bio 305 exam 3
1/91
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
92 Terms
processes by which bacteria exchange/take up DNA
transformation - DNA uptake from environment
conjugation - plasmid transfer or partial genome transfer
transduction - intake from bacteriophage
lytic cycle
phage attaches to host and injects DNA
phage chromosome replicates
new phage components produced and assembled
phage particles release by lysis from host bacteria
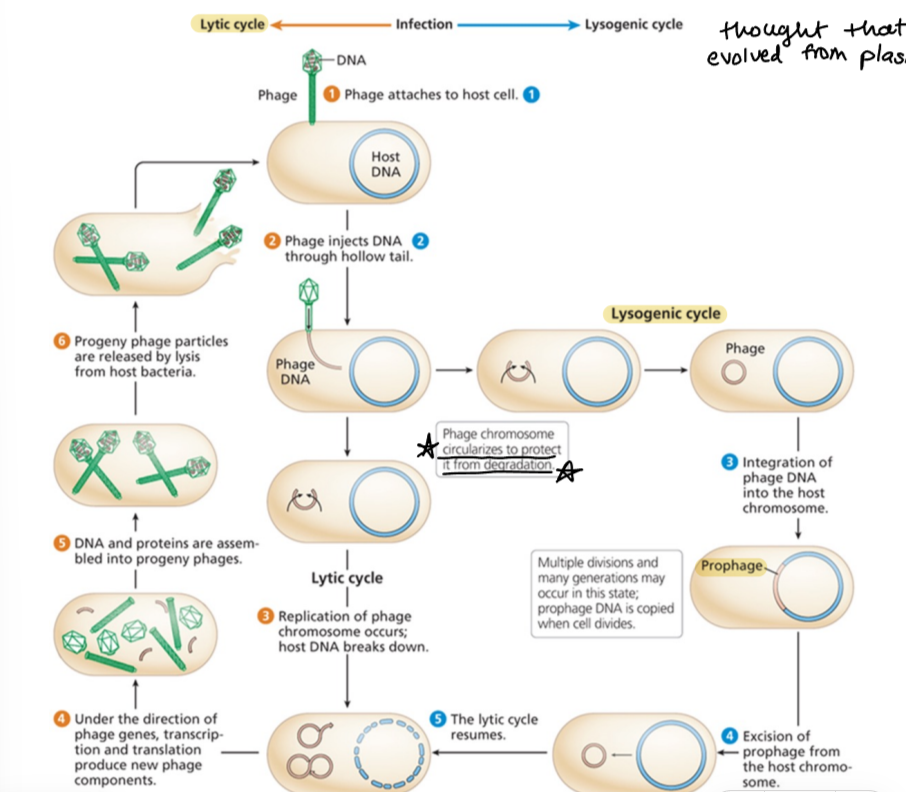
lysogenic cycle
phage attaches to host
phage DNA circulates and is incorporated into bacterial DNA as a prophage
prophage excised and lytic cycle resumes
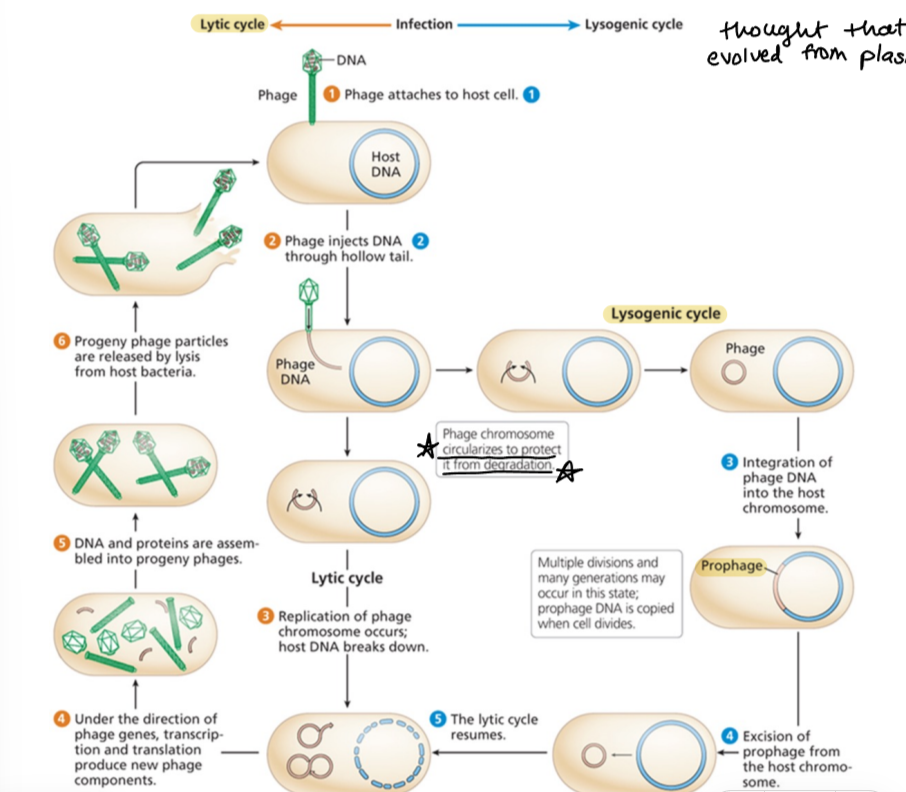
culturing lytic phages on plates in a lab
