Anticancer - oncogene specific therapies
1/48
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
49 Terms
Designing drugs to target cancer
Identify the difference between cancer cells and normal cells:
Molecular events
Phenotypic changes, as a consequence of molecular events
Current drugs we have target these 2 areas:
Cytotoxic drugs usually target the faster-growing nature/phenotypically different cells
Molecular target drug targets the changes that take place leading to cancer and tend to be more specific
Cancer cells rely on oncogenes and the production of their “surviving signals” —> known as oncogene addiction
if we remove these surviving signals, this is one of the methods to selectively treat cancer
But cancer cells will try and reestablish and use other surviving signals, known as resistance
For some patients, we don’t know the molecular mechanism that leads to their cancer, so treatment can be less specific and more challenging
Cell culture
Use 3D chromatogel culture to mimic the cell in vivo better compared to 2D culture
Breast epithelial cells in culture usually look round and symmetrical (top right); however, the cancerous cells are asymmetrical with lots of protrusions (bottom left)
The other two samples are not normal either: the one on the top right has a tumour suppressor gene knockout while the bottom left has an activation of an oncogene
Shows that there is more than one types of mutations needed to induce cancers and more than one type of cancer
For example: If you wanted to treat the bottom left sample you could simply knock out the oncogenes as they’re reliant on this
Oncogenes and Tumou supressor genes
Two types of cancerous genes
Oncogenes – genes which drive the tumour phenotype expression that the cells are reliant on
Tumour suppressor genes – prevent the formation of tumours
To fully transform into a cancerous cell, you need two events to fully transform cells from normal to cancer cells
This also allows for selective targeting of cancer cells
Selective Toxicity
Diseased cells have similar properties to normal cells (especially those in bone marrow, hair, GI mucosa and skin)
If we target these fast-growing sites we will have consequences in the form of side effects:
If we target bone marrow cells will impact the immune system —> anaemia or susceptibility to infection
If we target hair cells will cause hair loss
We need to look for “windows of opportunity” where cancer cells are especially vulnerable to avoid damage to our cells
Precision medicine
Instead of grouping the patients by the organ of their cancer, can group patients by their molecular signature, indicative of the driving events leading to the cancer
Can create more efficacy and selective toxicity
Can also apply this to other diseases, such as diabetes
How can we selectively target cancer cells
PTEN tumour suppressor genes
PTEN+ cells normal cells with tumour suppressor, PTEN- cancerous cells with no tumour suppressor
Mixed in a 1:1 ratio and can knock down genes individually, can treat the cells with a drug or siRNA library and observe the outcome
The siRNA targets specific mRNA and degrades it creating a knockout of each gene
Can see if the specific gene knockout affects the cancer cells, the normal cells or both
Additionally, you can do this with a drug library instead of knocking out the genes with siRNA libraries
Through this technique was able to identify the gene WDHD1 if knocked out, it selectively inhibits or kills PTEN. Targeting these genes can selectively target this type of breast cancer
Transcriptomic Profiling
Another technique of categorising patients for selective treatment, but instead of wet lab can use bioinformatics
Can group cells from their molecular signatures
Examined neuroblastoma patients and examined the overall survival rate
Can help you categorise the molecular markers into high or low risk cancers
The green profile patients had a high overall survival rate without any treatment —> suggests that their cancer type is low risk
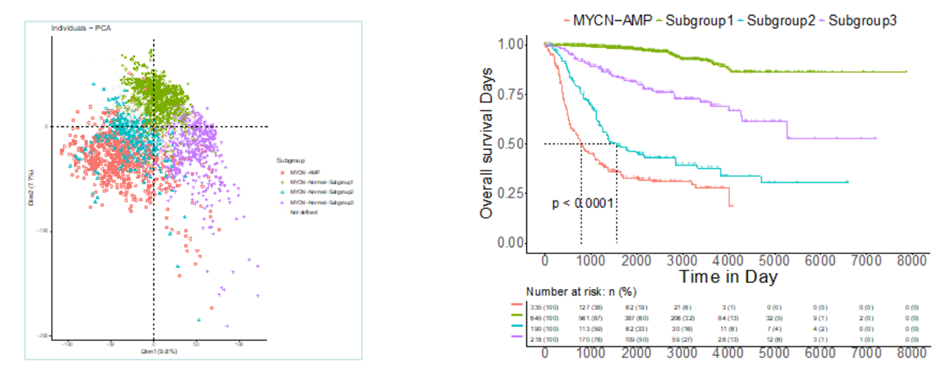
Can also create maps which hotspot the molecular events which occur, leading to that cancer.

