Intro to Cancer Genomics
1/20
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
21 Terms
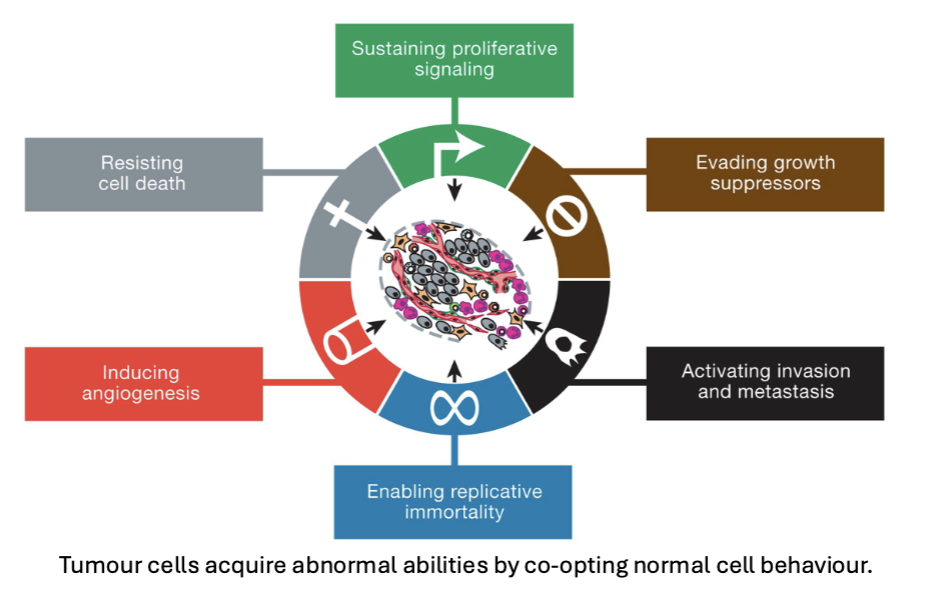
Hallmarks of Cancer
tumor cells acquire abnormal abilities by co-opting normal cell behavior
Karyotyping of Colorectal Cancer Cell Lines
numerical and structural chromosomal instability
translocation b/w chromosomes
extra chromosomes (genome may be doubled)

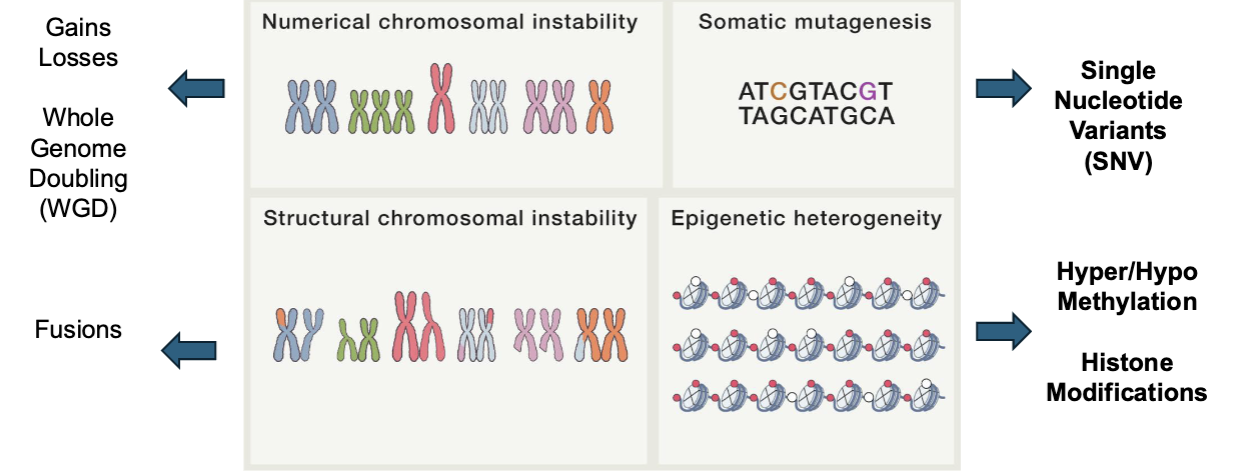
Cancer is a Genetic Disease
