From Raw Sequences to Assembled Genomes
1/40
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
41 Terms
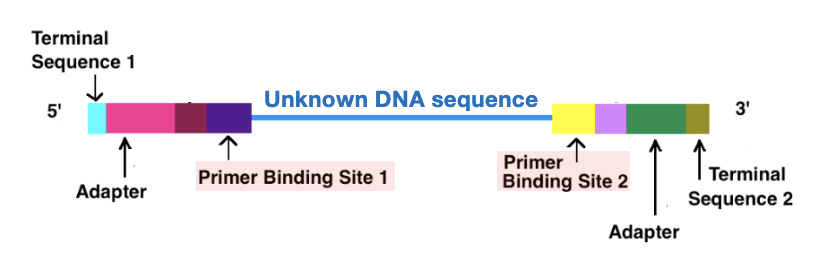
Library Construction
involves ligating known DNA sequences (adapters) onto unknown genomic fragments, so that the fragments can bind to the flow cell, be amplified and sequenced from both ends
terminal sequences: complementary to oligos on flow cell; allow DNA fragments to attach (tether) to sequencing surface
adapters: short know DNA sequences ligated via enzymes, enables amplification and sequencing
primer binding sites: where sequencing primers bind, required to initiate DNA synthesis
unknown DNA sequence: can obtain ~150-250 bp from the ends of each fragment
Library Construction Figure
Why Sequence Both Ends?
helps resolve repetitive DNA
places fragments more accurately in the genome
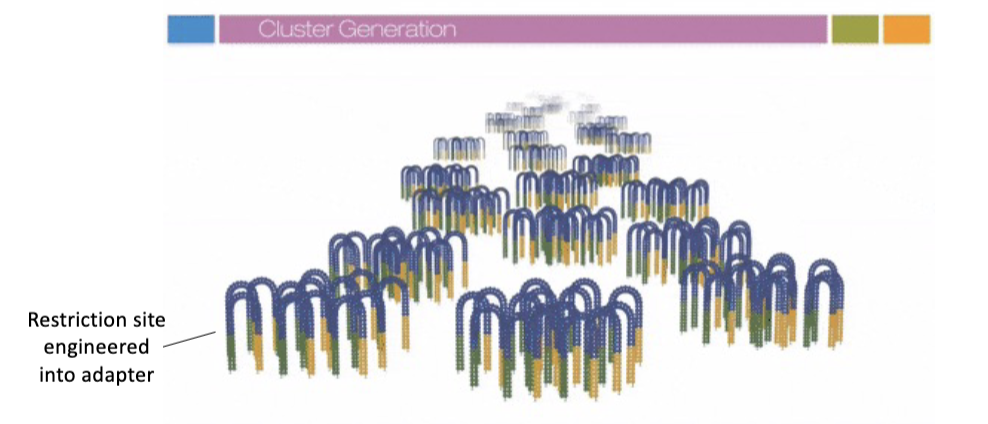
Cluster Generation
steps: 2) generate clusters of amplified DNA sequences