Red signals indicate oncogenic signals, and were associated with the aggressive, high mortality rate cancers, supporting the idea that we can group patients based off their gene expression profiles
Can then create therapeutic targets which help to specifically target these molecular events that lead to cancer
Success of current treatments
Surgery is not likely a viable treatment method for melanoma as it spreads all over the body in many tumours
Can understand the oncogene activation and block the oncogene pathway to target the cancer cells using a drug
However, after a few rounds of treatment, the cancer returns due to their ability to reestablish surviving treatments (called drug resistance)
Molecular basis of cancer
Normal cell growth and cancer
Cancer cells have altered genomes
Mutations
Oncogenes and Tumour supressing genes
Hallmarks of cancer
sustaining proliferative signals
evading growth supressors
avoiding autoimmune destruction
enabling replicative immortality
tumour promoting inflammation (enabling characteristric)
activating invasion and metastasis
inducing or accessing vasculature
genome instability and mutation (enabling characteristic)
resisting cell death
degrading cellular metabolism
these are some of the commonly observed features of cancer cells which give them the enabling characteristics that enable their development and survival
plasticity of cancer cells
unlocking phenotypic plasticity is a crucial component for the cancer pathogenesis and allows cells to escape normal differentiation programmes —> which gives cancer cells the ability to initiate, metastasise, develop therapy resistance
ways which this occurs:
loss of transcription factors such as HOXA5 allow cells to dedifferentiate
Blocking differentiation, for example, through the expression of SOX10
trans differentitaion where the cell is able to transform into an entirely different lineage
epigentic changes
mutations in gnes which organise chomatin architecture can drive epigentic changes known as “nonmutational programming”
these epigentic changes can drive the cancer to enter drug tolerant states where they reduce proliferation, alter their surviving signals and increase their stress tolerance —> resistance
Can also drive them to reduce drug penetration, drive them to undergo plasticity mechanisms or gneerate intratumoural heterogeneity which is a bet hedging technique which allows cancer cells to adapt rapidly in the prescence of a stressor
Normal cell growth and cancer
Usually, there's a fine balance between apoptosis and proliferation for normal cell growth
In cancer the events of oncogene activation result in more proliferation or less apoptosis
The removal of the tumour suppressor genes prevent these cells from being targeted

Classification of cancers
Neoplasia – excess proliferation without relation to normal growth or repair. Growth may be fast but rarely exceeds that in the fastest growing tissues
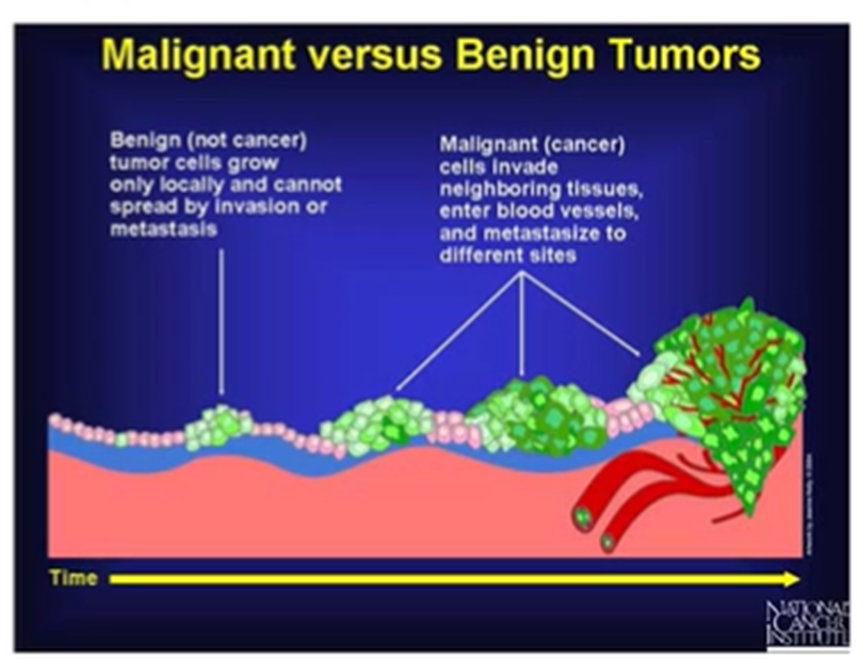
Benign – called a tumour rather than cancer. Proliferate locally and retain characteristics, have a defined boundary.
Malignant – Not encapsulated, ill defined edge, projections extend into surrounding tissues, less well differentiated than the cells of origin. Spread to other sites by invading surrounding tissues
Can also develop from benign to malignant:
Altered genome of cancer cells
Karyotypes can detect the changes in genomes that occur in cancer
Karyotypes of cancer genome have:
Different number of chromosomes —> more or less copies
Translocated DNA across the chromosomes causing mutated chromosome shape
Lost DNA
Chromosomes might start with small chromosome altering events but as the cancer develops accumulate changes leading to chromosome instability

Mutations
Germline mutations
A change in the DNA sequence that can be inherited from either parent
Occurs in all the cells
Somatic mutation
A change in the DNA sequence in cells other than sperm or egg
The mutation is present in the cancer cells and its offspring but not in the patient's healthy cells
Occurs only in the tumour cells
Most mutations causing oncogene activation will be point mutations, while in tumour supressor genes will be frameshift
Hereditary Predisposition
If generations before you in your family had cancer doesn’t mean you’ll inherit it —> some familial mutations that you will inherit, but only 5% of cancers are hereditary
Loss of tumour suppressor genes will result in a higher chance of cancer —> For cancer to occur you need multiple events and this is one of them
Don’t necessarily have cancer, but they have a higher chance of having cancer later in life as we have 2 tumour suppressor genes, it's likely that you’ll have at least one healthy copy.
