amino acids + urea cycle
1/30
Earn XP
Description and Tags
week 2
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
31 Terms
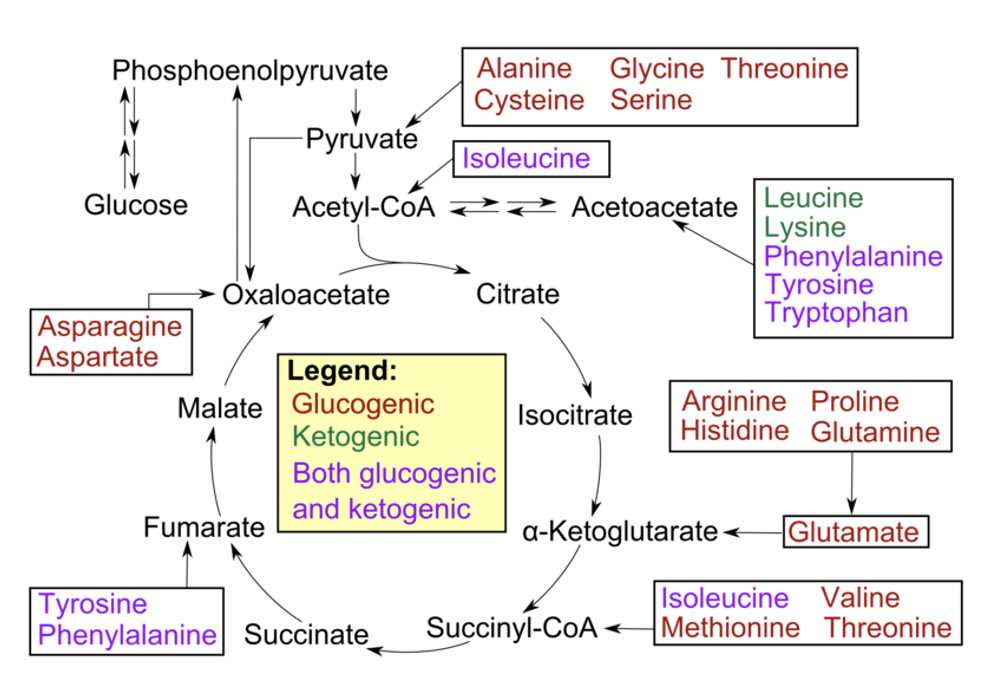
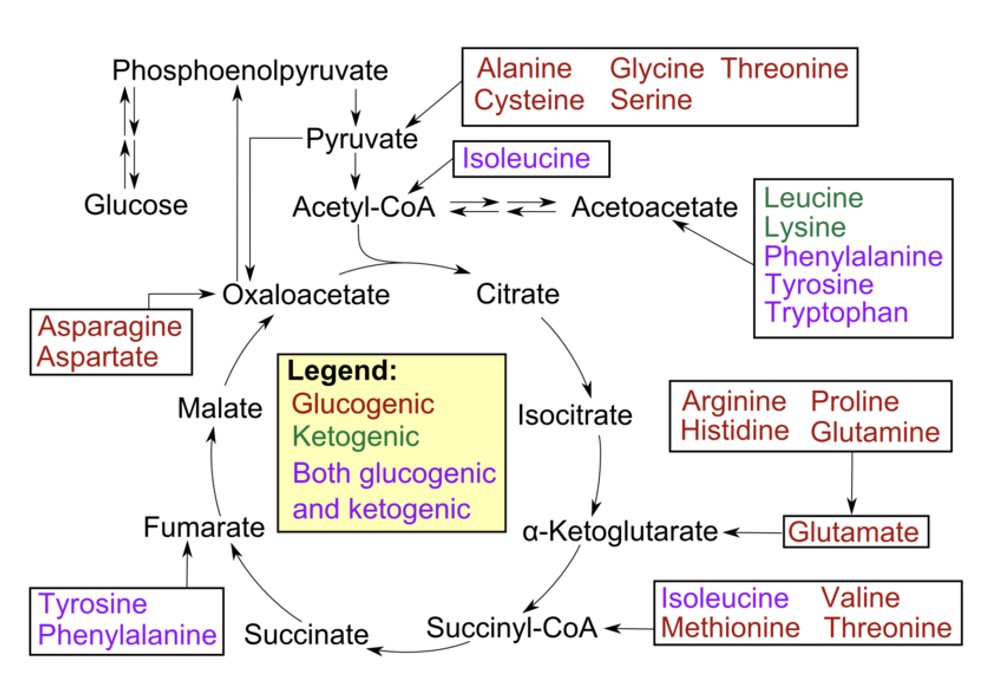
glucogenic
used to provide glucose via the process of gluconeogenesis
Most amino acids are glucogenic
during their breakdown the remaining carbon skeleton can be converted → oxaloacetate → glucose, via gluconeogenesis, if needed
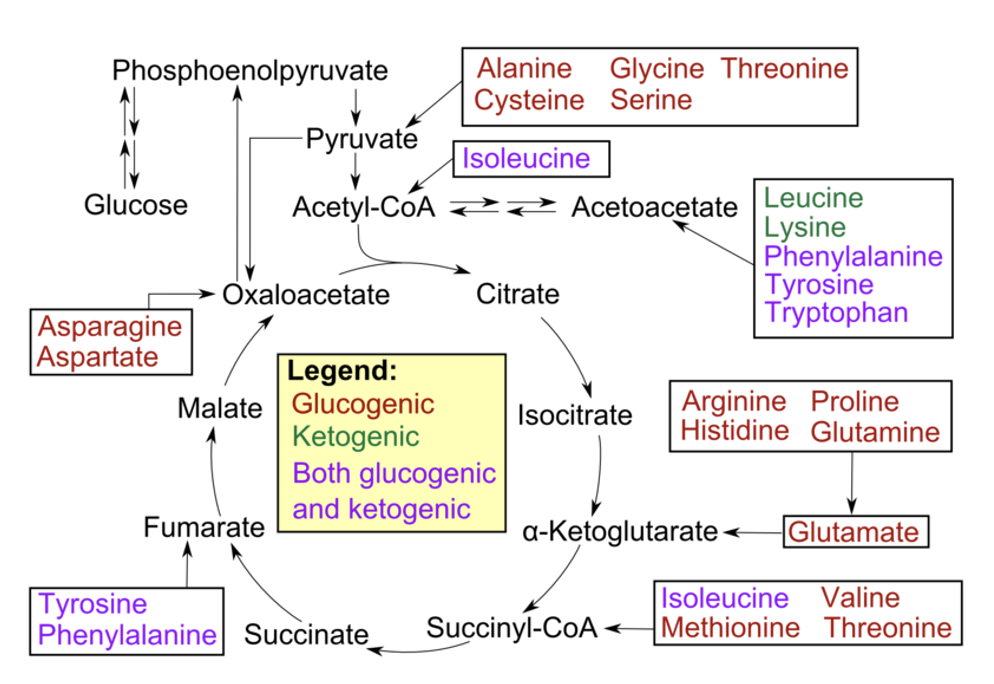
ketogenic amino acids
used to provide acetyl CoA or acetoacetate equivalents
amino acids with carbon skeletons that cannot be converted to glucose (because they are broken down to either acetyl CoA or acetoacetyl CoA)
are shunted towards fat synthesis or ketosis
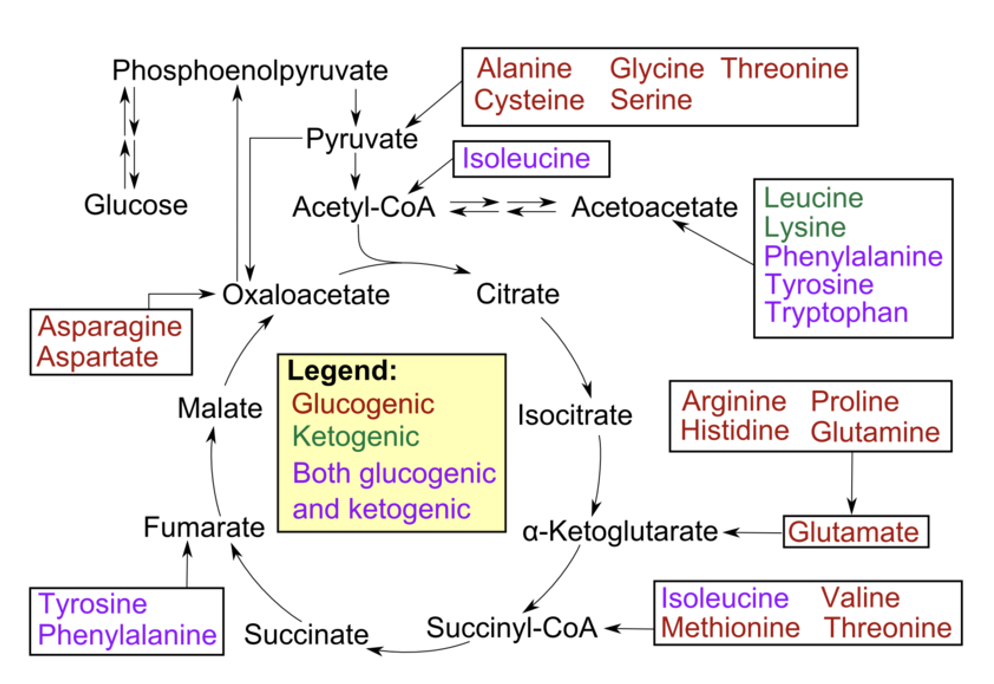
list of ketogenic amino acids
Phenylalanine, tyrosine & tryptophan (aromatic)
Lysine, threonine
Leucine and Isoleucine (2 branched chain amino acids)
amino acid biosynthesis
To synthesise non-essential amino acids we need 2 basic components:
A source of carbon (a ketoacid)
An amine group donor (NH3+)
Glutamate/Glutamine
Aspartate
Carbamoyl-P