genomic and epigenomic alterations
somatic copy number alternations (SCNA)
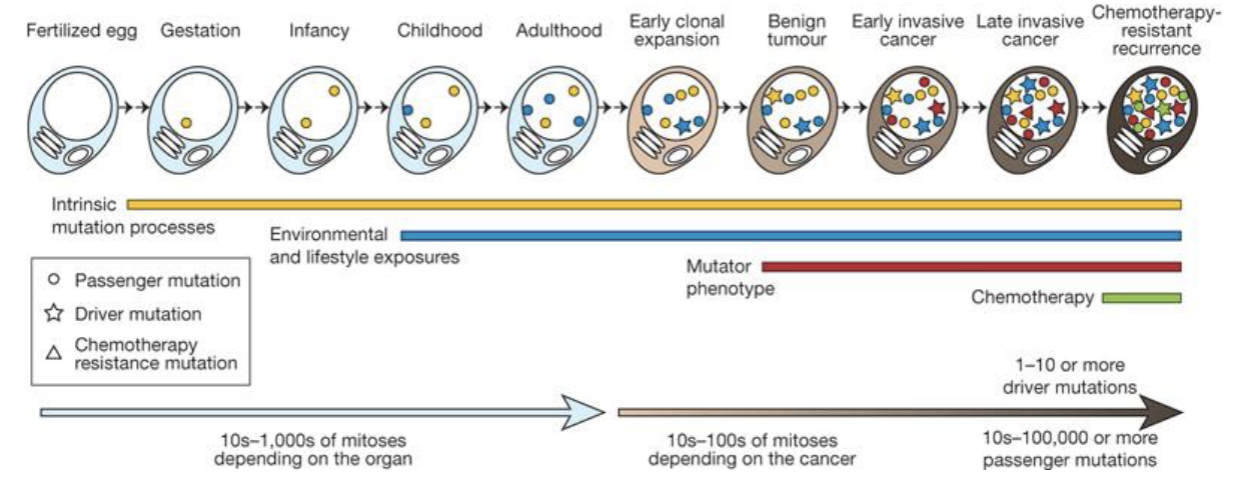
Cancer Cells accumulate somatic alterations over time
chemotherapy might induce mutations over time
mutations might break down cell functions
mutations in specific places might cause cancer
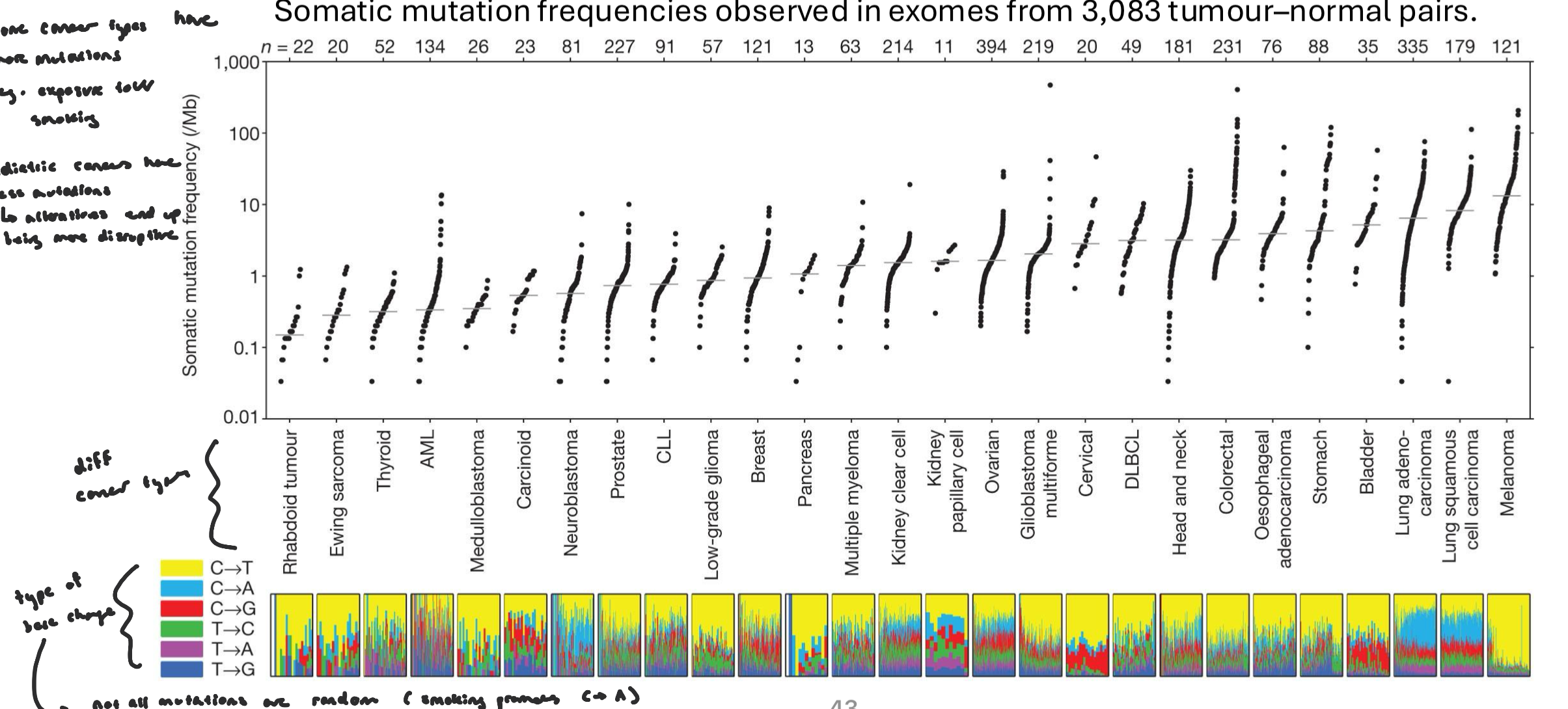
Mutation Burden
mutation burden varies by cancer type, exposure, age of onset, DNA repair ability, etc.