Transformation of cells to cancer cells
For normal cells to transform into cancer cells 2 events need to happen (hallmarks of cancer cells)
Activation of oncogene
Inactivation of tumour suppressor gene
This offers the cancer growth advantages compared to other cells
Also creates therapeutic targets —> once we understand these therapeutic events we can target these mechanisms
We won’t target normal cells because they’re not addicted to the oncogenes
Events leading to transformation of cells BRCA
Somatic or germline mutations in the tumour suppressor genes
BRCA important for damage repair in the DNA, therefore loss of BRCA results in high chance of breast or ovarian cancer
Once the tumour cells have lost BRCA they can rely on POP to repair the damages —> we can target POP with AntiPOP inhibitors
In most cases those with BRCA mutations have no family history —> developed over their lifetime.
Origins of cancer
Most tumours require multiple mutations, but this can vary
E.g: white blood cell cancers only need one mutation whereas epithelial cell cancers need multiple —> this is because for an epithelial cell they need multiple mutations to develop the properties to proliferate, then grow out of the space etc. however, white blood cells don’t need many events as they’re already highly proliferative and are already circulated around the body

Car Crash Theory
Oncogenes are like the engines which drive the cell cycle (the car)
Tumour suppressor genes are like brakes that prevent the crash – in the absence, these mutations are not repaired and allow them to be passed onto other cells and accumulate
For cancer you need both events
Events causing colon cancer (adenocarcinoma)
Loss of APC (tumour suppressor) involved in Wnt signalling activates the oncogenic pathway
At this point is not a cancer, only a tumour as its not yet invasive
Next is the activation of KRAS is the activation of the oncogene and drives the progression from an early adenoma to a late adenoma
Next is the loss of p53 which allows the tumour cell to acquire multiple mutations to drive the cancer.
Progression of late adenoma to adenocarcinoma which has invasive properties
We need the loss of the tumour suppressor early on to allow the mutations to accumulate

p53
Can decide whether damage can be reversed or repaired —> whether the cell survives or dies
Most cancers actually have increased rates of apoptosis but due to the sheer amount of proliferation the net result is growth
Loss of function p53 results in more cell survival, whereas overactive p53 results in unnecessary cell death, resulting in ageing, neurodegenerative disease or radiation sickness
Elephants have 40 copies of p53 while we only have 2, so despite having more cells can better control the progression of cancer
p53 is only activated under severe stress which is why the mutations have been allowed to accumulate up to this point. Additionally, p53 favours DNA repair and cell cycle arrest over apoptosis —> when it’s lost the cell cycle no longer arrested and mutations passed onto daughter cells
Examples of key oncogenes - Ras
Mutations in Ras responsible for a majority of cancers
3 isoforms of ras – k,h,m Ras
Ras activity is controlled by GDP or GTP bound
When GTP bound it will be able to bind to Raf or PI3K
PI3K causes activation of AKT
While activation of Raf causes activation of ERC signalling pathways
Ras can also be activated by upstream factors like EGFR
Very regularly mutated pathway in cancer
EGFR highly mutated in lung cancer
Raf oncogene for melanoma
K Ras oncogene in pancreatic cancer

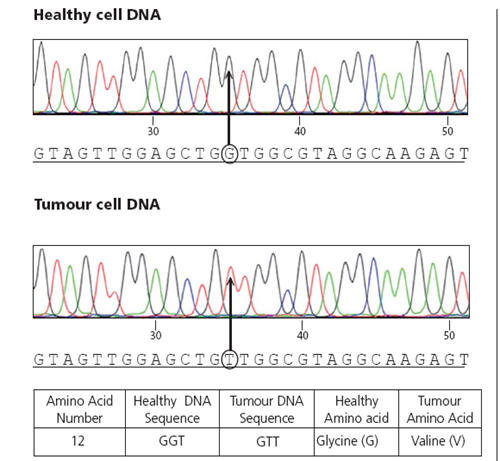
Identifying mutations using sequencing
Each colour represents a nucleotide, can compare healthy DNA to mutated DNA
Can then figure out which amino acids in the proteins will be changed
For example at position 12 a glycine is changed to a valine, which will lock the Ras into the GTP format due to a higher affinity –L> doesn’t require upstream signalling for activation
Called a G12V mutation as its G—>V at position 12
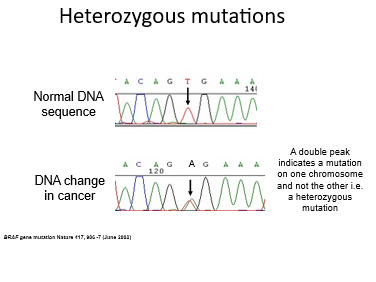
Heterozyous muations
As we have two copies of the genes, we can have a mutated gene and a normal one which looks like this: representing a normal and mutated allele
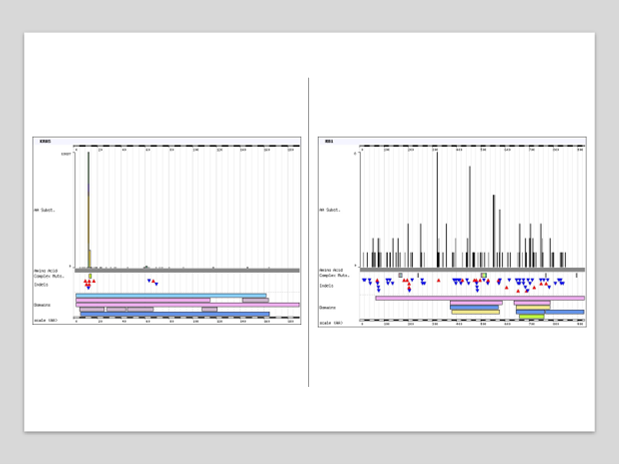
How common are Kras mutations
There are specific hotspot mutations which are more common
These are in position 12, 13 and 61
The particular mutations will create a unique binding site in the tumour KRAS which we can target
Therefore, the wild-type Ras will be unaffected —>selectivity
Impact of Kras mutations
Growth signals, released when cells are damaged stimulate Kras
Kras activity is important for the proliferation of cells when they’re damaged
Ras has GAP, an enzyme which can inactivate it by exchanging GTP for GDP
Comparison of Kras and RB1 tumour supressor gene
Mutations in the RB1 (tumour supressor) are a lot more evenly dispersed, don’t necessarily have specific hotspots
Also deletion mutations a lot more common in the tumour suppressor genes
On the right is RB1 and left is Kras mutations
For the tumour suppressor genes, as long as there is a loss of function of the tumour suppressor gene, it will be inactive. However, mutations in the oncogene must be more specific as it must affect the GTP binding site.
