BIN300 W10: Genotype imputation
1/13
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
14 Terms
what is genotype imputation
estimating missing data

applications of imputatoin
SNPchip gneoytping reuslts in some no-calls (5%), impute the missing genotypes
use different SNPchips from different venders, denser SNPchip comes on the market, do I need to re-genotype all the previously genotyped aniamls, not necessary to to imputation
new cheaper SNPchip comes on the marker, many SNPs overlap but not all, impute the missing SNPs, both ways: SNPs on old-chip → impute on new chip, SNPs on new chip → impute on old chip
use expensive dense chip on
important ancestors of populations
use cheap parse SNP chip on
production animals
0 impute missing genotypes of the sparse chip, impute all genotyped animals up to high density, impute all genotyped animals up to whole genome sequence
example in fish breeding
genotpe all parents with dense SNP (200~generation)
genotype all offspring and test-sibs with sparse chip (many thousands/generation)
impute genothypes of offspring up to high density
general method
training set animals: high density genotypes
no/few missing genotypes
e.g. 1000-bull genomes: to impute whole-genome-sequences in cattle
imputation set: animals whose genotypes need to be imputed
some methods use pedigree
reduces the set of relevant haplotypes (to those within the pedigree)
however: mostly imputation methods are well able to find the most relevant haplos, pedigree does not help (much)
most genotype imputation is entierly based on linkage disequilibrium
2-step approach
phasing: which alleles are next to eachother on chromosome
imputation of missing genotypes
often: prephased/imputed reference population (used for training)
one-step simultaneous phasing and imputatoin is more accurate, but computationally more costly (speed is important especially with WGS (whole genome sequence) data)

haplotype library approach
split chromosome into pieces/segments
set up library of haplotypes for each segment (prephased reference)
match sample
how large should the segments be → art of imputation
need some overlap between the segments: how much overlap
Hidden Markov Model (HMM)
HMM: estimate probabilities for unoversved (hidden) states (S)
states are the haplotypes on previous slide
which haplotype does the animal have at its paternal chrom, and which at its maternal chrom, states can change (somewhat) from one position to the next due to recombination, new haplotype
observations are the genotypes which are sometimes missing (G)
estimating probabilities ofr animal i
where G = all (known) genotypes at all loci at chromosome; summation is over all possible states at all loci on chromosome (only computational possible by forward-backward algorithm)
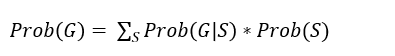
HMM
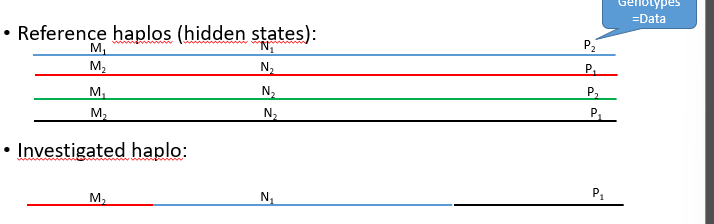
Haplo is a amosaic of reference haplos
HMM: finds mosaic given some/few known marker genotypes (minimises number of crossovers between reference haplos)
Imputatoin software output

measuring imputation accuracy
discordance between estimated and true genotype, highly dependent on frequency, SNP with freq(A)=.01 is easy to impute (low MAF (MinorAlleleFrequency)): P(AA)=0.98 Ifrom Hardy Weinberg Freque)
squared correlation (r²) between estimated and true allele score
allele score is 0,1,2 for genotypes 00, 01, 11
less dependent on allele frequency
hard to get high r² for low MAF SNPs
mask some known genotypes and impute them
i.e. some SNPs on sparse-cip in imputation set
genotype imputation generally based on LD
Haplotype library method
hidden markov model method
useful for
imputing occasional missing genotype, imputing between different versions of SNPchips, systematically imputing low density genotypes up to high density, possible up to sequence data
output
genotype and allele probabilities, phased genotyeps, genotype calls