DNA strands bind to flow cell via base pairing to 1st terminal sequence
flow cell oligos are extended using DNA polymerase
3’ ends of the extended sequences hybridize w/ nearby oligos
flow cell oligos are extended using DNA polymerase (extension, bridge formation)
repeat to generate clusters
Remove Reverse Strands
after cluster generation, each cluster contains forward + reverse strands but sequencing-by-synthesis method requires a single stranded template, all oriented the same way
one strand must be removed
restriction sites are included into the adapter; the site is only present in one strand orientation
restriction enzyme cuts and removes the reverse strands
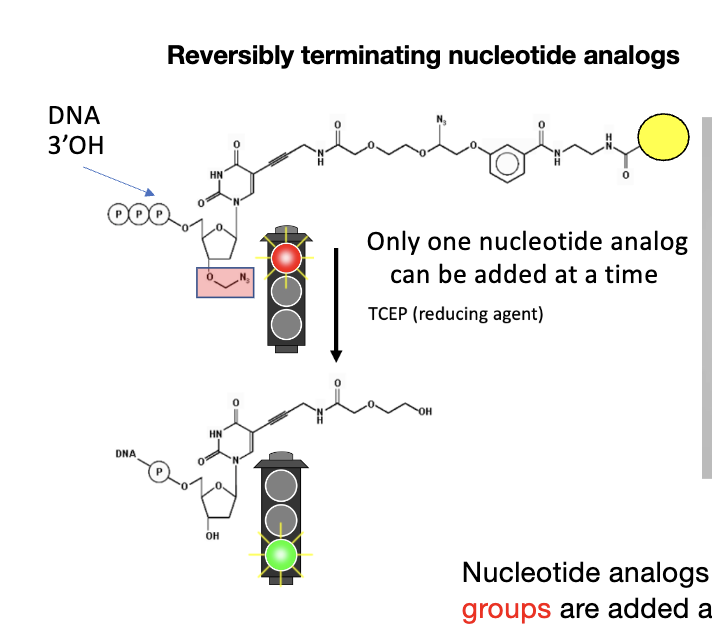
Sequencing by Synthesis
steps: 3) massively parallel sequencing of clusters
nucleotide analogs w/ cleavable fluorophore and terminating groups are added and then terminator/dye are cleaved each cycle
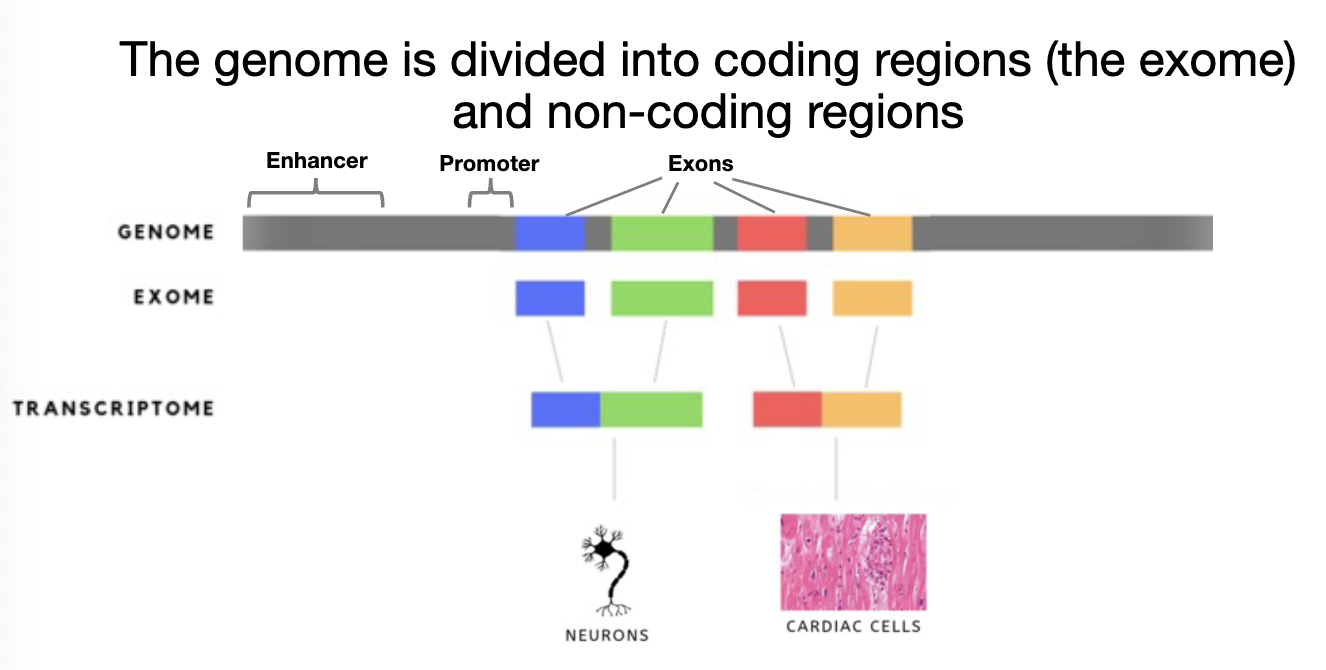
How is the Genome Divided?