Carcinogens
Introduce mutations to key genes
Can activate oncogenes
Herb treatments
Some can be carcinogens
Unique mutation patterns in Asian cohorts linked to carcinogens in herbal medicines
UV light
Linked to malignant melanoma
Signature C—>T or G—>A mutations
If these DNA changes occur in critical genes like BRAF this can lead to inappropriate cell growth and melanoma
Tobacco smoke
Tobacco smoke carcinogens such as PAH (polycyclic aromatic hydrocarbons)
Signature G—>T mutations
HPV
Linked to cervical cancer
The most high risk (HPV16) the viral protein can activate oncogenes and cause inactivation of tumour suppressor genes
Eg: E6 viral protein produced by HPV virus can promote degradation of p53
Oncoproteins integrated into the cells
Now a vaccine to reduce cases of cervical cancer
Diet and obesity
Linked to bowel and stomach cancer
Inflammation from the diet linked to damage to the cells lining the stomach/bowel
Viral and cellular oncogenes
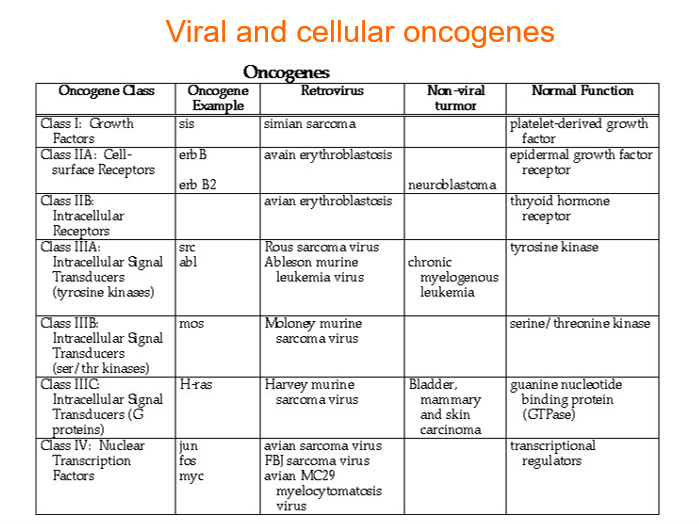
Viral and cellular oncogenes
Some cancers linked to viruses, were previously thought that it was caused by the oncoproteins from the virus causing cancer
Cellular oncogene will only be activated when necessary but when infected with viral infection the virus controls the expression of oncogene —> may cause loss of function of overexpression
some viruses can also cause cancer due to the chronic inflammation which leads to increased ROS which damages cells and induces mutations
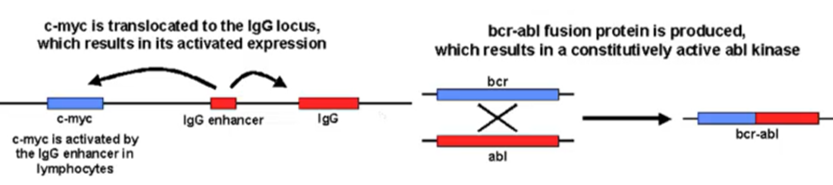
Translocation - Example C-Myc
CMyc an important oncogene which is controlled by its own promoter
Will only be expressed when it needs to proliferate
A single translocation event can cause Myc to be under control of IgG promoter rather than its own promoter
IgG more regularly produced than Myc
Targeting cancers
Loss of tumour suppressors gives cancer cells growth advantages:
Sustaining proliferative signals
Evading growth suppressors
Depending on which molecules provide these advantageous signals, we can create selective targets
Very challenging for us to restore tumour suppressor functions, as there are often more mutations in the tumour suppressor gene and you don’t know which mutation is causing loss of function
Oncogenes are good targets as they often have mutation hotspots —> relatively easy to design a drug to stop oncogene signalling
These targets can be applied for different types of cancer —> targeting based off the driving mutation not location
Specific examples of gene mutations
BCR-ABL
EGFR
BRAF
PD-1/PDPL-1
VEGF
Precision medicine
BCR-ABL oncogene
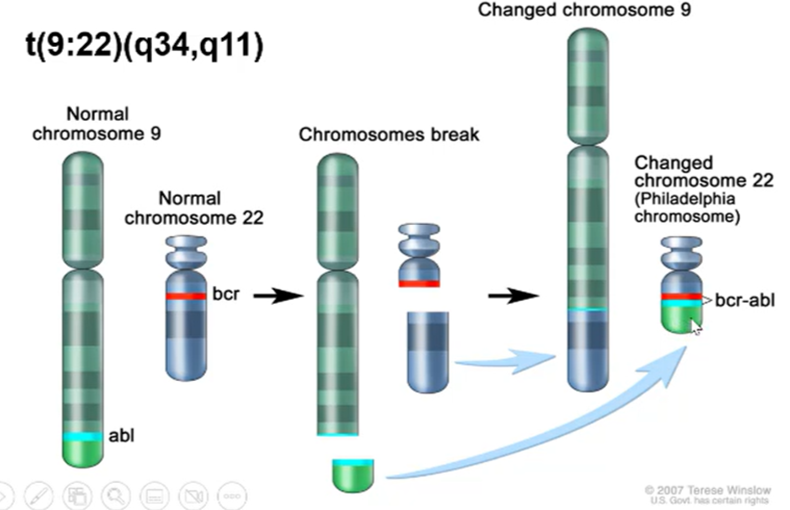
Results in a Philadelphia chromosome eliciting a fused protein