some cancer types have more mutations (e.g. exposure to UV/smoking)
pediatric cancer have less mutations, but alterations end up being more disruptive
Cancer Genomics Pipeline
comparing to person’s own blood instead of reference blood (for detecting somatic mutations)
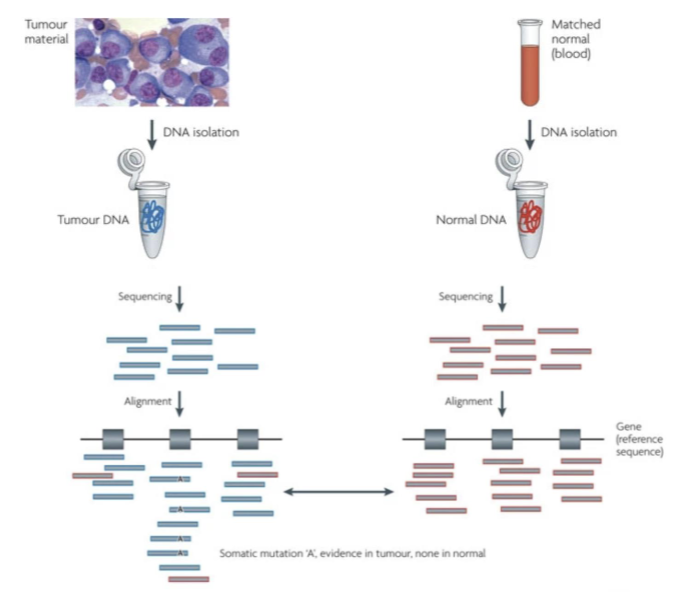
Cancer Genomics Pipeline: nf-core/sarek
nf-core/sarek is a workflow designed to detect variants on genome sequence data
can work on any species w/ a reference genome
can handle tumour/normal pairs
built using nextflow (a workflow tool)
uses Docker/Singularity containers making installation trivial and results highly reproducible
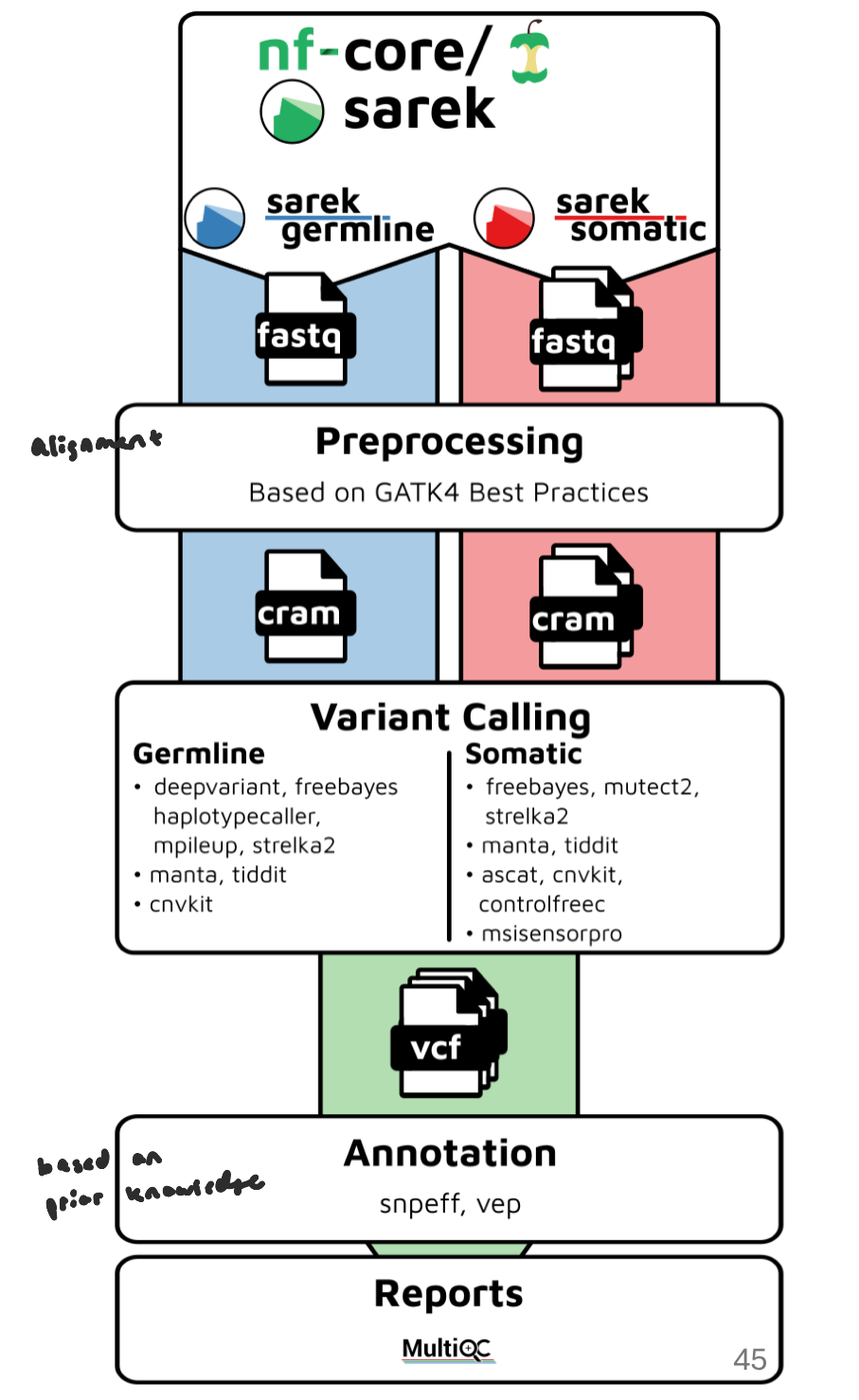
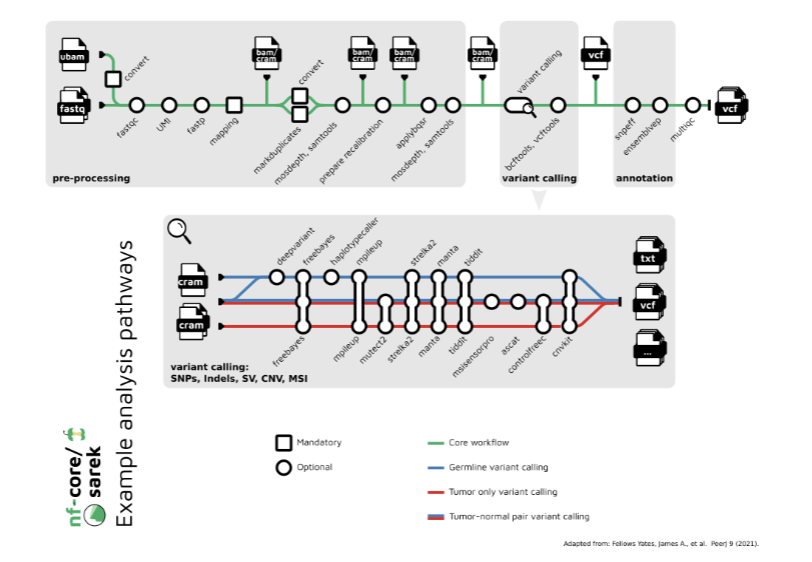
nf-core/sarek Overall Workflow
input raw sequencing files (FASTQ) or pre-aligned BAM files and reference genome
Align reads to the reference genome
Sort BAM files and mark duplicates
Call variants: identifying positions in a genome where a sample’s DNA sequence differs from the reference genome
Variant Filtering: Remove low-confidence variants
Annotation: translates raw genetic differences into biologically and clinically interpretable information.
