Tegay - Organic Acidemias
1/8
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
9 Terms
Organic Acidemias Background
All autosomal recessive
Primarily disorders of amino acid catabolism
Branch chain amino acids or Lysine
Major toxicity is from organic acid accumulation causing a metabolic “anion gap”
Secondary toxic effects of acidosis on
Mitochondria → Lactic acidemia
Urea Cycle → Hyperammonemia
Bone Marrow → Bone marrow suppression
CNS Function → Encephalopathy / MR
Major toxicity is from Organic Acid Accumulation
Causes metabolic acidosis with increased anion gap due to decrease in bicarbonates
Major presentation is neonatal encephalopathic acidosis, late chronic / intermittent
Metabolic Acidosis / Anion Gap
Blood pH low from excessive acid vs based
AG = [Na+] - ([Cl-] + [HCO3-]
Multiple etiologies for Metabolic Acidosis
Lowered HCO3- from diarrhea, medications, RTA
Elevated H+ from DKA, starvation, lactic acidosis, acid ingestions, organic acidosis
Causes
Neonate: lethargy, vomitting, tachypnea, hypotonia, seizures, coma, death
Adult: DD, ataxia, neurological deficits, lethargy, vomitting, tachypnea, seizures, coma, death
Organic Acidemias Treatment
Restrict dietary protein
Prevent catabolism via sufficient protein free calories
Reverse acidosis ± hyperammonemia
Hemodialysis
Ammonia and lactic acid scavengers
Cofactory therapy for specific disorders
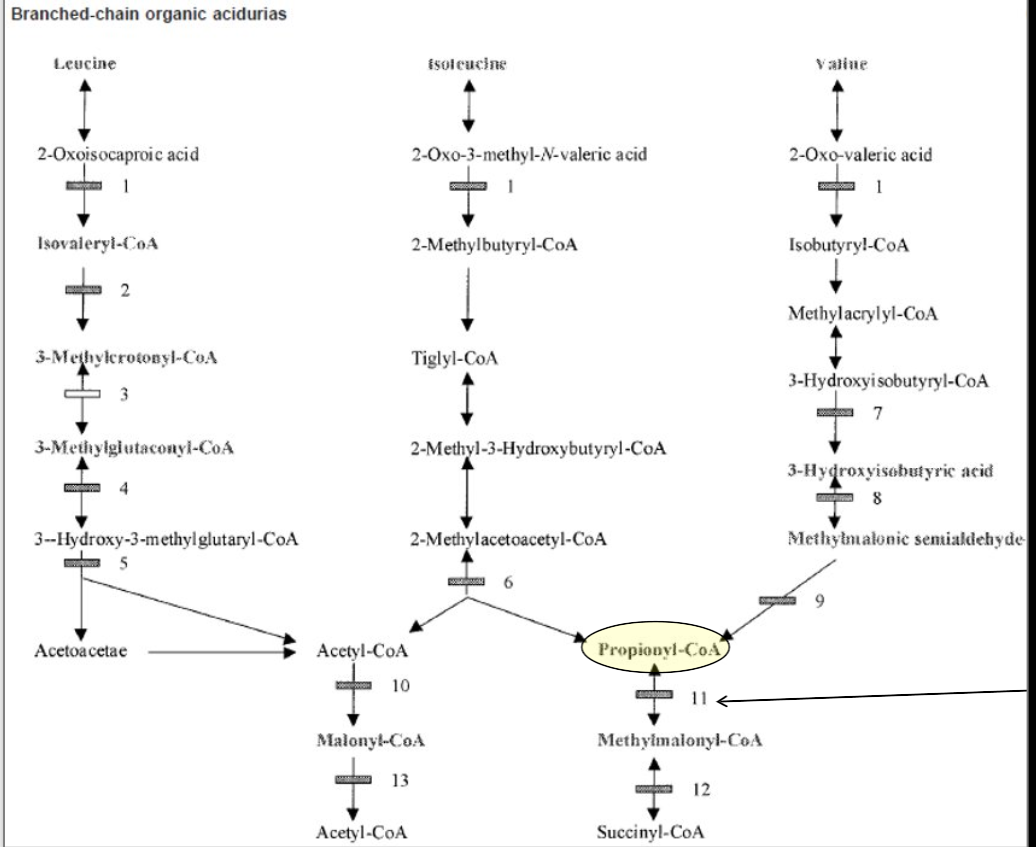
Proprionic Acidemia (PA)
Autosomal recessive
Proprionyl-CoA Carboxylase (PCC) alpha or beta subunit genes
Gene sequencing with deletion / duplication analysis
Aka
Classic organic acidemia with neonatal toxic encephalopathy
Proprionyl-CoA Carboxylase (PCC) alpha or beta subunit genes cofactor is biotin
Proprionic acid accumulation from MET/THR/VAL/ISO catabolism, gut bacteria, odd chain fatty acids
Symptoms
Classic
Normal at birth
Within a few days, poor feeding, lethargy, vomiting, hypotonia → encephalopathy, seizures, coma, death
Late onset
Developmental delay / regression, cyclic vomiting, protein intolerance, growth impairment, hypotonia, metabolic basal ganglia strokes, cardiomyopathy
Acute episodes of toxic encephalopathy
Rare cardiac subtype
Isolate cardiomyopathy