BCR gene is usually present in the q11 region of chromosome 22, ABL gene usually present on q34 region of chromosome 9
This translocation is written as t(9:22)(q34,q11)
T before brackets indicates a translocation
Q indicated that the mutation is on the long arm, p indicated the short arm
forms a fused protein which drives chronic myeloid leukaemia in all cases and acute lymphocytic leukaemia in 10% of cases
When you have the fused protein present there’s a coiled-coil domain which is always dimerised, leading to constant activation of the cell cycle
The wild type proteins are essential for the normal function in the cell but the difference is that signalling is only activated when required.
Most effective drugs against this mutation is the tyrosine kinase inhibitors —> this inhibits the ABL protein allowing for selective destruction of the cancer cell
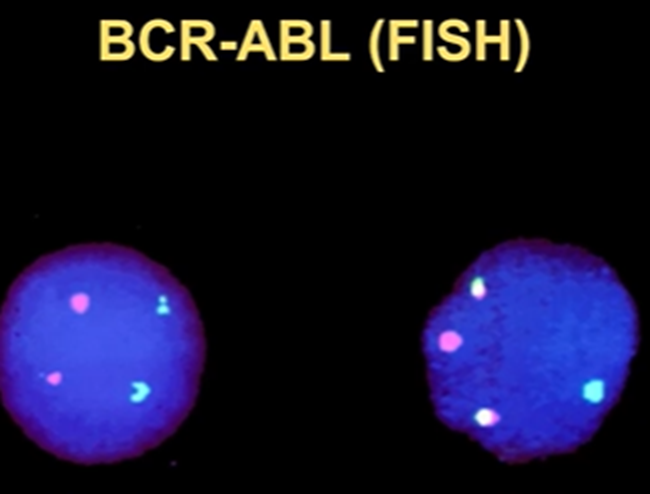
Visualisation of fused proteins using FISH
These fused proteins can be visualised by FISH (fluorescent in situ hybridisation)
The green tag indicated the ABL protein, and the red indicates the BCR
In the normal situation, the two colours are distinct, but when there’s a translocation, the colours are mixed
Useful for diagnosis/ identifying mutation occurring in the cancer
Targeting these proteins
Imatinib is a tyrosine kinase inhibitor
Any drugs which end in -ib means it’s a small molecule inhibitor
Holds the activation loop in the inactive conformation which stops signalling of both w/t and cancerous ABL
Kinases are relatively easy to inhibit so tend to be most successful anticancer agents
Mutations and persistence of cancer
However, cancer cells have an unstable genome so can relatively easily acquire mutations
If a mutation occurs in the binding site for the imatinib, then this drug is no longer effective, and the cancer will persist
After a few rounds of treatment around 1/3 of patients will develop a mutation. Common mutations which change the structure include: T315I (most common), E255K, H396P
Other mechanisms of resistance include
Use of an alternative pathway to create the survival signals
Use a different mechanism to activate downstream events
Amplify the ABL to overcome the inhibition
2nd generation drugs
Bind more tightly to the binding site
Can't cope with the T315I mutation —> both 1st and 2nd generation are not successful at all
All of them successfully bind to wild-type ABL
Examples: Dasatinib, nilotinib
3rd generation
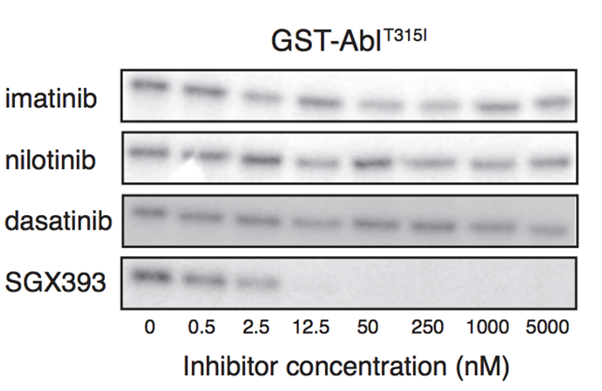
SGX393 a 3rd Gen BRC-ABL inhibitor
We can see from Western blots that this is the only drug which can inhibit mutant T315I
Since this is the only drug which works against this specific mutation, it’s essential to know which kind of mutation is causing the patients cancer.