carbon skeleton of amino acids can be derived from a variety of metabolic precursors
(from pathways such as glycolysis, TCA Cycle, pentose phosphate pathway or breakdown of important biomolecules)
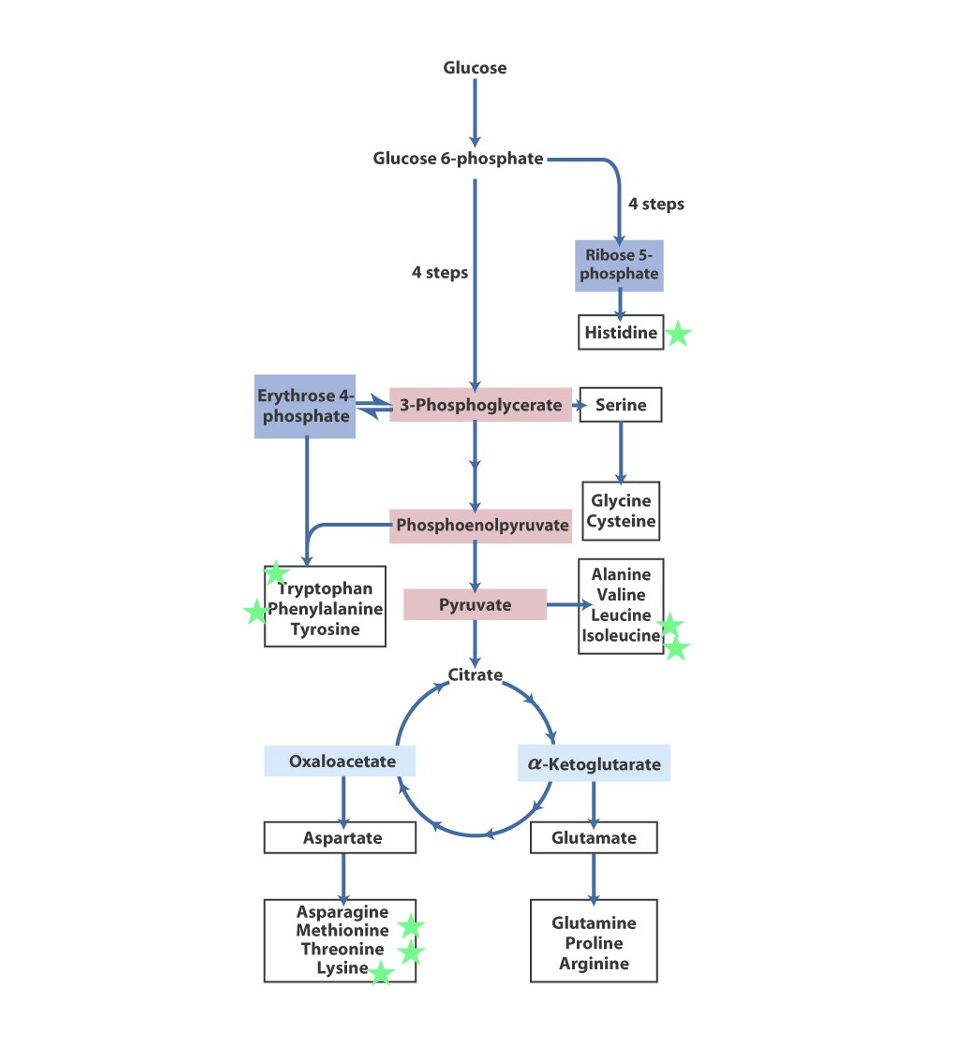
role of amino acid biosynthesis
formed amino acids can be used to synthesise protein or other important biomolecules (neurotransmitters, hormones, haeme etc.)
Metabolic Fate of Dietary & Intracellular Protein
All cells can ‘remodel’ amino acids but most amino acid metabolism occurs in liver
Remodelling: removing the amino group & recycling the carbon skeleton
To metabolise amino acids, the amino group must first be removed (deamination)
Toxic ammonia is converted, in the liver, to the less toxic compound urea, which is excreted in the urine
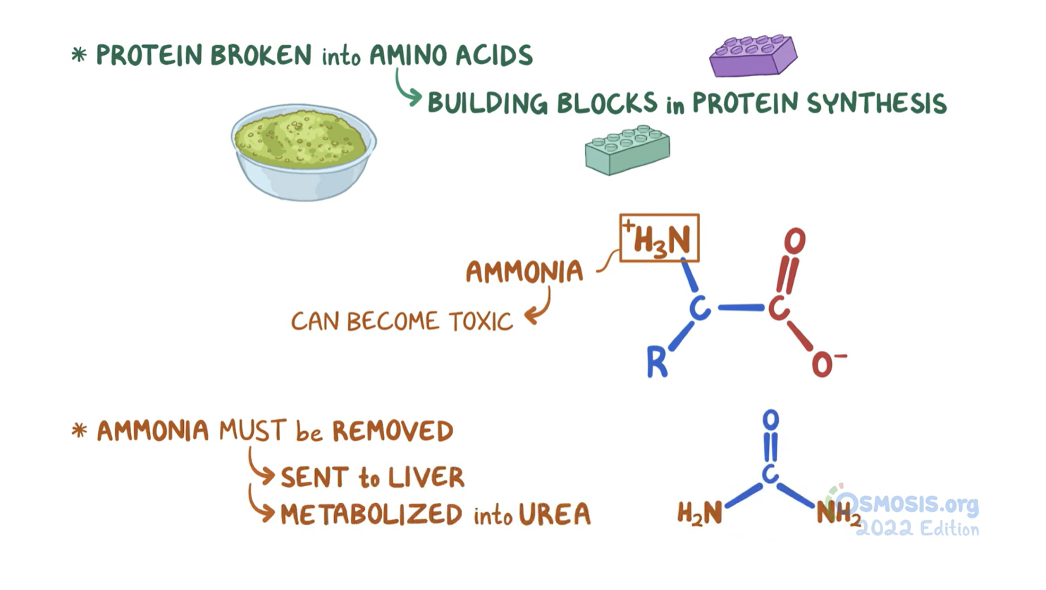
catabolic fate of amino acids
Nearly all carbon skeletons from amino acid metabolism can be converted into intermediates in glycolysis, TCA or lipid metabolism
Only lysine doesn’t undergo transamination
generally, deamination is followed by direct metabolism in a central pathway or interconversion to a metabolite in one of the central pathways
