching with proteases relevant?
you can tell if your drug is acting at the right protease, ex. to inhibit it, if you don't see fluorescence
importance of the dipole of fluorophores
light exactly parallel to the transition moment excites the fluorophore the best
what direction does a fluorophore emit light?
in the direction dictated by its structure, related to its transition moment
transition moment
dipole, basically
photoselection
in a solution of fluorophores, only those aligned with polarized light will get excited the best, meaning the emitted light will also be polarized
how do you polarize light?
tiny screen with slots
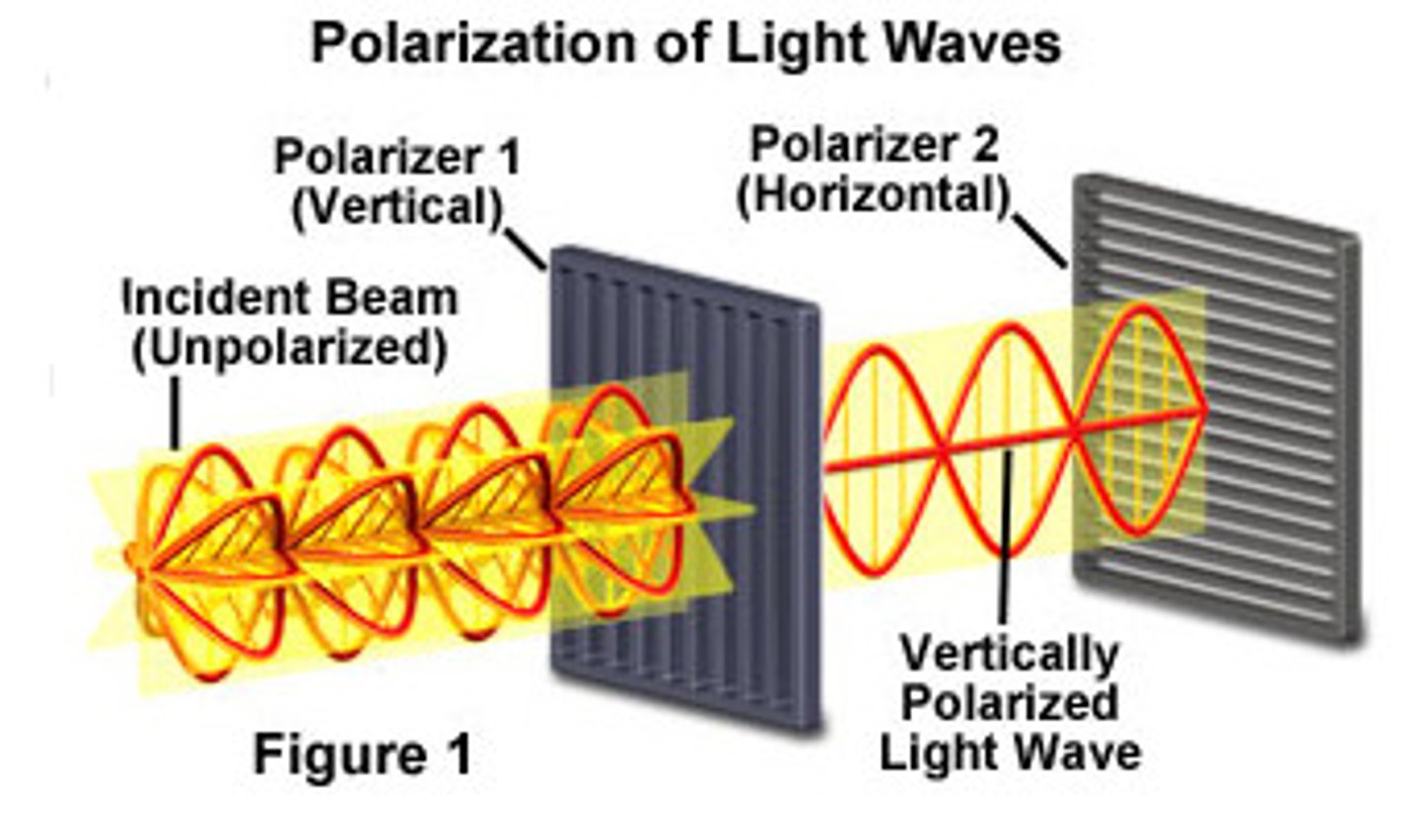
anisotropy
directionality of properties;
describes how much emitted light is still polarized
what does anisotropy indicate about a protein?
how fast it's moving, i.e., if it's bound to something
how does anisotropy indicate the protein is bound to something?
proteins will move slower if they're bound to something (more cumbersome) and so the polarization will be lost less fast
possible movements of a fluorophore
- rotational diffusion of the fluorophore
- mobility of the protein segment (ex. on a hinge)
- rotation of the whole protein
anisotropy emitted light polarization depends on what?
movement of the protein; transition moment
when will emitted light be polarized?
when movement is slow
when will emitted light be unpolarized?
when movement is fast
applications of anisotropy
compound screening assays to see if protein is bound to drug
label at selected proteins in target to see if a target is mobile
which amino acid is intrinsically fluorescent?
tryptophan
low anisotropy
means shit is moving fast and fluorophore is not bound to a protein = light is not polarized
high anisotropy
light is polarized because shit is not moving fast and fluorophore is probably bound to a protein
how did we used to discover drugs?
natural medicines were investigated, then refined
biologically observed agent -> asssay -> hit -> optimization -> studies -> drug
this was great because we already knew it worked!
genes-to-drugs target-based screens
in the 90s, gene sequencing revealed new targets
molecular genetics target identification -> target-based assay -> target hit -> lead optimization -> preclinical and clinical studies -> drug
this was not great because a lot of targets are not biologically activated—you can make a drug that doesn't work in vivo
phenotype-based screens
targeting a phenotype, so biology first!
human cell system -> biological hit (find something that works) -> lead optimization -> preclinical and clinical studies -> drug
this works because you're looking for something that has an effect phenotypically
cell-based assay cell lines
immortalized: human embryonic kidney, HEK293, HeLa
primary cell cultures (die quickly)
induced pluripotent stem cells
issue with primary cell cultures?
not immortalized; die quickly
target-based screen
you're targeting a specific protein—shit may not work because you don't know the full MOA of the protein, but you'd do this for example in cases you know for sure a target is bad