bacteria is the “lawn” and lysed cells are “plaques”
titer
[(number of plaques)(dilution)]/(volume), with answers in p.f.u (plaque forming units)
multiplicity of infection (MOI)
average number of phage particles that infect a single bacterial cell in an experiment
low MOI (<1 phage/cell) is used to phenotype and genotype a phage
high MOI (>2 phage/cell) is used for phage crossing/recombination and complementation testing
general transduction
a phage picks up host genome on accident (same fragment length as viral genome) and transduces it to another host. . . can transfer wt allele and rescue mutant allele in host cell
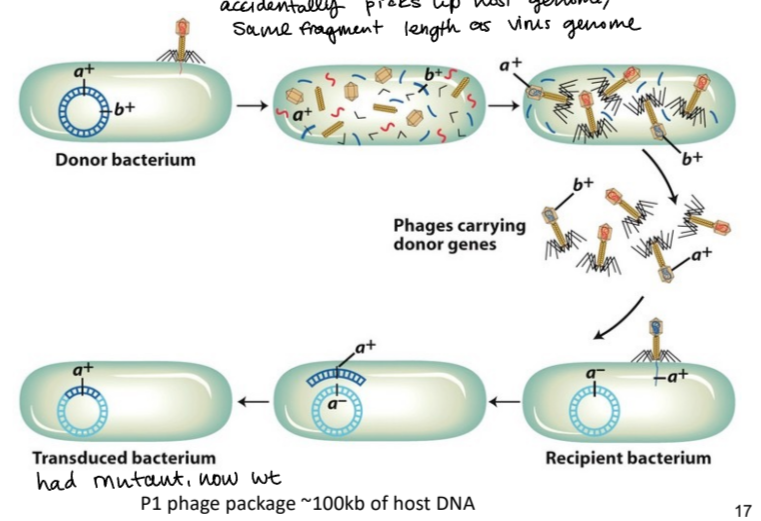
cotransduction frequency depends on. . .