Diagnosed via elevated C3 (Proprionyl) Acylcarnitine (and ratios)
Arterial blood gas, Complete metabolic profile, Ammonia, Complete blood count
Urine organic acid analysis
High 3-OH-proprionate, methylcitrate, tigyl / proprionylglycine but not MMA
Plasma amino acid profile
Elevated glycine and glutamine but not homocysteine
Acyl-Carnitine profile
PCC Enzyme Activitiy
Can measure PCC enzyme in leukocytes or fibroblasts
Useful prenatal when measuring enzyme activity from amniotic fluid, CVS, amnio
Treatment
Improved outcomes with treatments but still some degree of MR, metabolic basal ganglia stroke, pancreatitis, growth impairment, AA deficiencies, renal failure, visual deficits
Acute
Hemodialysis
Protein restriction
IV glucose and lipids
IV Carnitine
IV Ammonia scavengers
Antibiotics
Biotin
Chronic
Protein restruction
Oral L-Carnitine, Biotin, Antibiotics
Avoid decompensation (Infex, surgery, fever, protein)
May consider liver transplant
Isovaleric Acidemia (IVA)
Autosomal recessive
IsoValeryl-CoA Dehydrogenase (IVD) Gene Mutation
IVD Gene with deletion / duplication
Symptoms
Disorder of leucine metabolism, increased isovaleric acid
Sweaty feet odor
Severe neonatal onset form
Normal birth
Within a few days, poor feeding, lethargy, vomiting, hypotonia, encephalopathy, seizures, coma, death
Mild / Late Onset Form
Unexplained failure to thrive and DD, at risk for catastrophic decompensation with fasting, protein loads, or illness
Benign form
Typically asymptomatic but may mildly decompensate
Diagnosed via elevated C5 (Isovaleryl) Acylcarnitine
Arterial blood gas, Complete metabolic profile, Ammonia, Complete blood count
Urine organic acid analysis
High IVA and Isovaleryl Glycine but no 2MBG
Plasma amino acid profile
Elevated glycine and glutamine
Acyl-Carnitine profile
Enzyme Activitiy
Can measure enzyme in fibroblasts
Useful prenatal when measuring enzyme activity from amniotic fluid, CVS, amnio
Treatment
Outcome normal if detected and treated before severe symptoms
Acute
Hemodialysis
Protein restriction
IV glucose and lipids
IV Carnitine
IV Ammonia scavengers
Glycine supplementation
Chronic
Protein restruction
Oral L-Carnitine, L-Glycine