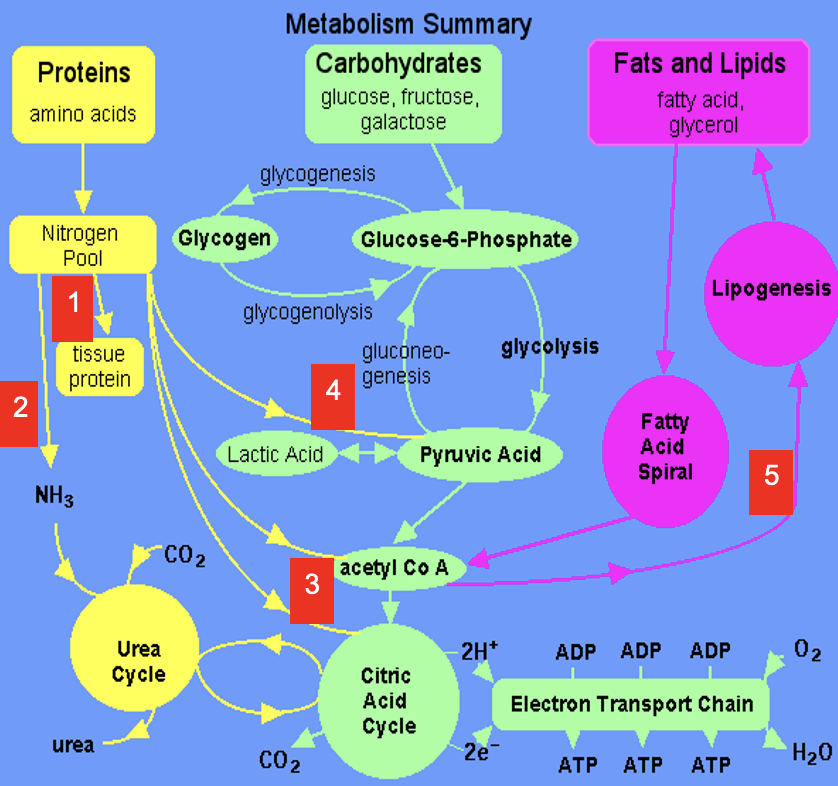
1
Dietary amino acids contribute to tissue protein
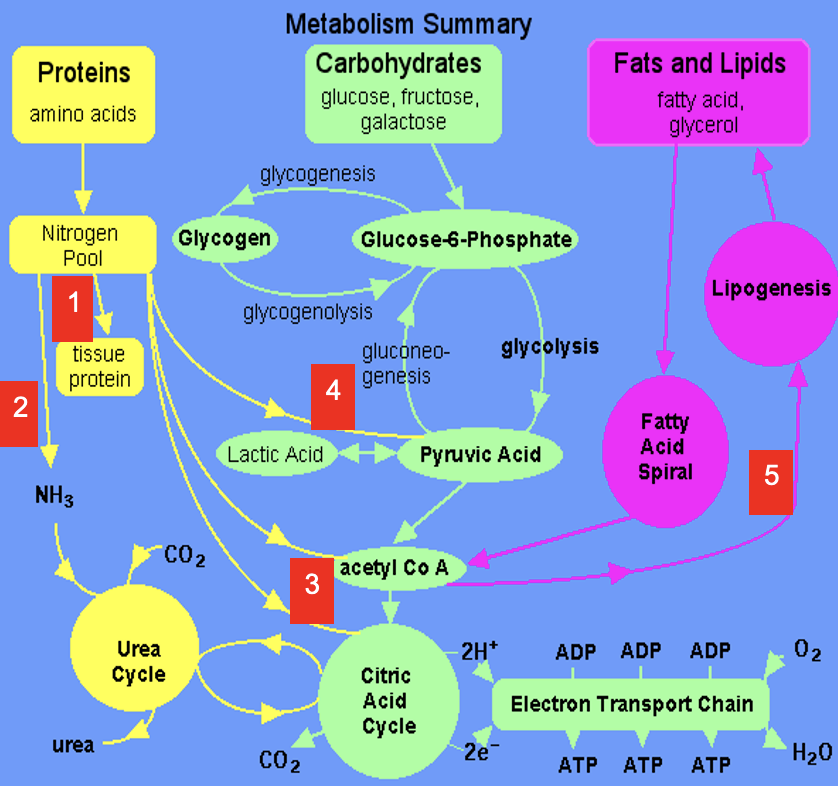
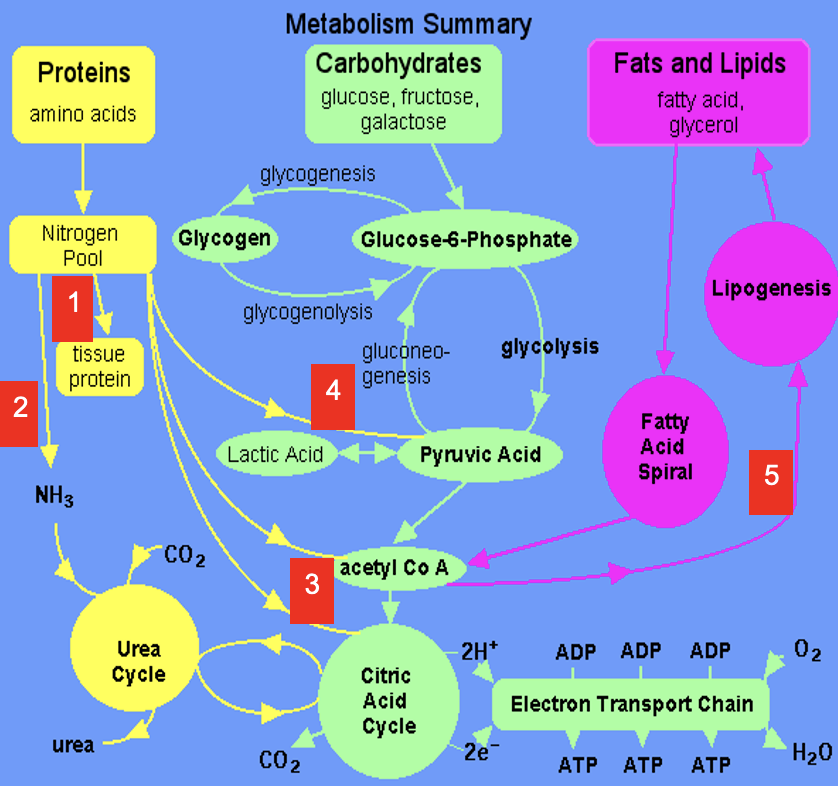
2
Excess amine converted to urea via Urea Cycle
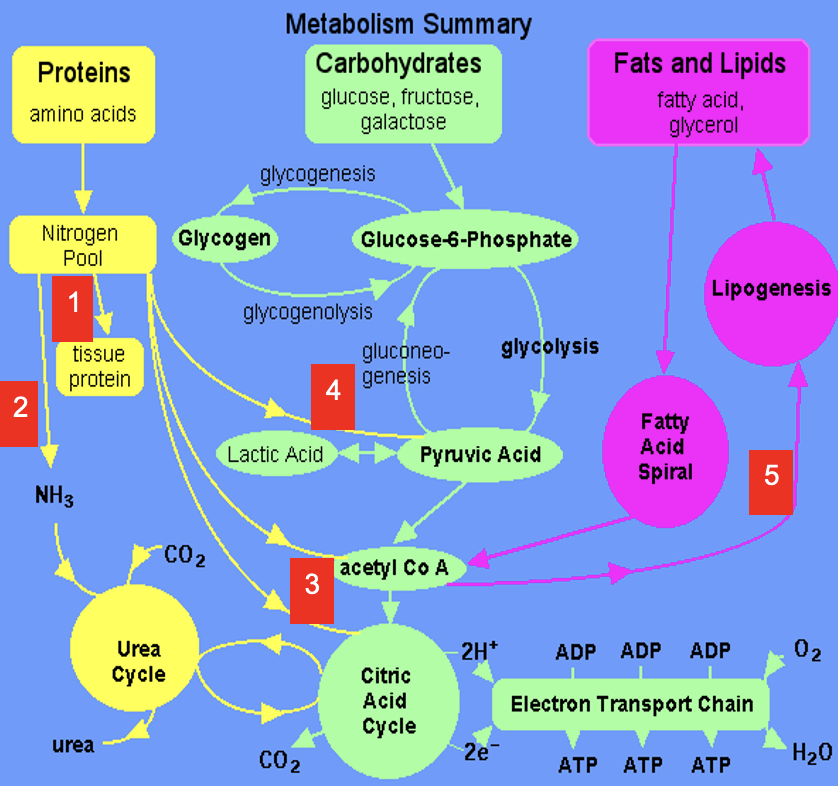
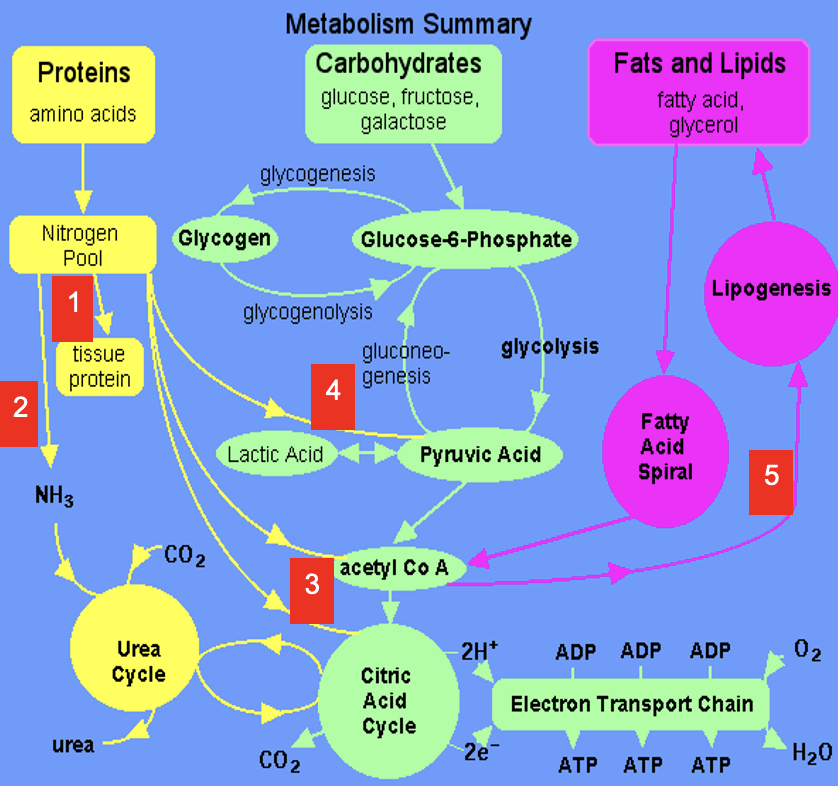
3
Carbon skeletons feed into TCA Cycle or converted to acetyl CoA which is a precursor for lipids (& ketone bodies, see 5)
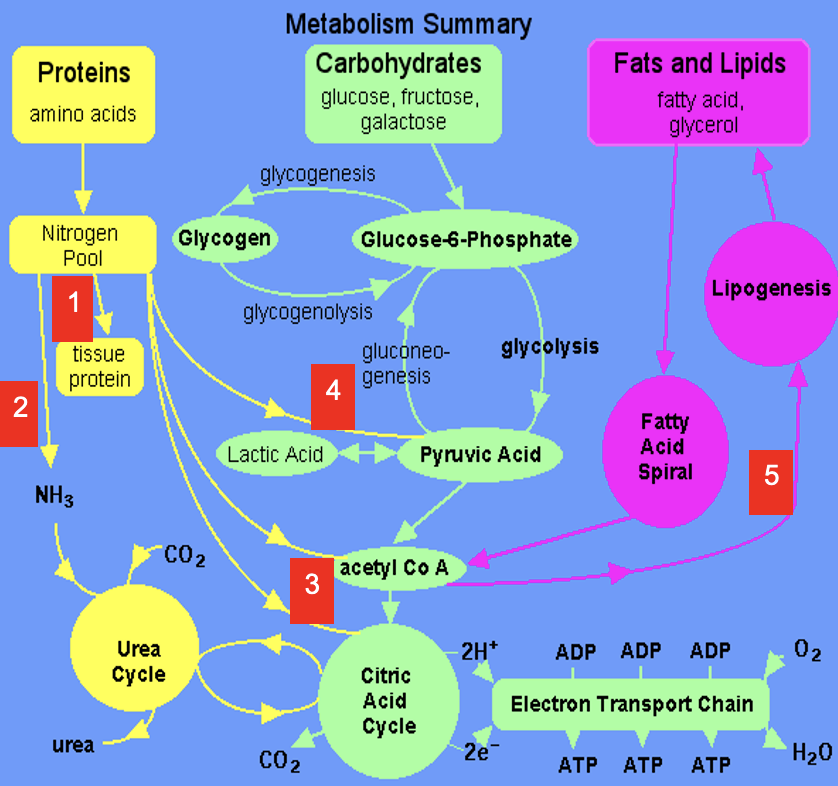
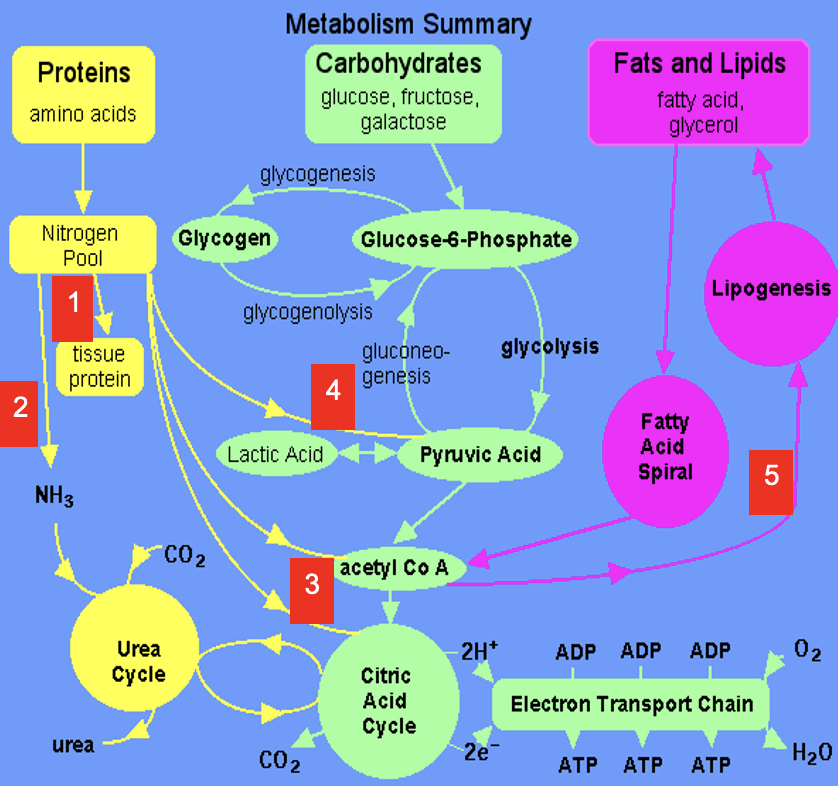
4
Carbon skeletons can be broken down to pyruvate (precursor for a variety of molecules & gluconeogenic substrate)
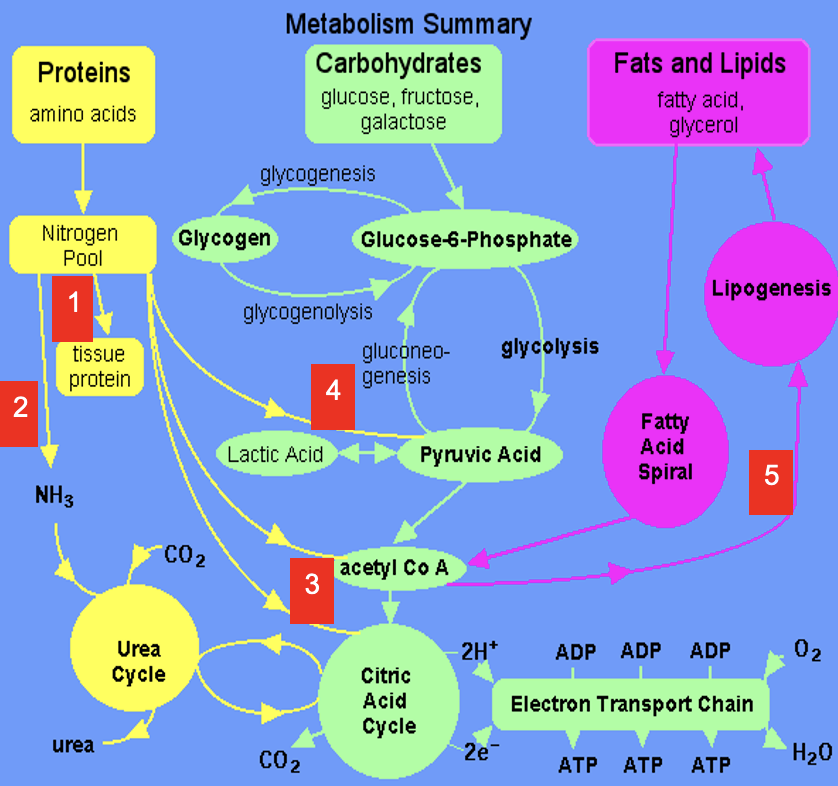
deamination of amino acids in liver
most amino acids undergo deamination in the liver
can occur by the action of a range of enzymatic reactions:
aminotransferases
glutamate dehydrogenase (oxidative deamination)
glutaminase
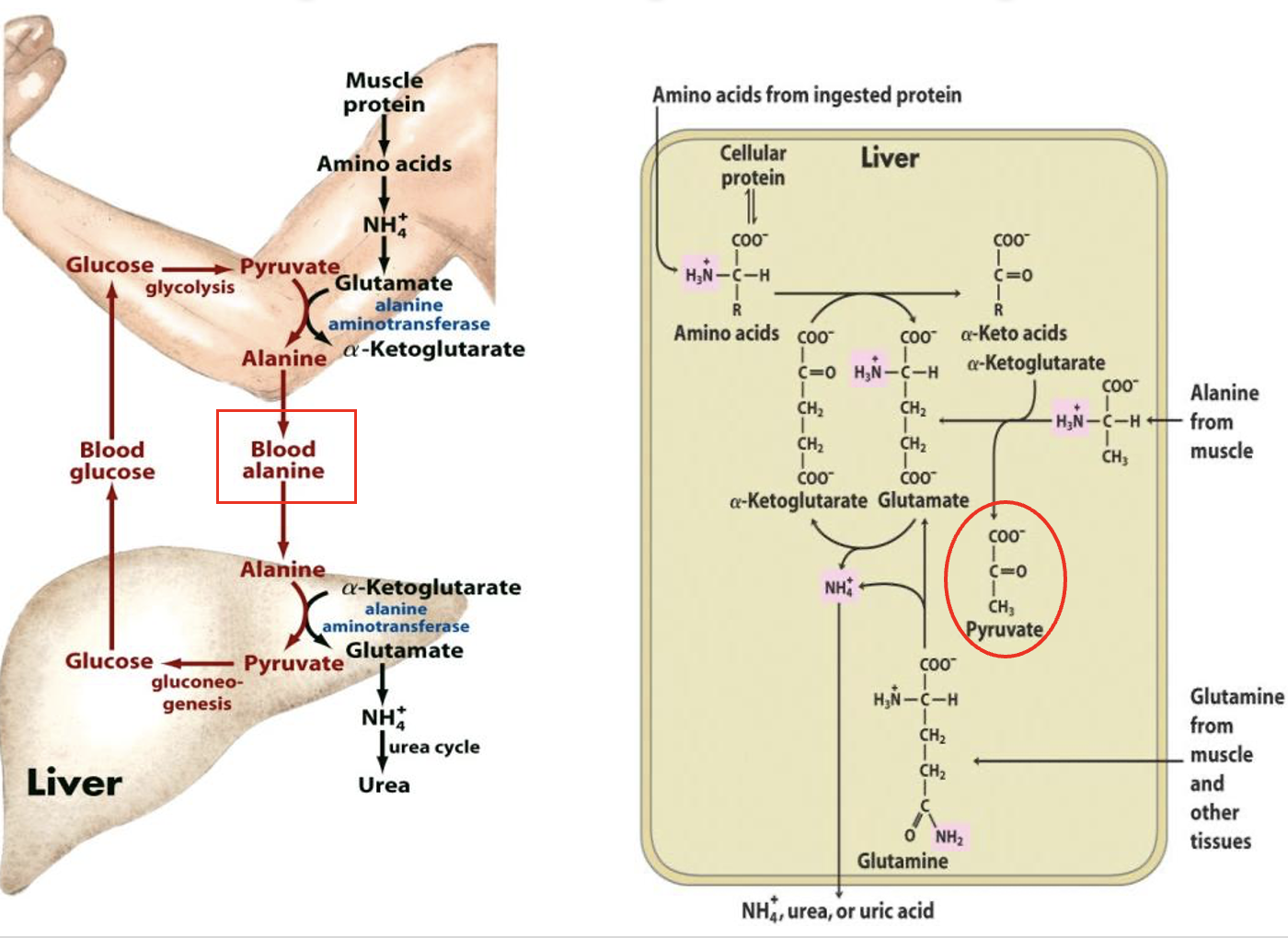
deamination of amino acids in skeletal muscle
since muscle cannot make urea, the amino groups must be transported (SAFELY) to the liver:
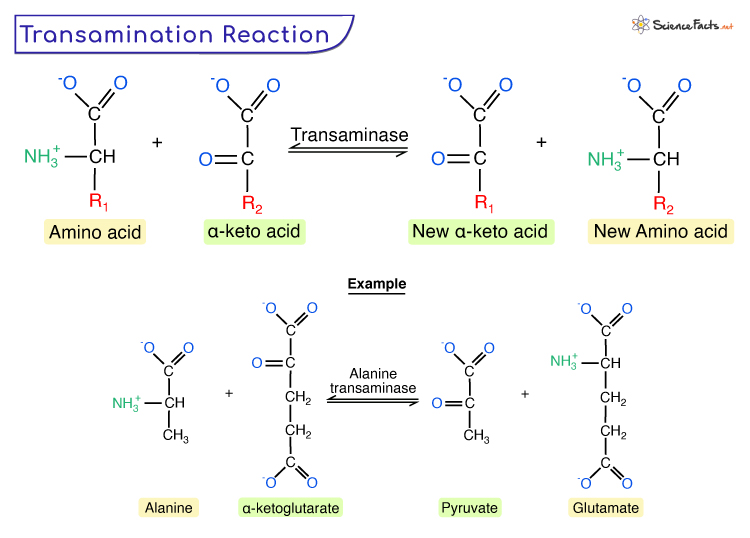