distance between two genes
higher frequency = closer together and vice versa
testing for complementation vs. recombination
complementation testing: one infection at high MOI
recombination testing: first infection with high MOI for recombination, second infection with low MOI to phenotype and genotype virus
seymour benzer
changed scientists understanding of genes, revealed existence of genetic fine structure, did work with lambda phage
esther lederberg
discovered fertility factor F+, lambda phage, replica plating, and genetic mechanisms of specialized transduction
made these discoveries but was consistently overshadowed by her husband joshua lederberg
continuous traits
on a scale, like height
variance
standard deviation, basically what the distribution of a trait looks like
Vp= Vg + Ve
quantitative traits determined by genes and environmental effects, variance in phenotype is due to variance in genotype and variance in environment
heritability
how much of a trait is determined genetically. . . phenotypic variance attributable to genetic variance
H² = Vg/Vp
if H² = 1, all due to genotypic variance, and if H² = 0, all due to phenotypic variance
midparent phenotype
average of two parent phenotypes
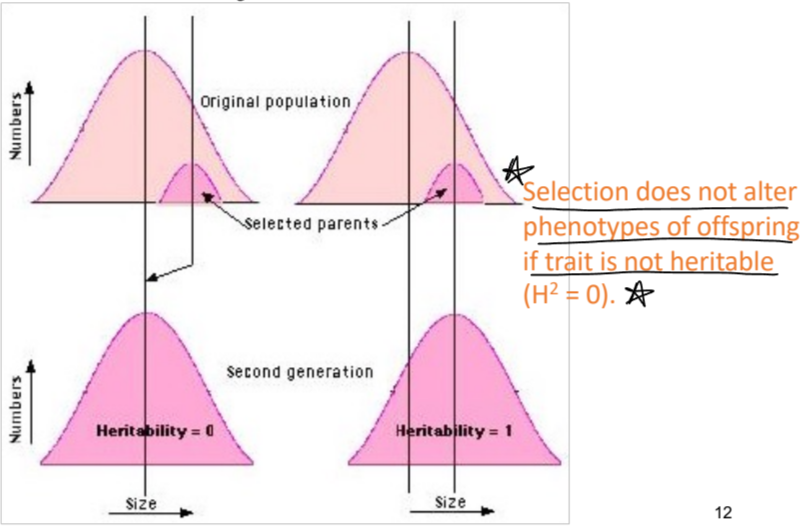
heritability and selection
selection doesn’t alter phenotypes of offspring if trait is not heritable
identifying QTL
looking for association between trait (which can be treated as another gene) and markers
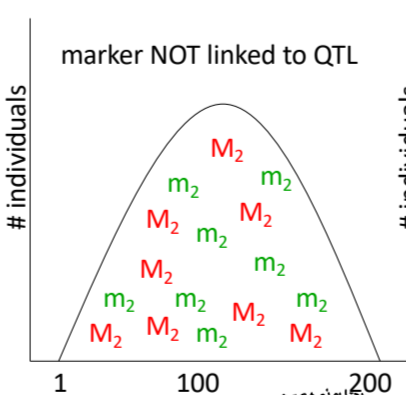
marker not linked to QTL
no association between certain marker and trait
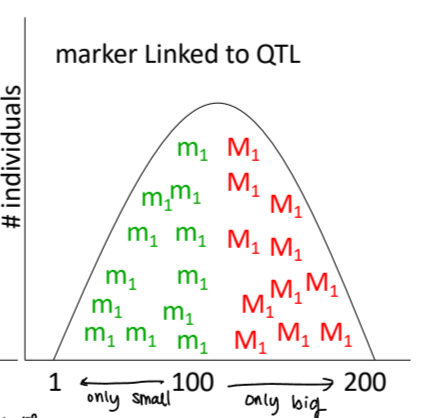
marker linked to QTL
association between certain marker and trait
HWE assumptions
infinitely large population
non-overlapping generations, no mixing
randomly mating populations
no mutation
no migration
no selection
genetic drift
change in allele frequencies due to chance
for alleles at low frequencies, most are found in. . .