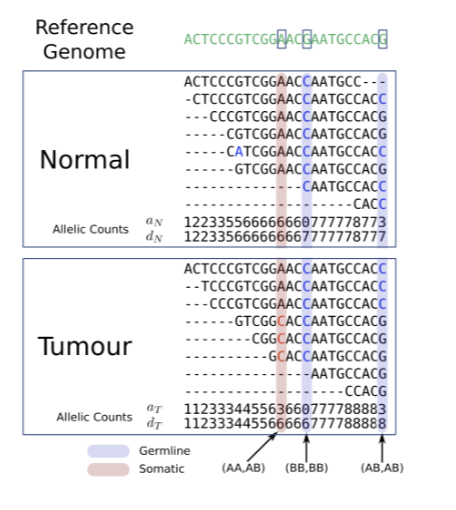
Calling of Single Nucleotide Variants
germline mutations are also detected (we ignore); only looking at somatic mutations
sequence both samples (tumor +normal DNA)
align reads to reference genome (generate BAM files for tumor and normal)
compare at each genomic position
tumor = variant, normal = no variant → somatic mutation
tumor = variant, normal = variant → germline variant
statistical modeling: evaluating read depth, variant allele fraction, base quality, tumor purity
estimating the probability that the variant exists only in tumor and not sequencing noise
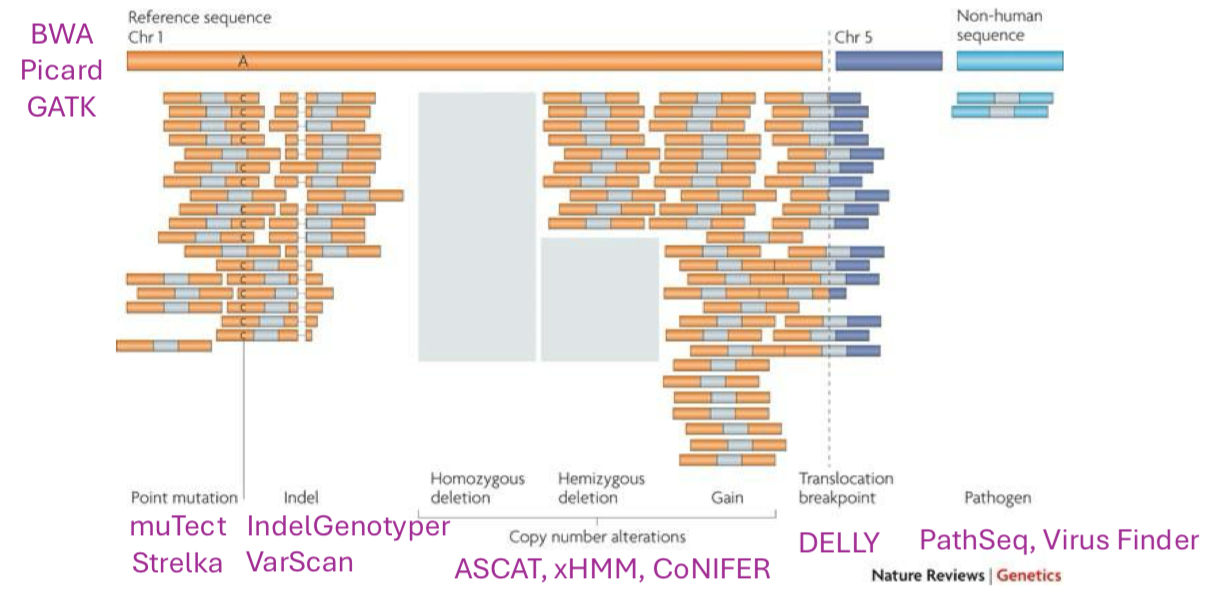
Cancer Genome Variation: Sequencing Read Alignments
multiple types of cancer genome variation may be inferred from sequencing read alignments
Sample Purity on Coverage
if a sample was completely pure, variants are detectable at low coverage (don’t have to cover genome very deeply)
every read at a variant site comes from a cell that actually has the mutation
even a low number of sequencing reads can reliably detect the variant
variant allele fraction is higher in pure samples: heterozygous mutation → 50% of reads show the variant; homozygous mutation →100% of reads show the variant
in mixed samples (tumor+normal), normal cells dilute the signal and the variant allele fraction drops → requires higher coverage to confidently detect variants
Are tumor samples pure?