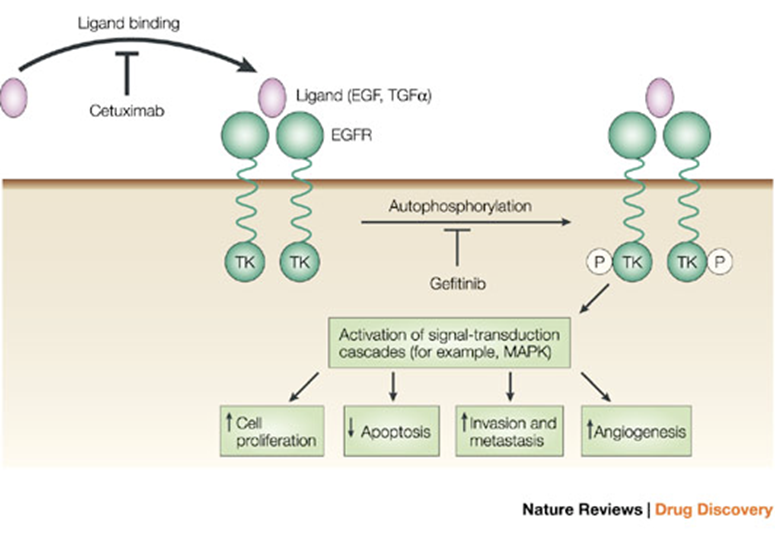
EGFR as target
Epidermal Growth Factor Receptor
Important signal for tissue repair and wound healing
Controlled by ligand binding to the receptor, causing a dimerisation which activates the Ras signalling pathway (which goes onto activate Raf and Erg)
Unlike BCR-ABL, EGFR mutation is mainly driven by point mutations
Illicits lung cancer
Cetuximab prevents the ligand from binding to EGFR, however, this will not work in cancer types where the mutation is in the EGFR causing it to be constantly dimerised
Useful in cancers which are caused by amplification of ligand (EGF) or overexpression of the receptor
Gefitinib is another drug which binds to the centre of EGFR to prevent the signalling —> successful in cancers which are caused by mutations in EGFR which result in constant dimerisation
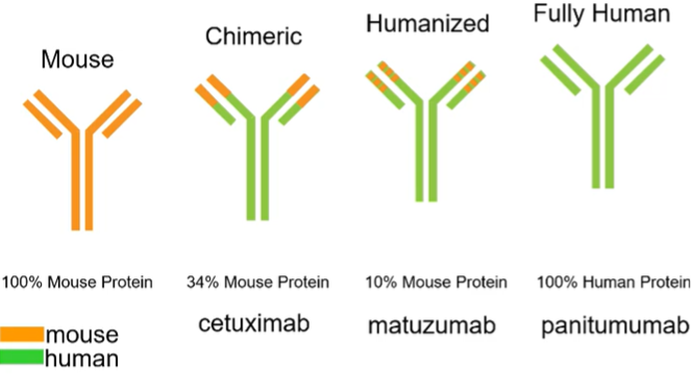
Monoclonal antibodies as targeted therapy
Can be used in some cases E.g: EGFR
Need to humanise the antibodies so they’re more stable
However, again, when we tray and treat the mutation the cancer will try to develop other mechanisms to overcome this
T790M mutation accounts for 55% of mutations resulting in the re-establishment of this pathway.
Other mechanisms to re-establish this pathway
BRAF: Overexpression of Raf (downstream)
MAPK1: downstream of the Ras
PI3K: downstream of EGFR
MET: upstream of the Ras pathway
HER2: mechanism to activate Ras signalling pathway (upstream)
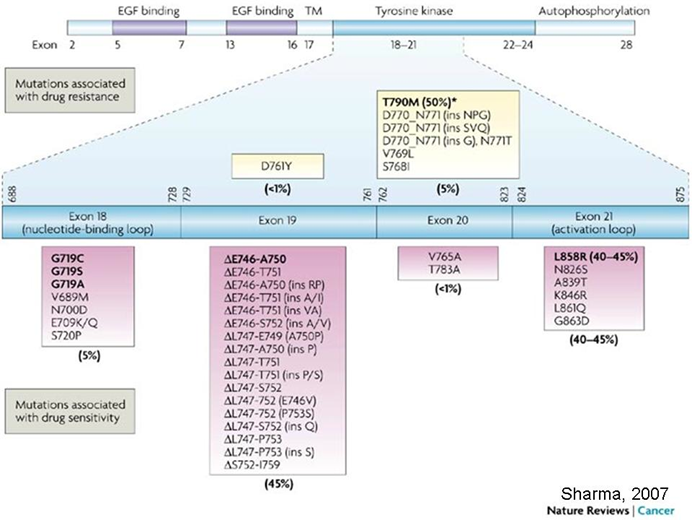
EGFR mutations
Big protein, mutations frequently occurring in exons 18,19, 20,21
examining where the mutation lies is important for determination of treatment —> sequencing of EGFR necessary for this
Below the diagram is the index of mutations associated with drug sensitivity so can give anti-EGFR treatment
L858R mutation In exon 21 is an activation mutation
Above the diagram is the index of drug resistance mutations, which usually happens after several cycles of treatment.