heterozygotes
HWE for X-linked genes
males:
p = X^AY
q = X^aY
females:
p² = X^AX^A
2pq = X^AX^a
q² = X^aX^a
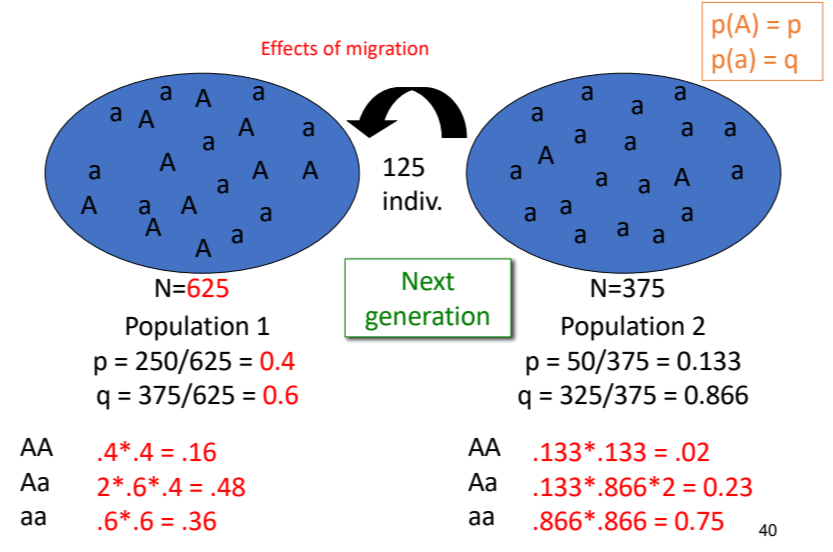
effects of migration in violating HWE assumptions
migration between populations with different allele frequencies will alter the allele and genotypic frequencies of both populations
assortative mating consequences in violation of HWE
in general, alters allele and genotypic frequencies
inbreeding consequences in violation of HWE
decreased frequency of heterozygotes, increased frequency of homozygotes, no overall change in allele frequency but change in genotypic frequency
haplotype
collection of mutation sequencies on a single chromosome. . . each haplotype is a unique allele
effects of mutations are. . .
situation dependent! could be beneficial, deleterious, or neutral! but always increases variation
natural selection
one of the processes that contributes to phenotypic evolution, survival of the fittest and the basis of darwin’s theory of evolution
modern synthesis
provides a theory about how evolution works at the levels of genes, phenotypes, and populations
in response to paradigm shift in biology in 1930s with morgan, fisher, dobzhansky, etc.
where does phenotype variation come from?
environmental and genetic variation, Vp = Vg + Ve
what are the two main sources of genetic variation?
recombination and mutations
fitness (w)
differential ability of individuals to survive and reproduce in a particular environment
selection coefficient (s)
1 - fitness (w)
neutralist - selectionist debate
argues over relative importance of selection and chance
random genetic drift. . .
can have large impact on allele frequencies, more than selection for low frequency alleles
founder effect
type of genetic drift where small population is established from larger population
allele frequencies in new population will differ from original population, such changes are due to chance
population bottleneck
when a relatively large population is reduced by a catastrophic event, with allele frequencies in the new population differing from the original population
founder effect is a version of this
molecular evolution
uses comparisons of DNA variability within and between species to make inferences about evolutionary relationships and past evolutionary processes
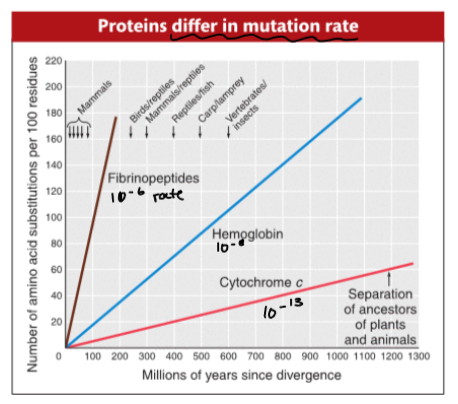
molecular clock
mutations are always happening, but the type of mutation affects the mutation rate of each protein
ex. if it’s a deleterious mutation with a strong negative effect, it will be tightly regulated and selected out of the population, creating a very slow mutation rate due to constraints