Aminotransferases catalyse the transfer of amine groups from amino acids to amine acceptors (ie alpha keto acids like pyruvate) producing the amino acid, alanine
Alanine → bloodstream → liver —transaminated→ pyruvate + glutamine
glutamine → NH4+ → urea
Pyruvate —gluconeogenesis→ glucose
“Glucose-Alanine cycle”
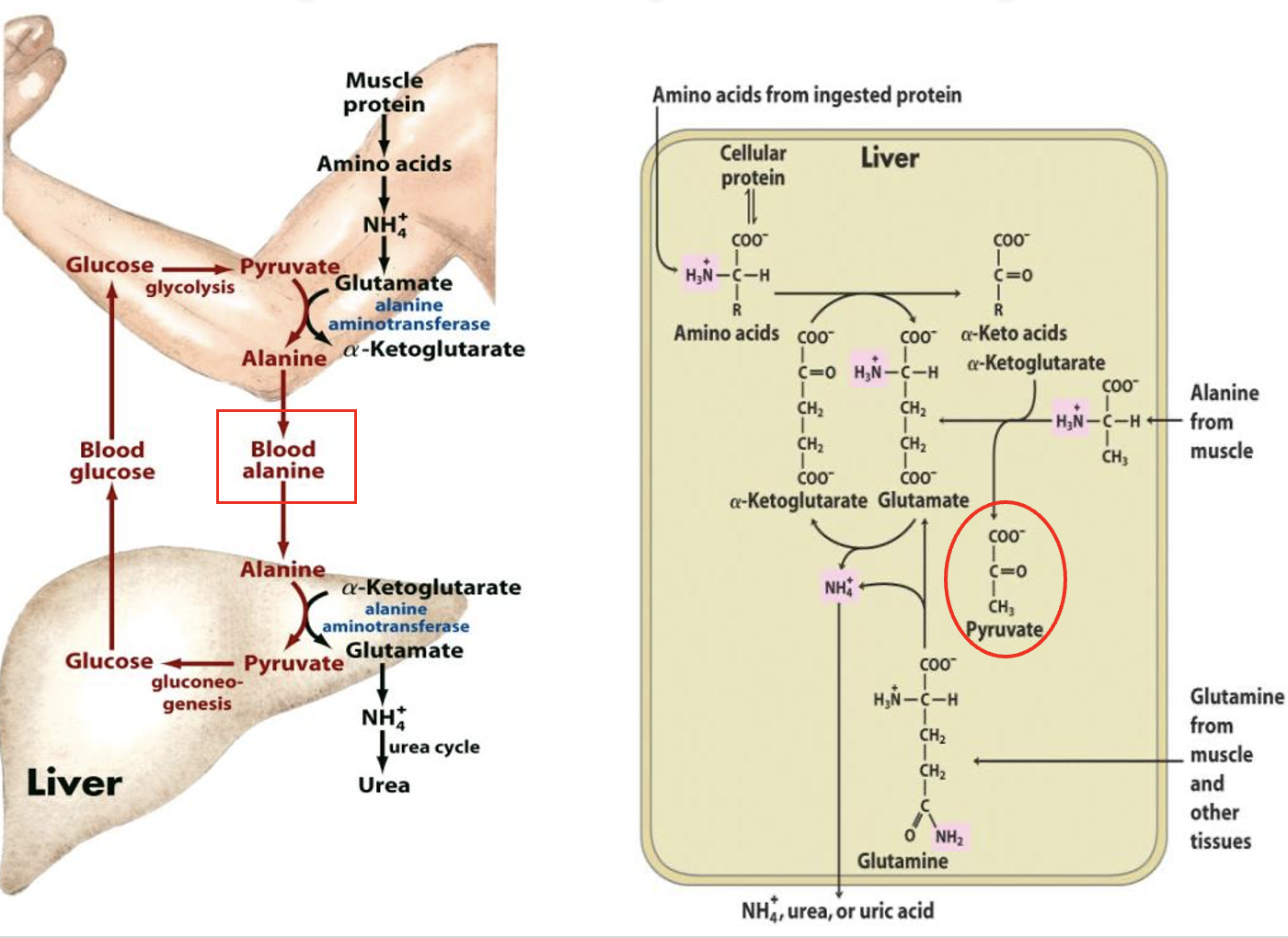
Glucose-Alanine Cycle
involves the transport of excess nitrogen from muscle (via alanine) to the liver
In the liver it is converted into glucose via gluconeogenesis which is then exported from liver back to muscles for energy etc.
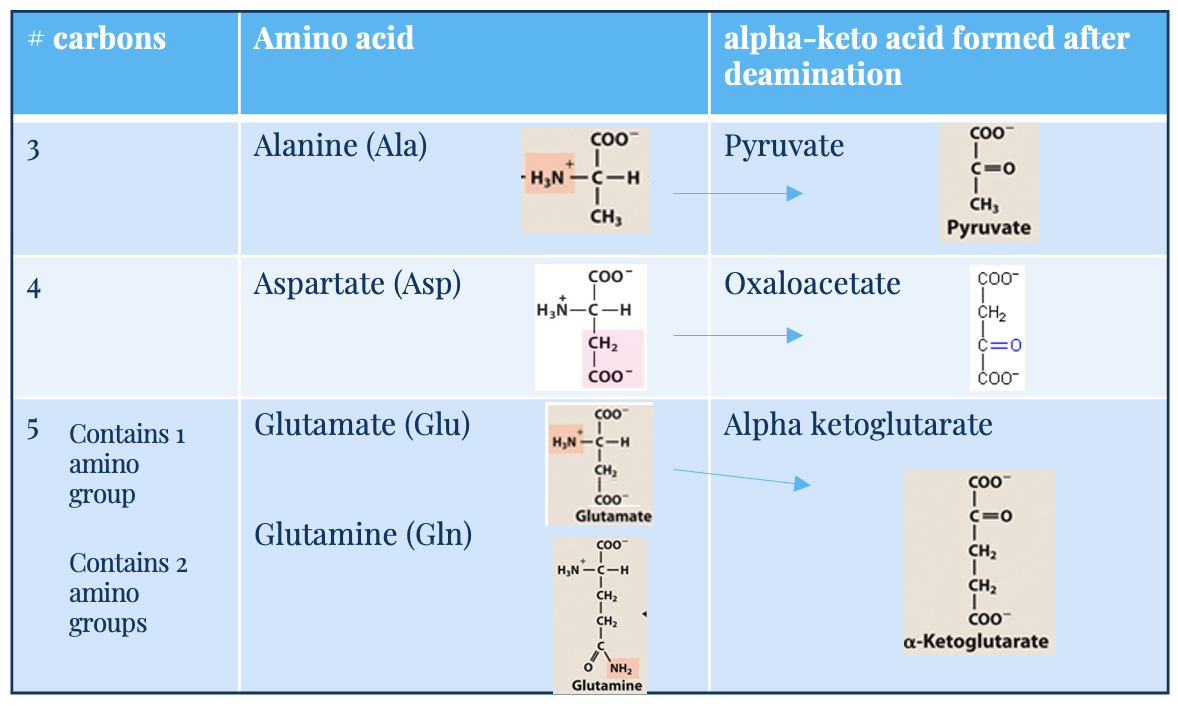
Glutamate (Glu)
5C, one amino acid group
acts as –NH3 acceptor (in AA degradation, accepts –NH3) forming glutamine
acts as –NH3 donor (for biosynthetic pathways/excretion) forming alpha-ketoglutarate
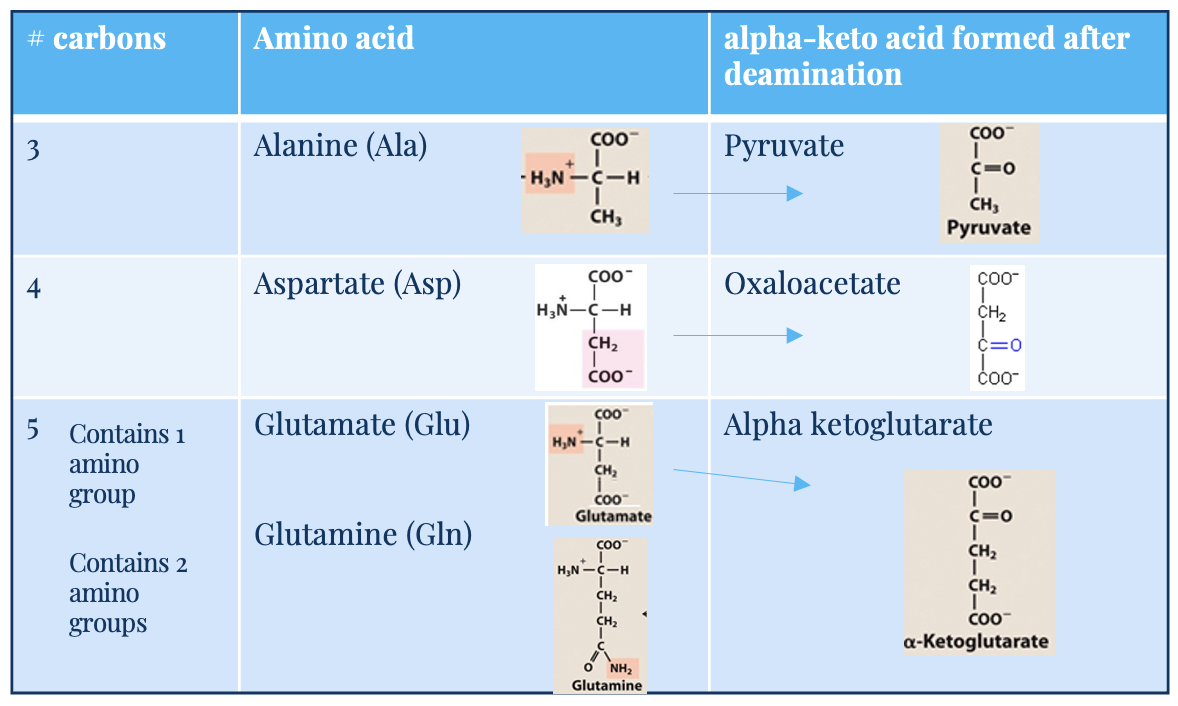
alpha keto acid
carbon skeleton
glutamine (Gln) and alanine (Ala)
key transporters of amino groups between tissues and liver
levels of these amino acids in blood is higher than all other amino acids
alanine (3C) → pyruvate (after deamination)