into coding regions (exome) and non coding regions
exons: differential splicing can occur
transcriptome: complete set of RNA transcripts produced in a cell (or tissue) at a given time and condition
can be used to compare differences between cells
tells us which genes/region of genome is transcribed
ome ending: studying all at once
enhancers: regions that affects the activity of a promoter (can be far from gene)
promoters: where core RNA polymerase machinery binds to initiate transcription (has to be close to gene)
mutations in regulatory non-coding regions can affect gene expression
mutations in exons can affect the underlying amino sequence as well as gene expression
How is the Genome Divided? FIGURE
What does Exome Sequencing Identify?
exome sequencing identifies causal mutations even in a small patient pool
mutations in embryonic myosin MYH3 are thought to cause Freeman-Sheldon syndrome, a severe congenital joint disorder
NS/SS/I = potential damaging mutation
exome sequencing: MYH3 was the only damaged coding region in all 4 affected patients and not found in a public database of common SNPs, or in 8 control individuals
figure: as you compare patient genome and reference genome, you narrow down which gene is the problem (where the mutation is)

What does Whole Genome Sequencing Reveal?
whole genome sequencing reveals the role of non-coding mutations
mutations affecting enhancers, promoters, silencers, insulator elements can have substantial affects on gene expression, and cannot be captured by exome sequencing
Conclusions so Far
the development of new technologies have dramatically lowered the cost of DNA sequencing
number of genes hasn’t increased that much in more complex organisms – complexity during animal evolution must be driven by increasingly sophisticated gene regulation
the ability to obtain massive amounts of sequencing data have helped to discover the causes of many rare genetic syndromes (even w/ very few patients)
Why has it taken ~20 years to complete the Human Genome Project?
Going from raw sequences to an assembled genome is not simple
putting short reads in the correct order requires there to be sufficient read coverage
sequenced genome may have deletions, insertions, rearrangements, substitutions, etc. compared to a reference genome
genomes have many regions that are highly repetitive
From Reads to Contigs
contig: a consensus sequence that is result of assembling together overlapping fragments