No! In reality, tumors are a mix of cancer and normal cells
in cancer genomics, we need to consider “tumour content” or “sample purity”
tumour purity (or lack of) makes calling cancer mutations difficult
as such, sequencing a cancer genome requires sufficiently deep coverage, especially for samples w/ low tumour content
increases the chances that alternate alleles (mutations) are detected
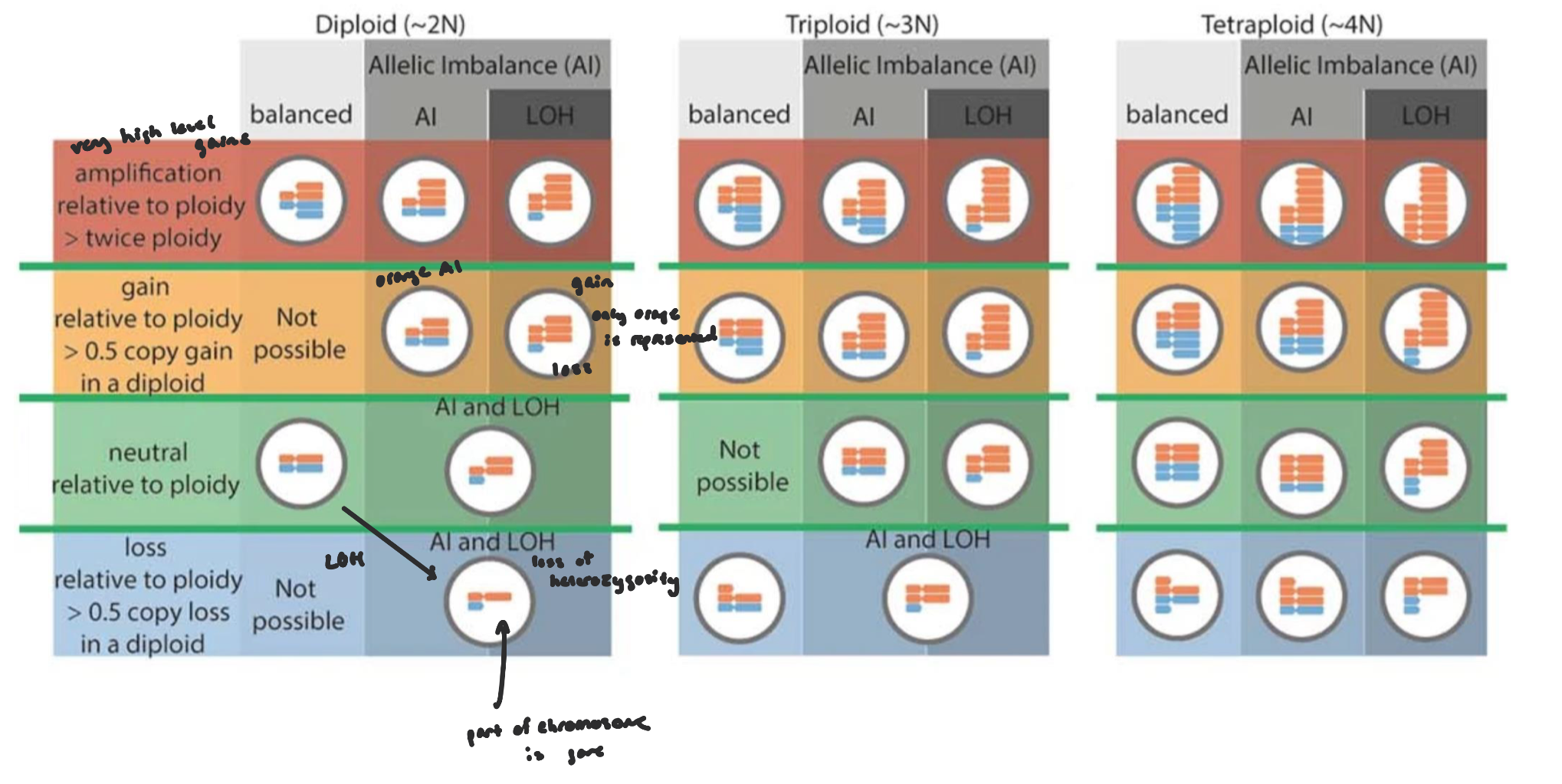
Somatic Copy Number Alteration (SCNAs)
prevalent, acquired genomic changes in tumor cells (not inherited) involving the gain (amplification) or loss (deletion) of DNA, ranging from small segments to entire chromosome arms
changes relative to one’s ploidy (need to determine current ploidy state before determining SCNAs)
major drivers of cancer development, progression, and heterogeneity, affecting oncogene and tumor suppressor gene dosage
Compare tumor vs. normal at the same locus. Gain (amplification) → tumor has more copies than normal. Loss (deletion) → tumor has fewer copies than normal.