glutamine (5C, 2 amino acid groups) → glutamate → alpha-ketoglutarate
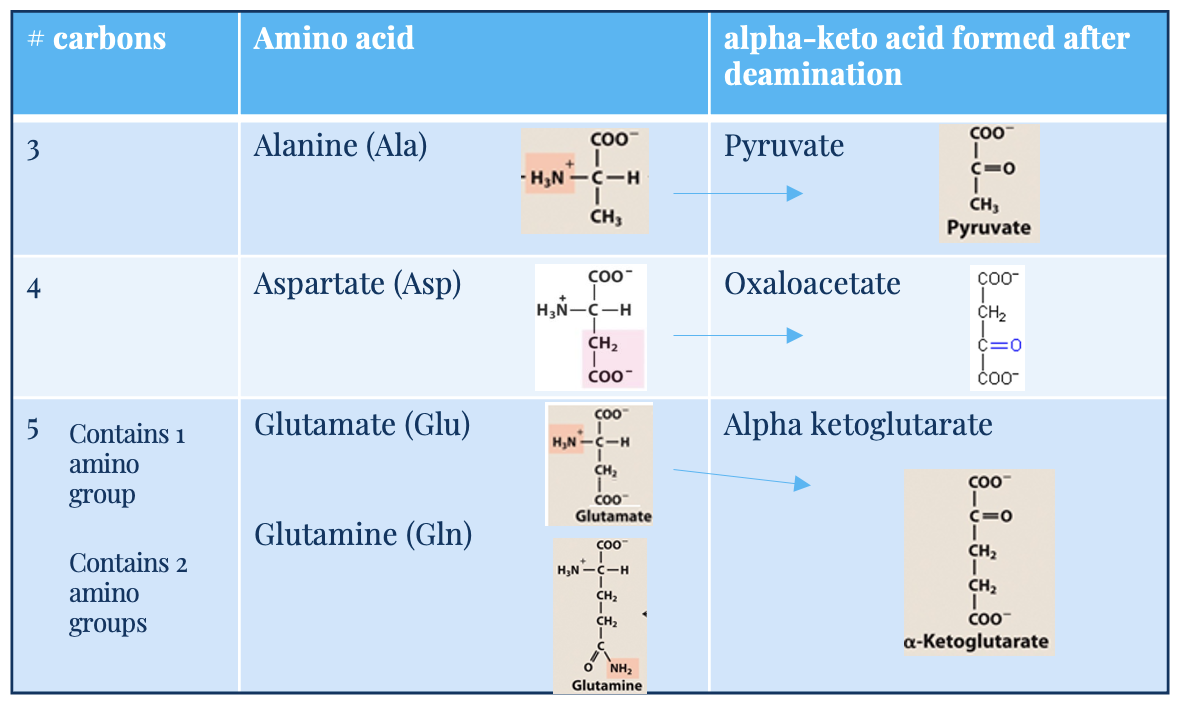
Aspartate (Asp)
4C
deaminates into oxaloacetate
2 key mechanisms for deamination
transamination
oxidative deamination
transamination
Transfer of amino group to a suitable keto acid acceptor (no free amine released)
involves aminotransaminase enzymes:
alanine aminotransferase (ALT)
aspartate aminotransferase (AST)
Reactions are reversible
enzyme requires vitamin B6/pyridoxine as a cofactor
oxidative deamination
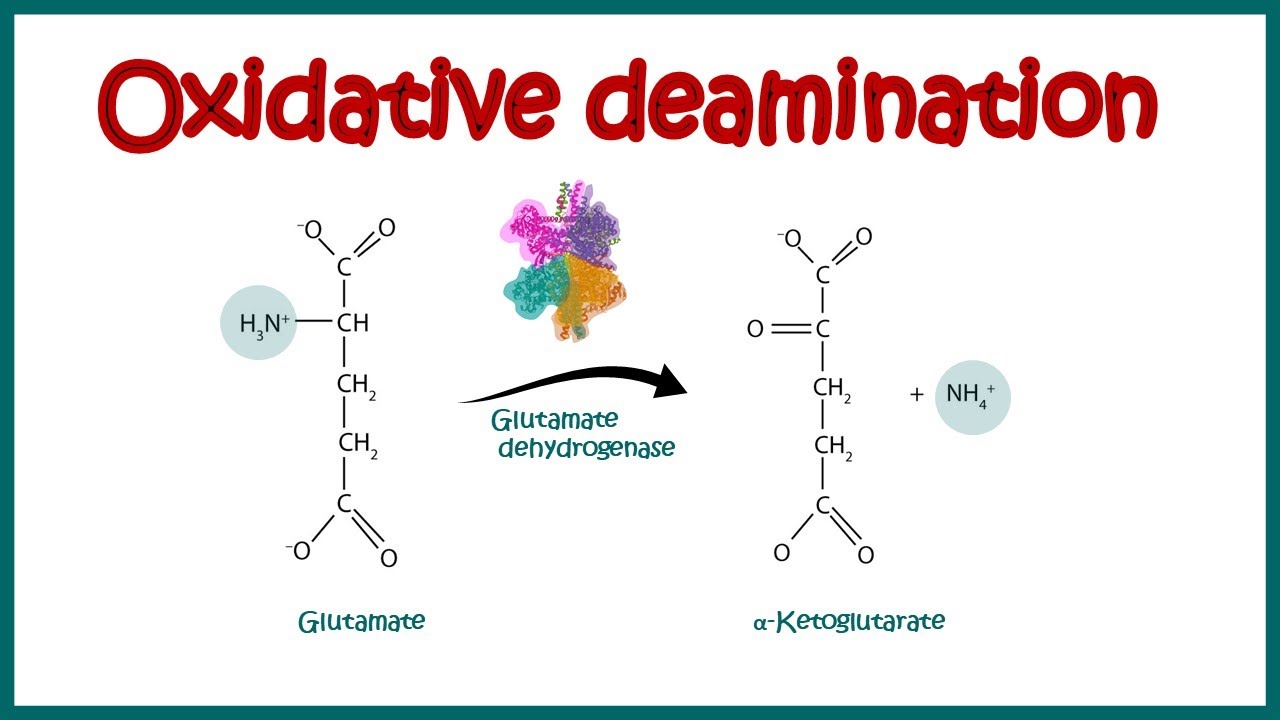
Oxidative removal of a free amino group forming an a-keto acid + free ammonia via glutamate dehydrogenase
deamination: removal of ammonia
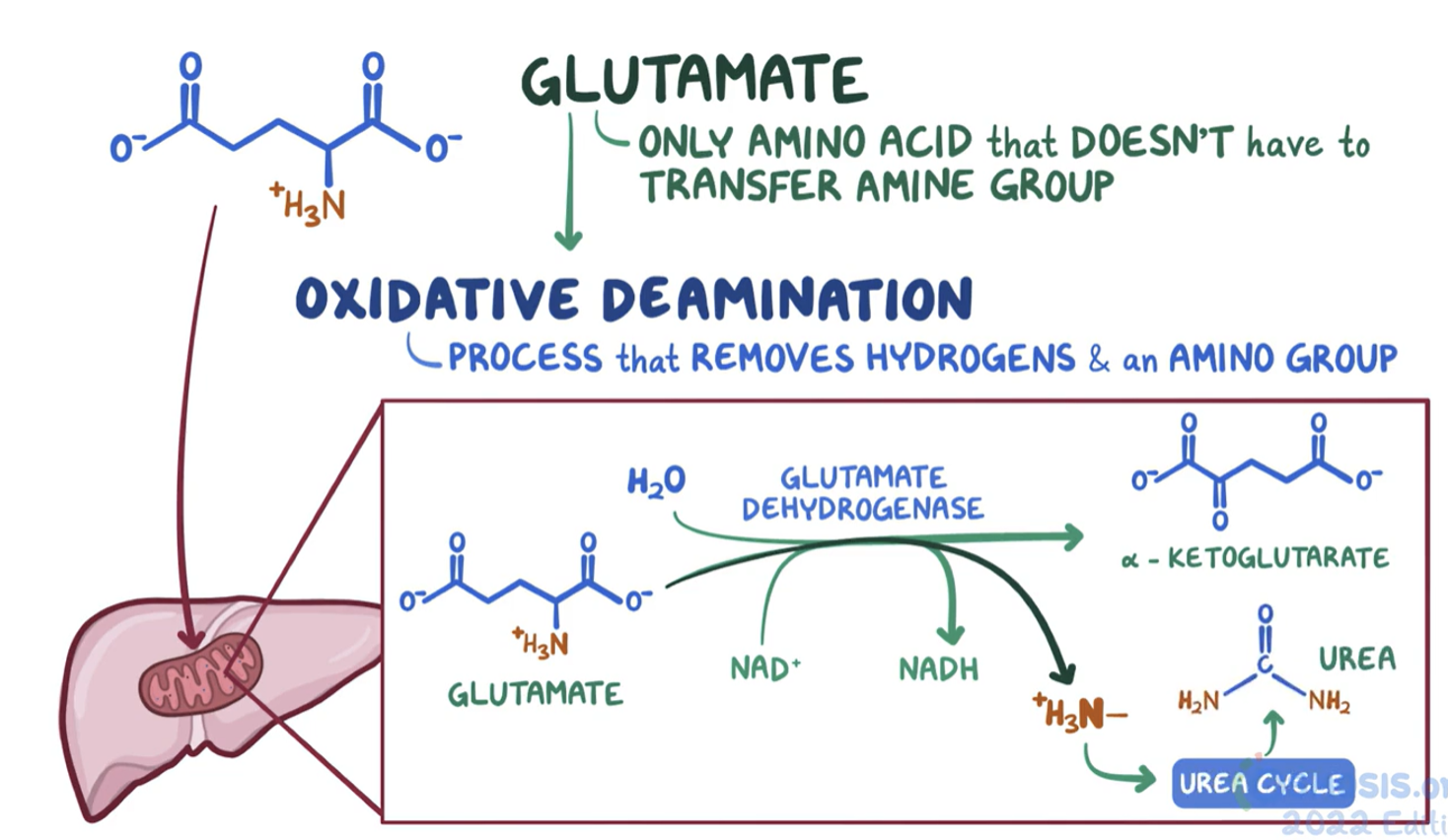
Glutamate is the only amino acid that doesn’t have to transfer its amino group to another molecule
Glutamate undergoes oxidative deamination - glutamate dehydrogenase removes the amine group and hydrogens
Glutamate + NAD+ + H2O
↔
α-Ketoglutarate + NADH + H+ + NH4+
ammonium produced is used to form urea
mechanism of toxicity of excess ammonia