T790M mutation is a drug resistance mutation
How can we treat patients with resistance induced by first gen treatments?
In EGFR mutations causing cancer L858R and E746-A750 mutations are oncogene activation mutations while T790M is a resistance mutations
First-generation treatment causes this mutation to arise, second generatuion drugs such as Afatinib and Neratinib are also ineffective against this type of cancer.
Typically second generation EGFR TKIs form irreversible covalent binds with the ATP binding site of EGFR as well as other members of the HER family of receptors
Third-generation drugs on the other hand, such as AZD99291, HM61713 and Co-1686 will be effective in approximately half of cases.
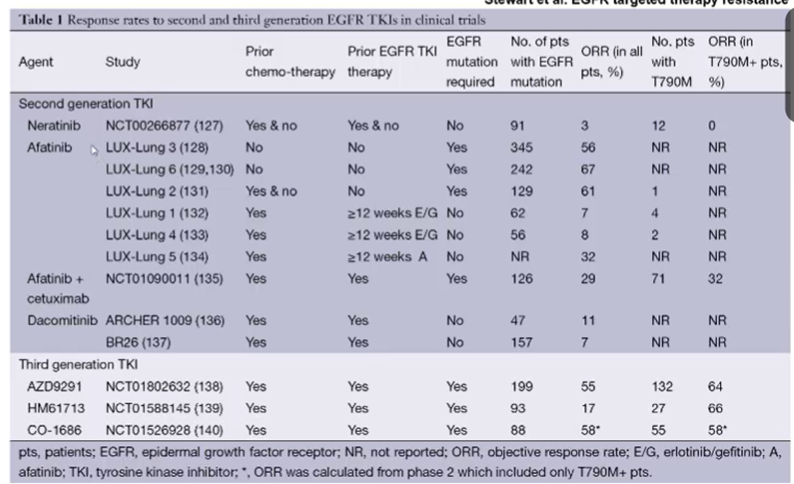
Vermurafenib
Small inhibitor targeting BRAF V600E mutation
Can no longer activate the MEK/ERK pathway
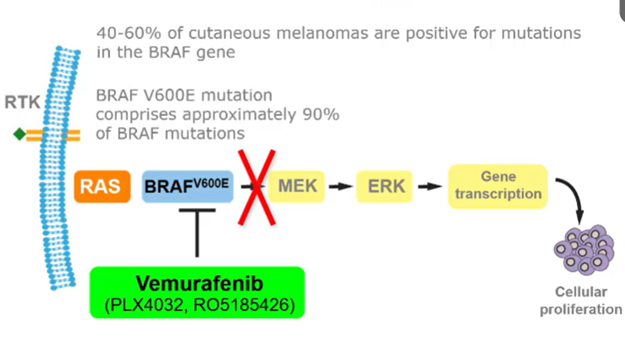
However, the cancer cells can reestablish the ERK surviving signals by:
mutations in MEK1 and MEK2
Upregulation of genes that can activate MEK
Overexpress another isoform of BRAF
Increase the expression of another RAF called CRAF
Introduce mutations in RAF genes/other upstream events
Once we understand these alternative mechanisms we can come up with another drug to treat them
Unlikely that we can treat the cancer completely but can keep the cancer at a low level, so is less of a burden to the patient —> cancer now considered a chronic disease
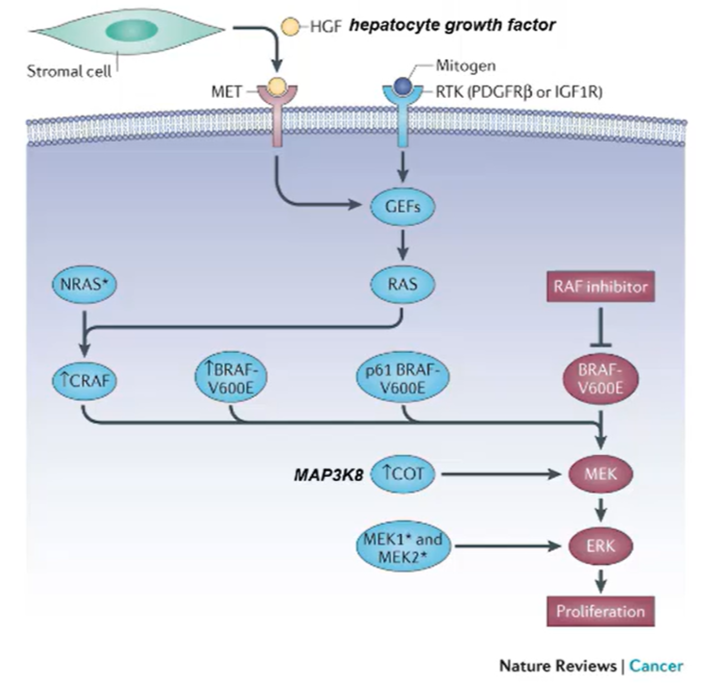
Immunotherapy
2 major checkpoints in our body
CTLA-4 in the lymph node
PD-1 in the tumour
These two signals act as surviving signals for cells and confer protection against the immune system
This interaction between PD-1 on the T cells and PDL-1 on the tumour cells dampens the immune response and protects the tumour against the immune system
However, we can create anti PD-1 antibodies to block this signal and selectively kill the tumour cells.