Detection: higher coverage than expected → gain, lower coverage than expected → loss
Calling Somatic Copy Number Alterations
most SCNA callers use a read-depth based approach (focus on SNPs)
two main input channels:
Log2 ratio (logR): relative depth between tumor and normal
B-allele fraction (BAF): allelic imbalance, gives the ability to call allele-specific copy number
Combining BAF with logR allows you to see:
Which allele is lost or amplified
If there is copy-neutral LOH (no change in logR, but BAF shows imbalance)
data are segmented into regions of constant copy number (i.e blue lines)
segments are classified into copy number events
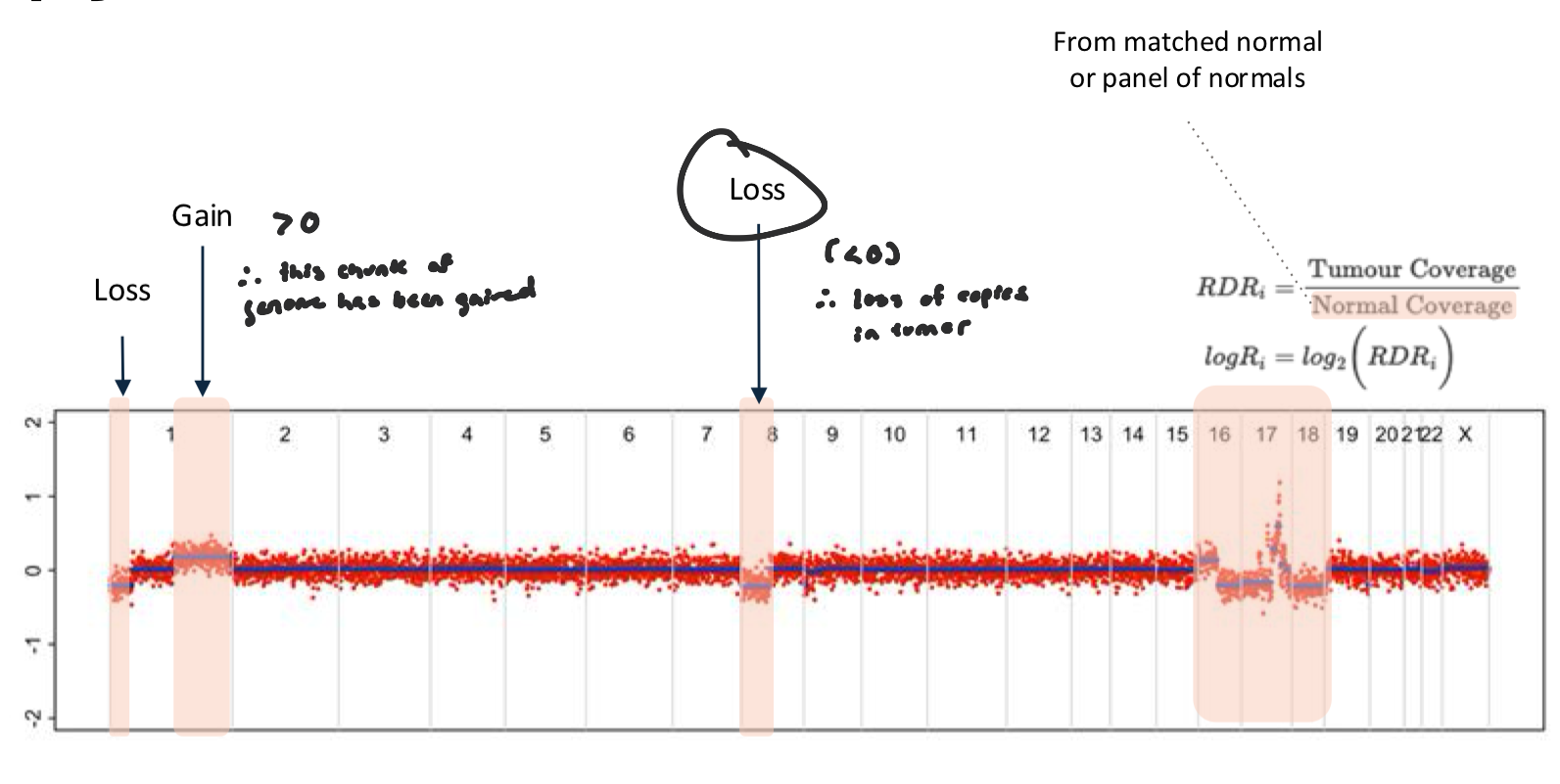
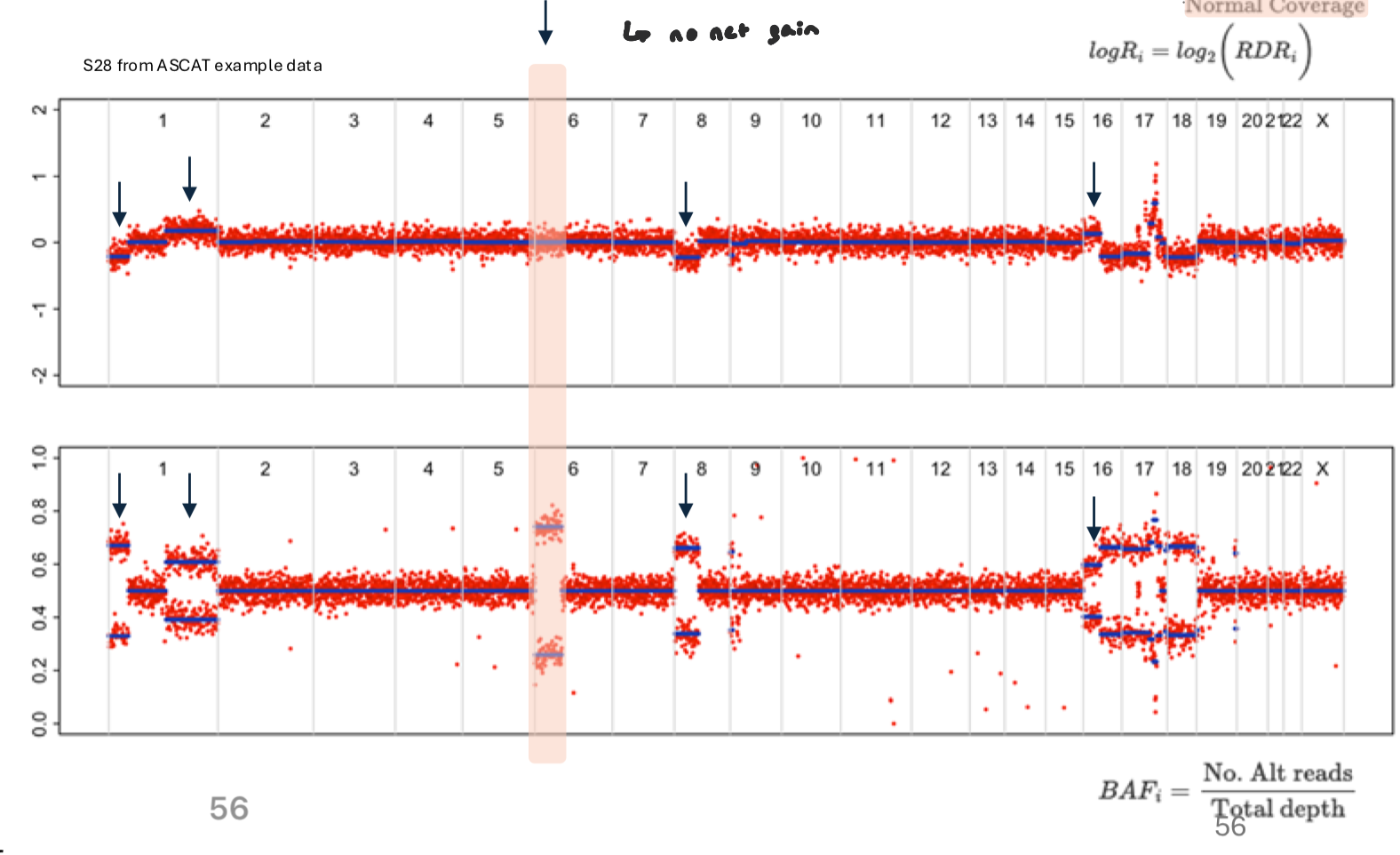
Log2 Ratio Figure
data from this plot is sufficient to say whether there is a change in ploidy (gain or loss)
compares sequencing coverage in the tumor to the normal at each locus
logR > 0 → tumor has more DNA than normal → gain/amplification
logR < 0 → tumor has less DNA than normal → loss/deletion
logR ≈ 0 → no change (copy number = normal)
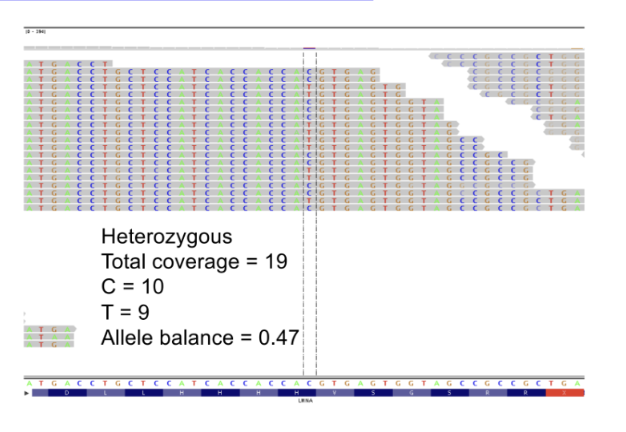
B-allele fraction Figure