Ammonia (NH3) readily crosses the BBB by diffusion so any process that increases serum ammonia is potentially dangerous i.e. hyperammonemia
ammonium toxicity:
Increased levels of glutamate (excitatory neurotransmitter/excitotoxin → brain cell damage)
Increased levels of glutamine (disrupts BBB letting water/plasma in → oedema)
Depletion of ATP (interferes with mitochondrial function, possibly through inc. free radicals)
Terminal stages → coma, brain swelling & death
urea cycle
liver & kidneys work together to ensure toxic levels of ammonia do not accumulate
Urea Cycle combines 2 amino groups into the urea molecule (one from glutamate dehydrogenase reaction, one from aspartate)
Urea is transported via the blood supply to the kidney where it is excreted, and excreted in sweat
urea synthesis occurs mostly in the liver
urea diffuses into the blood and goes to the kidney
High rates of amino acid breakdown result in elevated glutamate (glutamic acid) concentrations which increases the supply of substrate for the cycle
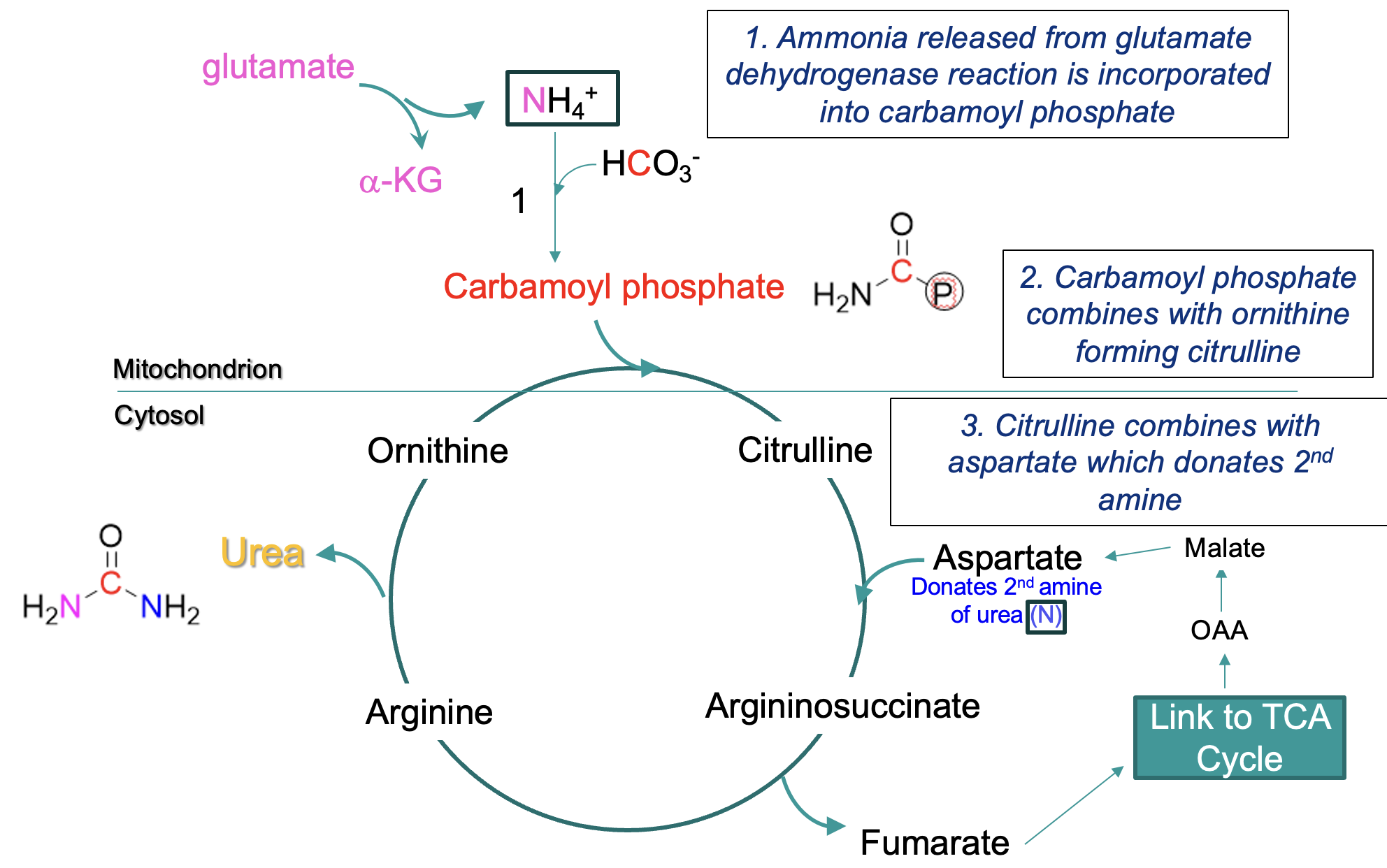
steps in Urea Cycle
mitochondrial matrix:
ammonia → carbamoyl phosphate
carbamoyl phosphate + ornithine → citrulline (crosses from mitochondrion into cytosol)