Avoid decompensation (Infex, surgery, fever, protein)
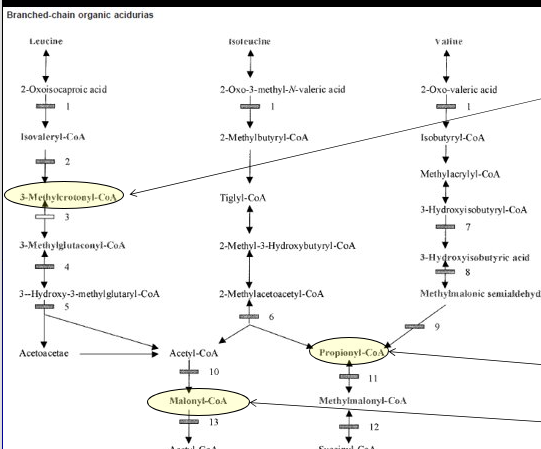
Biotinidase Deficiency (BTD)
Autosomal recessive
Biotinidase (BTD) Gene Mutation
Results in deficient biotin recycling and biotin deficiency
Biotin is a cofactor of multiple carboxylases
BTD Deletion / Duplication Analysie
Symptoms
Profound Deficiency (<10% enzyme)
Normal at birth
Symptoms develop after a few months
Developmental delay, seizures, hypotonia, ataxia, hearing loss, visual problems (optic atrophy), alopecia & eczemetous skin rash
With age develop visual loss, limb weakness & spastic paresis
Partial Delay (10-30% enzyme)
Intermittent symptoms with stress
Hypotonia, skin rash, hair loss or any of the above symptoms
Symptoms irreversible once present
Diagnosed via elevated C5-OH Acylcarnitine but many other conditions cause that
ABG, CMP, Ammonia, CBC
+/- High AG metabolic acidosis, elevated ammonia, low glucose
Urine Organic Acid Analysis
3-B-OH-isovaleric,3-methylcrotonic, 3-OH-propionic, Methylcitric, 3-OH-butyric acids, Acetoacetate, Propionyl glycine, Tiglylglycine
Acyl-Carnitine Profile
Elevated C5-OH acylcarnitine
Enzyme Activity
Can assay in blood (serum/plasma), if normal then it’s Holocarboxylase deficiency, useful for prenatal / preimplanation as can be measured in amniocytes
Treatment
Normal outcomes if detected and treated before symptoms, the best out of all organic acidemias
If after, some symptoms irreversible
Rarely Severely Acidotic or Hyperammonemic
May occasionally need sodium bicarbonate
May occasionally need ammonia scavengers
Institute Biotin therapy immediately
Chronic Treatment
Biotin therapy
No protein restriction or other treatment necessary
Avoid raw egg whites (avidin protein binds Biotin)
3-MethylCrotonyl-CoA (3-MCC)
Autosomal Recessive
3-Methylcrotonyl-CoA Carboxylase (3-MCCC) Subunit 1 or 2 Genes (Biotin is cofactor)
A Leucine metabolism pathway defect
Symptoms
May be asymptomatic
Some have episodic liver dysfunction, hypotonia, hypoglycemia
May cause developmental delay and seizures
Diagnosis
Newborn Screen & acyl-carnitine profile = High C5-OH (if only NBS may be maternal)
High Organic Acids: High 3-Methylcrotonylglycine and 3-hydroxyisovaleric acids