silent substitutions
aka synonymous, change in DNA sequence that doesn’t change AA sequence, usually neutral
replacement substitutions
aka non-synonymous, change in DNA that changes AA sequence and most likely subject to selection
macdonald-krietman test
developed to test for evidence of selection, only applied to protein coding regions
compares non-synonymous (dN) mutations with synonymous (dS) mutations, and if ratio of these two mutations between species is the same as within species, then there is no selection
macdonald-kreitman test results
between > within is evidence of positive selection
between < within is evidence of negative selection
selective sweeps
process by which a new advantageous mutation eliminates or reduces variation in linked neutral sites as it increases in frequency in the population
ex. teosinte and corn
experiments showing that genetic info is in DNA
griffith’s rough and smooth bacterial experiments, found evidence for a “transformational factor” changing RII to SIII after SIII bacteria had been heat-killed and infected mice still got sick and died
macleod, avery, and mccarty identified DNA as the transformation factor after eliminating lipids, proteins, sugars, and RNAs from extract with heat-killed SIII and RII
hershey and chase showed that DNA is the hereditary molecule with their bacteriophage experiment, having DNA with radiolabeled P and protein coat with radiolabeled S
DNA structure
right-handed antiparallel double stranded double helix
major groove for binding sequence specific proteins
minor groove
non-specific binding proteins binding the backbone
ribose phosphate backbone and nitrogenous bases
how long is human DNA?
almost 2 m
DNA supercoiling
can accumulate twists and supercoils if DNA fixed at both ends, important for replication because can disrupt it
negative supercoiling is behind the protein
positive supercoiling is in front of the protein
essential features of genetic material
sufficient information capacity
ability to replicate
ability to mutate
DNA replication direction and pattern
semi-conservative, with strands being read from 3’ to 5’ and built from 5’ to 3’
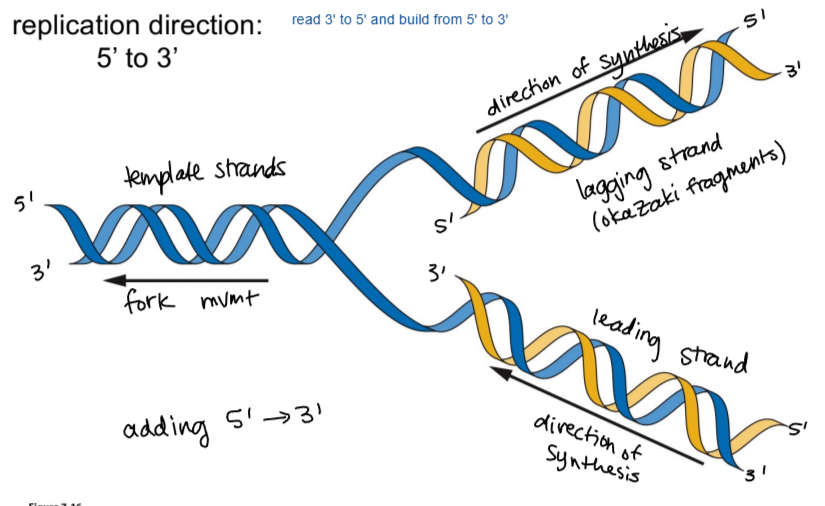
memorization hack for DNA direction
DNA is read 3’ to 5’ like how 3 years old is the age to start reading, should be earlier than 5 years old
DNA is built 5’ to 3’ like how 5 year olds can build better things with toys or with their imagination than 3 year olds
initiation of DNA replication
starts at ori-initiation, AT rich area
DNA helicase activity separates the strands and a replication bubble forms with two replication forks
what does DNA polymerase require to replicate?