citrulline + aspartate (donates 2nd amine) —eventually→ arginine, arginine → urea + ornithine
ornithine reused in cycle
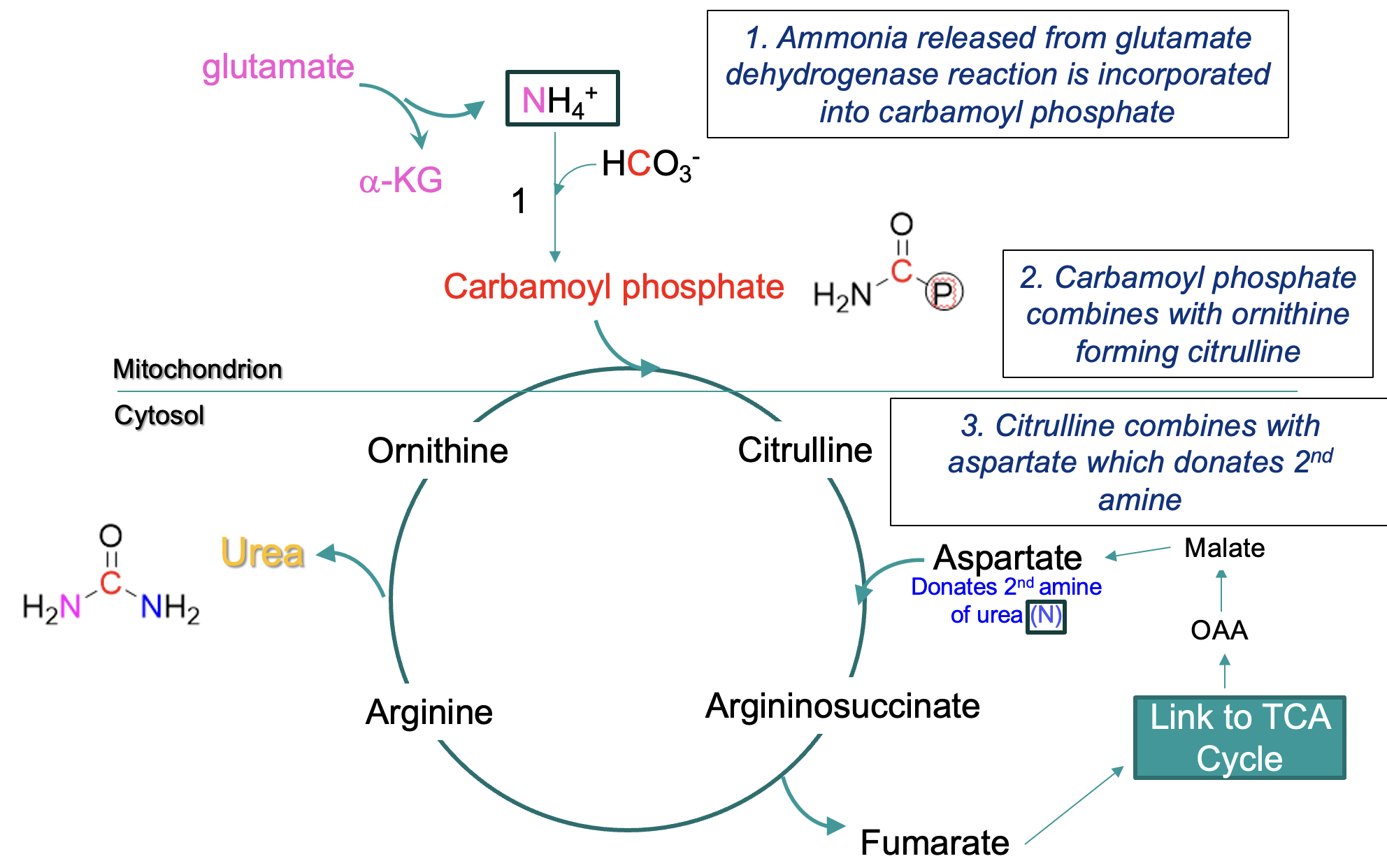
formation of urea -where the amines are derived from
One amine is derived from NH4+ (produced primarily from glutamate via deamination - the glutamate dehydrogenase reaction)
enters cycle via carbamoyl phosphate
Second amine NH3+ is derived from aspartate (formed by the transamination of the α-keto acid oxaloacetate)
fumarate is formed by this process & recycled via TCA cycle to oxaloacetate
urea cycle and TCA cycle overlap
Fumarate is formed from the cleavage of arginosuccinate (an intermediate of the citric acid cycle)
Fumarate → malate → oxaloacetate (an a-keto acid)
Oxaloacetate can acquire a second amino group to become aspartate – returns to the urea cycle
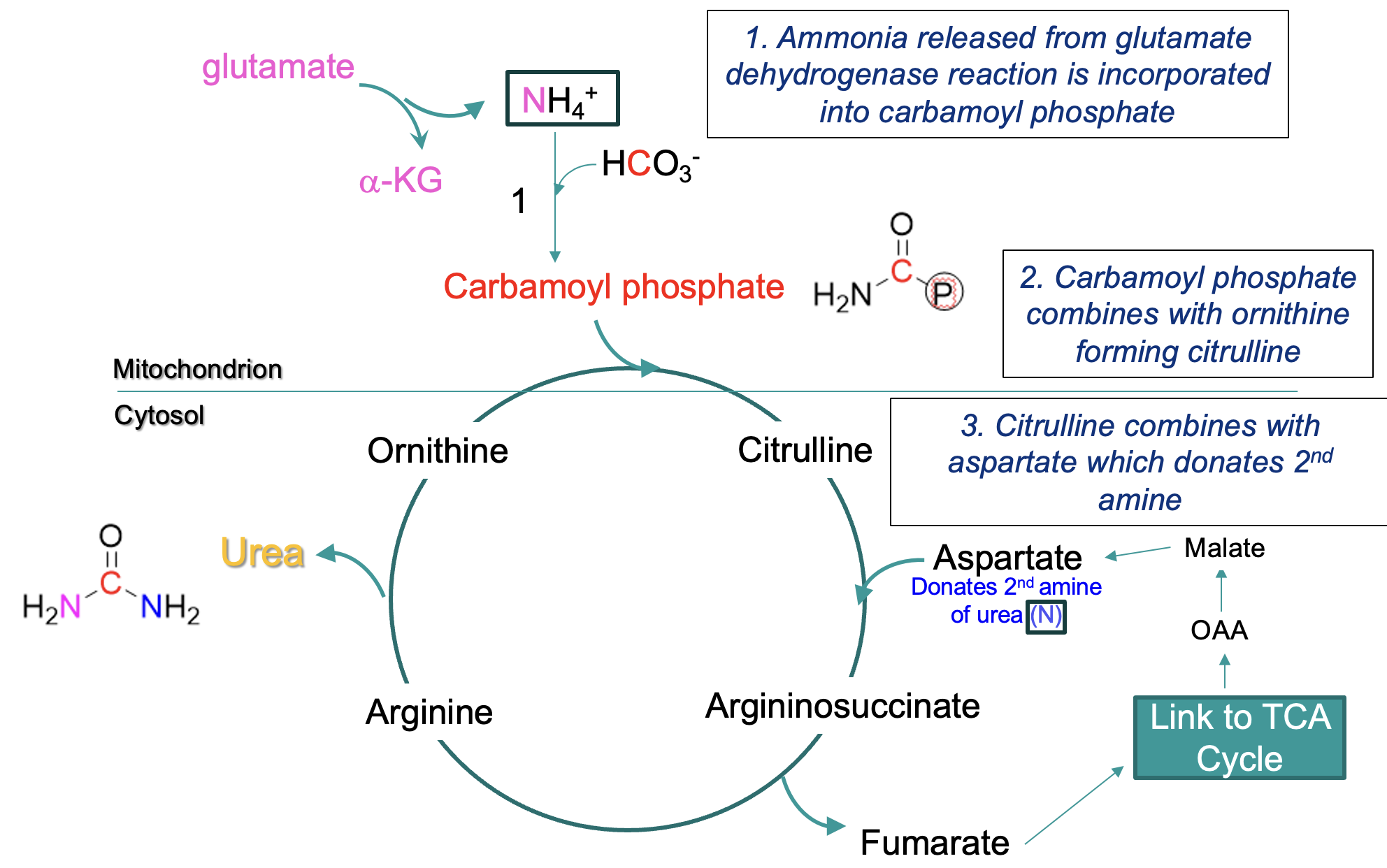
regulation of the urea cycle
Urea cycle increases or decreases in response to high/low protein diet (increases and decreases ammonia)
Regulation is at the level of the enzyme that synthesises carbamoyl phosphate
acid-base balance:
In acidosis urea synthesis is decreased and NH4+ excretion is increased to excrete protons
During fasting increased amino acid metabolism to fuel gluconeogenesis increase urea synthesis
Urea Cycle Disorders
Genetic disorders resulting from a defect in synthesis/function of one of the urea cycle enzymes
Symptoms arise in infancy (often triggered by switch from human milk to formula or introduction of solid foods – both higher in protein)
Symptoms:
hyperammonaemia
lethargy
seizures
vomiting
hypotonia (poor muscle tone)
respiratory alkalosis
coma (even death if untreated)
Severity depends on which enzyme is affected
Most common is ornithine transcarbamoylase deficiency - severe neonatal symptoms – X-linked inheritance
Blood tests would show increased blood levels of ammonia and/or build up of one or more urea cycle intermediates
increased serum urea
increased urea in bloodstream
caused by:
increased urea production
and/or
decreased urea elimination
seen in:
heart failure
dehydration
a diet high in protein)
decreased serum urea
decreased urea in bloodstream
caused by: decreased urea production
seen in: liver failure