Treatment
Restrict Leucine, supplement with LEU free metabolic formula
Carnitine Supplementation and Biotin supplementation (often responsive)
Prognosis
May have no symptoms or if symptomatic may respond to Biotin
If symptomatic but not Biotin responsive prognosis may be poor
3-Hydroxy-3-methlglutaryl (3-HMG) CoA Lyase Deficiency
Autosomal Recessive
3-HMG-CoA Lyase (HMGCL) Gene Mutation
Symptoms
Unknown prognosis
Another Leucine metabolism pathway defect
May be asymptomatic until decompensation, minimal acidosis in decompensations
Liver Dysfunction “Reye Syndrome” with hyperammonemia and hypoglycemia
Diagnosed via acyl-carnitine profile
High C5-Hydroxyacylcarnitine (C5-OH)
High Organic Acids: 3-hydroxy-3-methylglutaric, 3-methylglutaric, and 3-hydroxyisovaleric acids
Treatment
Restrict Leucine, LEU free metabolic formula
Carnitine Supplementation
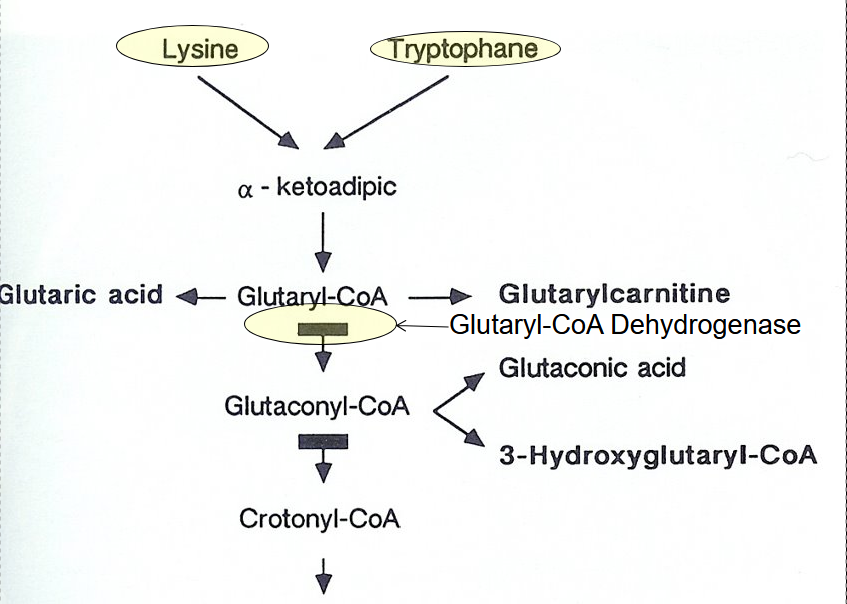
Glutaric Acidemia Type 1 (GA1)
Autosomal recessive
“Cerebral” Organic Acidemia
Glutaryl-CoA Dehydrogenase (GCDH) Gene Mutation
Deletion / Duplication analysis
Symptoms
High AG metabolic acidosis, elevated ammonia, low glucose, elevated glutaric acid and 3-hydroxyglutaric acid
Often normal at birth or only macrocephalic
Symptoms begin a 2 years
May start with neurologic decompensation
Often precipitated by fever, illness, surgery, metabolic stress
Stress-induced encephalopathy
Ataxia
Epilepsy
Myoclonus
Extrapyramidal symptoms
Stroke like symptoms
Hypotonia, irritability, motor delay, dyskinesias
Diagnosed via
ABG, CMP, Ammonia, CBC,
+/- High AG metabolic acidosis, elevated ammonia, low glucose
Plasma and Urine Acyl-Carnitine Profile
+/- Elevated C5-DC (glutaryl) acylcarnitine
Urine and Plasma (and CSF if available) Organic Acid Analysis
+/- 3-OH-glutaric acid and glutaric acid
Enzyme Activity
Fibroblast enzyme assay
CT/MRI:
Cerebellar & cerebral atrophy, basal ganglia infarct & hemorrhage
Treatment
Reverse/Prevent catabolism when sick
Portein free calories (IV dextrose and intralipids)
Dietary modification
Low lysine and tryptophan diet
Medications
Riboflavin B2 cofactor
Carnitine helps bind excess glutaric acid
Avoid valproate (binds carnitine)