DNA template, dNTPs, and 3’ OH from a primer (RNA primer from primase)
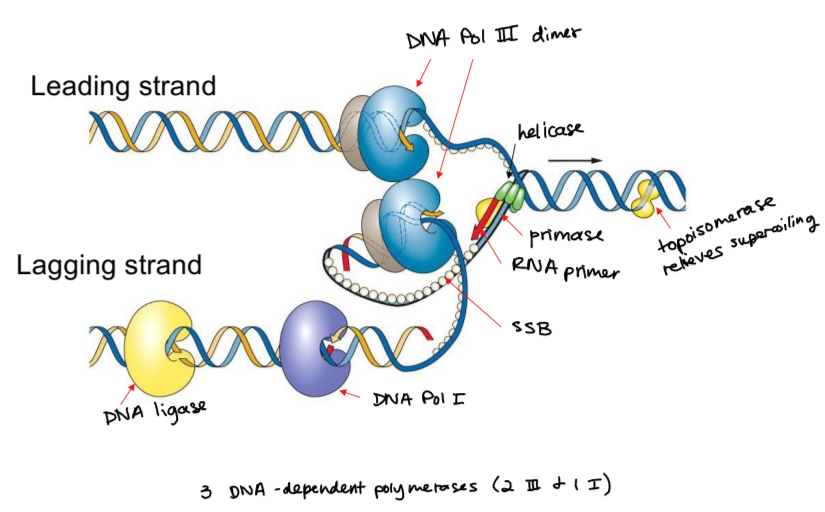
bacterial DNA polymerases
I is primer removing (exonuclease activity)
II is for DNA repair
III is for DNA synthesis
all of them proofread and can build/polymerize DNA
replisome and accessory proteins
composed of leading strand, lagging strand, DNA polymerases, DNA ligase, primase, RNA primers, DNA helicase, and topoisomerase
end replication problem
when RNA primer is removed from lagging strand, DNA gets shorter with every round of replication
telomeres
protective caps that prevent chromosome shortening, repeated DNA sequences
telomerase
RNA-dependent DNA polymerase that elongates telomeres
the RNA component acts as a template for telomere elongation and the protein component catalyzes the addition of nucleotides (reverse transcriptase)
works between rounds of replication, elongating the template strand that the new lagging strand is copying from
shelterin
protein that protects the 3’ overhang because ssDNA is bad
telomerase activity
tightly regulated
high in early embryonic cells, germ cells, stem cells, and cancer cells
low in somatic cells
directional selection
drives a population towards homozygosity, reduces variation within a population
effect of migration selection
homogenizes variation between populations
effect of balanced polymorphism selection
aka overdominance, selects for heterozygotes so that multiple alleles are stably present in a population, like in sickle cell anemia example
selection against heterozygotes
aka underdominance, allele frequency either goes to 1 to be a fixed allele or to 0 to become extinct. . . heterozygote does worse than either homozygote
balanced selection
selection that maintains multiple different allele frequencies within a population, occurs in cases of codominance and can be caused by overdominance or overdominance
stabilizing selection
aka purifying selection, when natural selection removes deleterious mutations and keeps phenotypes constant. . . keeps population the same
diversifying selection
aka disruptive selection, sometimes it’s advantageous to have many alleles of a gene within a population, and this type of selection keeps them around, like MHC antigen recognition protein
molecular cloning
vector DNA (plasmid) digested with restriction enzymes, resulting in removed fragment and exposed sticky ends
DNA of interest digested with restriction enzymes, those sticky ends combine with the vector to create a recombinant vector
recombinant vector is plasmid with sequence of interest
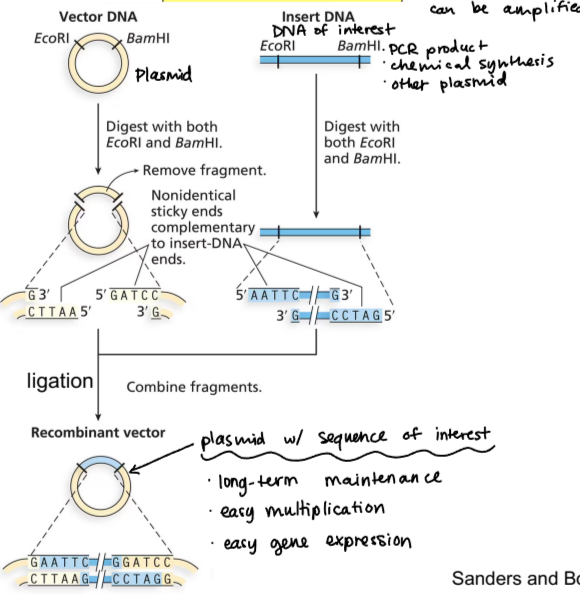
benefits of recombinant vector (result of molecular cloning)
long-term maintenance, easy multiplication, easy gene expression
composition of recombinant plasmids that code for expressed protein
promoter, protein coding sequence, epitope tag that interacts with antibodies to identify and purify recombinant protein, also other helper sequences
ex. production of human insulin in E. coli

northern blot
determines RNA length and quantity
steps:
separate RNA on gel via electrophoresis
transfer RNA to membrane
hybridize with probe
what is a probe?