fraction of reads supporting the alternate allele at heterozygous SNPs.
see if representation is equall b/w alleles
Normally, for a germline heterozygous SNP: BAF ≈ 0.5 (50% reference, 50% alternate)
allelic imbalance: Copy number changes can shift the balance between alleles.
Loss of one allele (LOH) → BAF → 0 or 1
Gain of one allele → BAF shifts toward 0.33 or 0.66
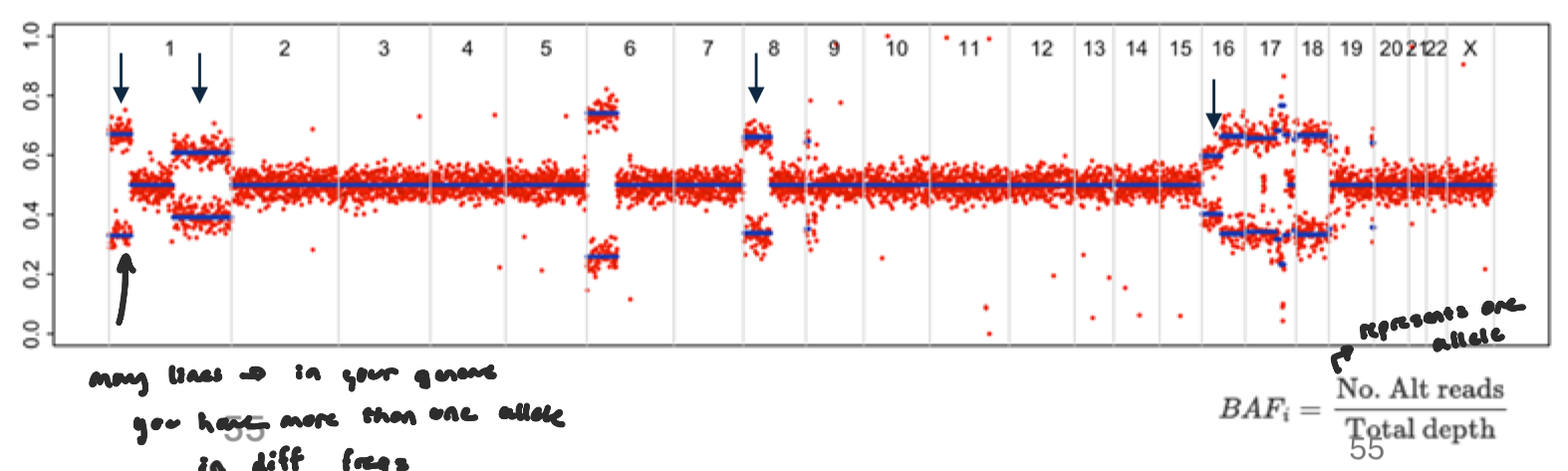
Neutral Loss of Heterozygosity
shift in allele representation, but no visible gain or loss
occurs when one allele is lost in replaced by other allele (still the same amount of copies, but there’s a loss in heterozygosity)
no net gain
wouldn’t show as a change in logR, but in BAF, deviates from 0.5 (allelic imbalance)
Instead of a single band at 0.5, heterozygous SNPs split toward 0 and 1, forming two “allele-specific” clusters
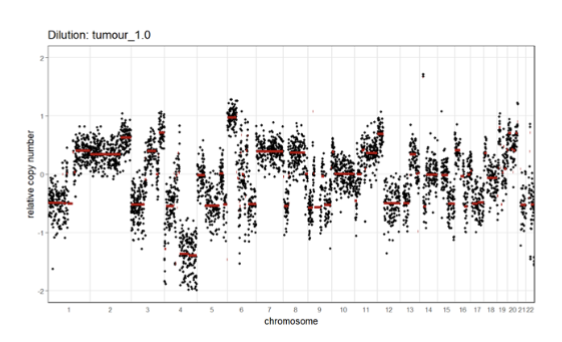
Tumor Purity on SCNA Signal
tumor purity affects SCNA signal
lower purity means fewer cells harbor the SCNA events (harder to see SCNAs)
weaker signal (signal to noise ratio is decreased)
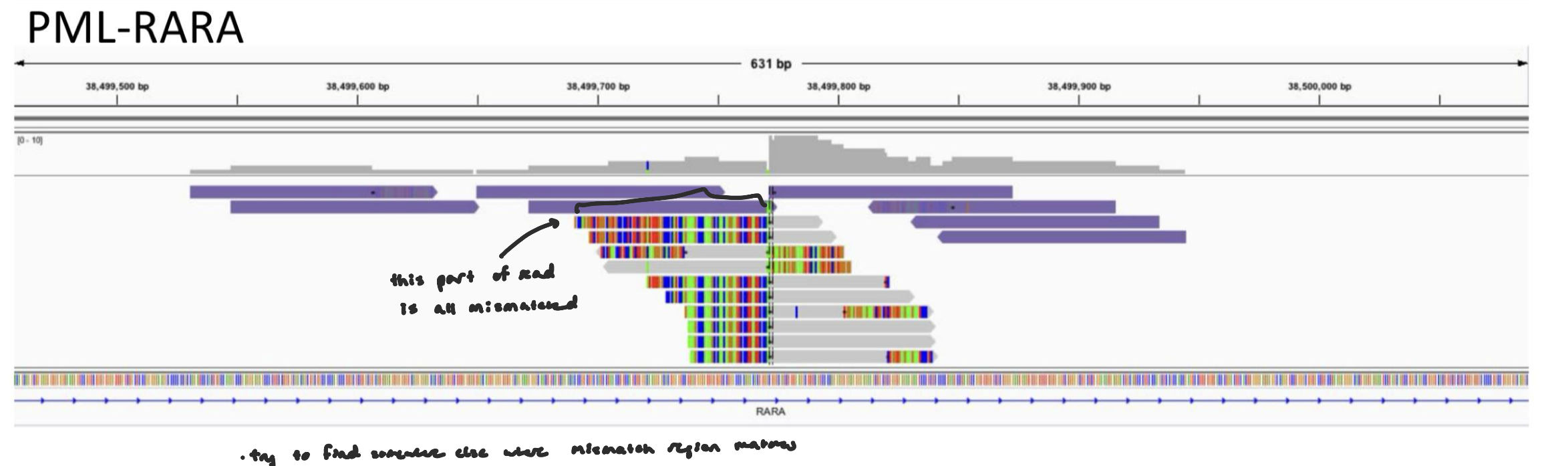
Translocations: Soft-Clipped Bases
A translocation occurs when a segment of DNA from one chromosome is moved and attached to another chromosome.
When reads are aligned to the reference genome:
Sometimes only part of a read aligns and the remaining portion does not match the reference at that location.
That unmatched portion is called soft-clipped.
translocations can be detected from soft-clipped bases
Soft-clipped bases can represent sequence that belongs to a different genomic location — potentially another chromosome.
Translocations: Soft-Clipped Bases FIGURE
Rearrangement Complex
rearrangements can be highly complex and detectable at a base pair resolution
genomic rearrangements include: translocation, inversions, deletions, etc.
in cancer, rearrangements can involve multiple chromosomes, fragmented DNA segments, etc.
tumors can show multiple breakpoints close together, chains of rearrangements, regions shattered and stitched back tgt, copy number changes intertwined w/ structural variants
With high-throughput sequencing: Split reads can pinpoint the exact nucleotide where DNA breaks and rejoins.
We can identify the precise breakpoint sequence.