Healthy cells are spared from this because their surface antigens are not recognised by the immune system, although this is not entirely selective
Anti-PD01 treatment shown better efficacy than the chemotherapeutics.
VEGFR
Tumour outgrowths creates a hypoxic environment which triggers the expression of VEGF (vascular endothelial growth factor) which will stimulate the creation of new blood vessels to supply nutrients and oxygen
VEGF antibodies removes the nutrient supply to the new cancer cells
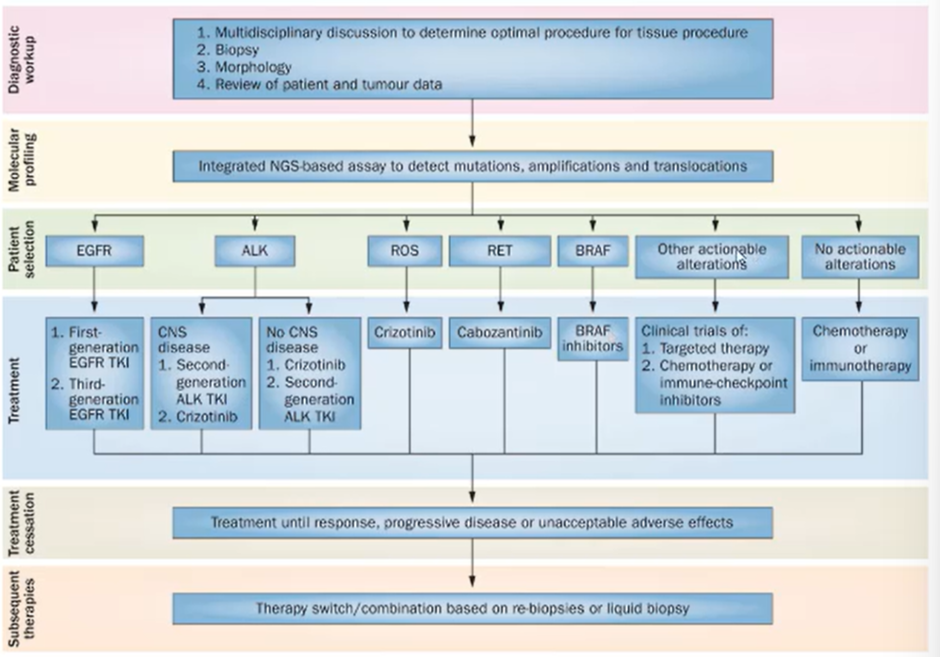
Precision medicine
Treating the patient based off the driving mutation
When we first diagnose the patient with cancer must decide if their cancer is squamous or non-squamous
In most cases squamous wont have a driving mutation and so will have to use traditional cytotoxic or chemotherapeutics
If the patient has a non-squamous cancer, such as EGFR mutation, can give erlotinib or afatinib —> if unsuccessful can use chemotherapy
If the patient has ALK translocation we give them crizotinib and if that doesn’t work certinib
If the patient is EGFR and ALK negative give them platinum based chemotherapeutics.
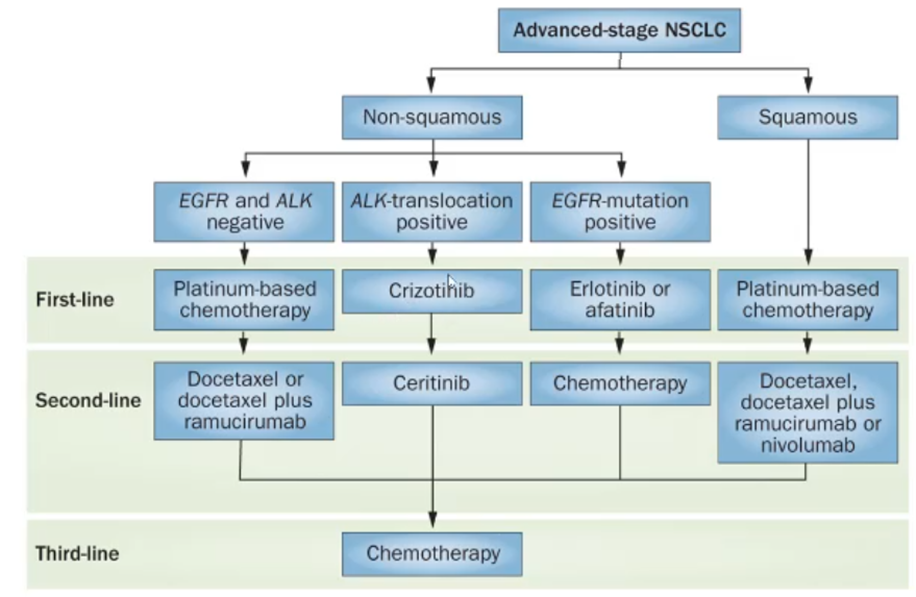
Potential algorithm for incorporating chemotherapies, immunotherapy and targeted therapies