short polynucleotide complementary to RNA of interest
binds to RNA on membrane by base pairing
typically radioactive, and radioactivity indicates presence and quantity of RNA
NOTE: CANNOT COMPARE BLOT INTENSITY ACROSS DIFFERENT PROBES
western blot
determines protein size and quantity
steps:
separate protein on gel
transfer to membrane
probe blot with antibody
secondary antibodies label and indicate presence and quantity of protein, detected by chemiluminescence
PCR
amplifies any DNA fragments you want
produce DNA
can quantify DNA or RNA (via reverse transcriptase)
fast, cheap, in vitro
limitations of PCR
need template DNA
need to know sequence of flanking regions to design primers
length limit of 10 kb
steps of PCR
denature DNA at high temps
anneal primers
extend primers, elongation step
cycles repeat, only products with no ssDNA overhang and length limited by primers are duplicated in subsequent cycles
typically 25-35 cycles
how do you choose Sanger sequencing primers?
sanger sequencing sequences a strand and allows inference of the complementary strand. . . to know the sequence of one strand, the primer should match the beginning of the sequence of interest, so that the strand of interest is built from the template of the complement strand
how do you make cDNA?
ss mRNA was transcribed via reverse transcription
product was mRNA/cDNA hybrid
RNase degraded RNA, so that only cDNA remains, and it can be used as a template in PCR
qPCR result interpretation
result appears as fluorescence curve
exponential curve occurring in earlier cycles shows high DNA/RNA concentration, and vice versa showing low DNA/RNA concentration
high throughput sequencing
sequencing on a massive scale
millions of sequences at once
limitations of high throughput sequencing
sequences within one sample usually anonymous
sequences usually short (up to ~600bp)
requires complex data analysis
genome (re)sequencing steps:
DNA extraction and fragmentation
library generation (mix of DNA fragments)
high throughput sequencing of each short fragment
aligned fragments to reference sequence - different from reference sequence means mutation at that point
genome (re)sequencing applications
find mutations causing a phenotype
find mutations associated with disease
determine best targeted treatment for a specific cancer
sequence a new interesting genome
genome (re)sequencing limitations
very hard in the absence of a reference genome
very hard on repetitive sequences
difficult bioinformatics required
RNA-seq
quantification of RNA
steps:
RNA from tissue transcribed to cDNA
cDNA sequenced and mapped to genome
many matching fragments show high RNA concentration and sometimes high expression (NOT ALWAYS), and vice versa for few matching fragments
advantages of RNA-seq
know RNA accumulation from ALL genes
analyze transcription and RNA processing
limitations of RNA-seq
need genome sequence
less abundant RNAs hard to study
often overinterpreted as a measure of gene expression, doesn’t have to mean this
ChIP-seq
shows protein-DNA interactions
steps:
cells lysed and bound with antibodies
DNA from these cells sequenced and mapped to genome
many matching protein to DNA regions means protein was bound and vice versa
advantages of ChIP-seq
find all binding sites of a protein
can study posttranslational protein modifications
limitations of chIP-seq
need an antibody specific towards protein
need a good negative control
limited resolution