polymorphism: common DNA sequence variation at a specific genomic position that exists in a population
alleles are different versions at that site
typically small differences (single nucleotide polymorphisms, small indels)
coverage: the number of times each position is sequenced in an experiment; poor coverage can lead to assembly errors
major challenge for sequencing is repetitive DNA
Repetitive Elements
make up the majority of human genome (66-69% is repetitive or derived from repeats)
Major types:
tandem repeats: short repeats including minisatellites (10-60 bps) found in centromeres, microsatellites (2-8 bps) found in telomeres
interspaced repeats: transposable elements
found throughout genome, enriched in centromeres, pericentric/intercalary heterochromatin
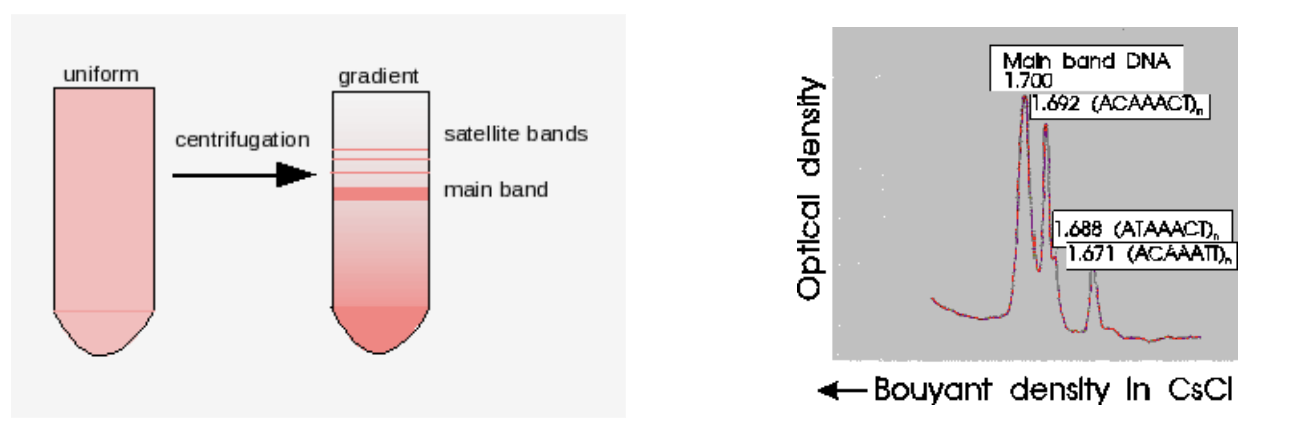
Satellite DNA Sedimentation
satellite DNA differentially sediments in a CsCl gradient due to different base composition than the “main band” DNA
don’t have same AGCT representation
satellite bands are repetitive DNA that don’t co-sediment w/ main band
AT DNA is less dense than GC DNA
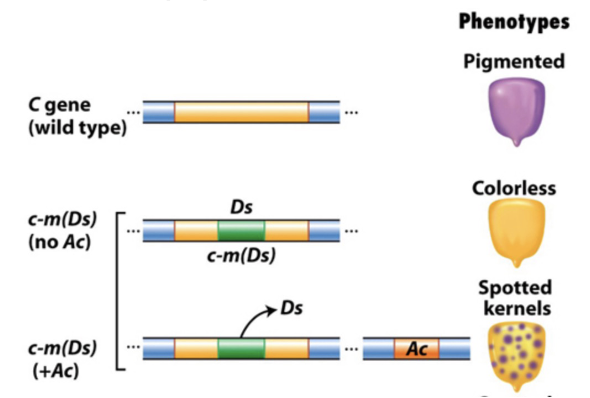
Transposable Elements in Maize
coloration of maize kernels are caused by the disruption of a pigment gene by a transposable element Dissociation (Ds)
if left in pigment gene → purple corn (WT)
dissociation transposition (jumping) requires an enzyme produced by a second transposable element Activator (ac)
this showed that the genome is much mroe dynamic than we previously thought
genes can literally jump out from and into genomes
Transposons Definitions
DNA transposons replicate by cutting and pasting themselves into different parts of the genome
LTR (long terminal repeat) retrotransposons: transcribed into RNA and reverse-transcribed into dsDNA in the cytoplasm using a tRNA primer (copy and paste)
Non-LTR retrotransposons: transcribed into RNA which is reverse transcribed into DNA used nicked host DNA as a primer in the nuclease (copy and paste)
both non-LTR and LTR use RNA intermediates → reverse-transcribe to DNA
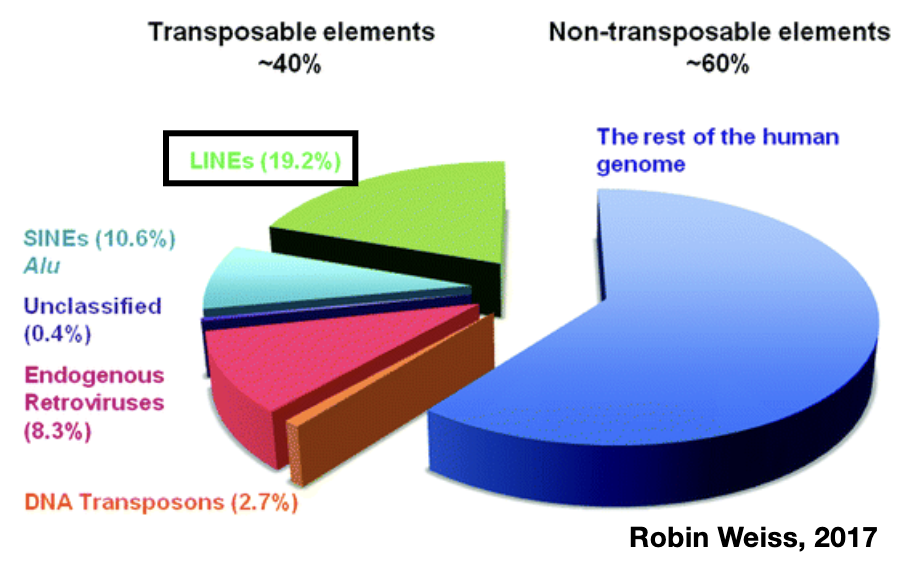
Transposable Elements in our Genome
transposable elements make up almost half our genomes
genomic parasites; impact on genes; important for evolution
~4,000 full-length LINE1 insertions and ~100,000 fragments in the human genome
initially thought ~40 LINE1 elements are still active in our genomes
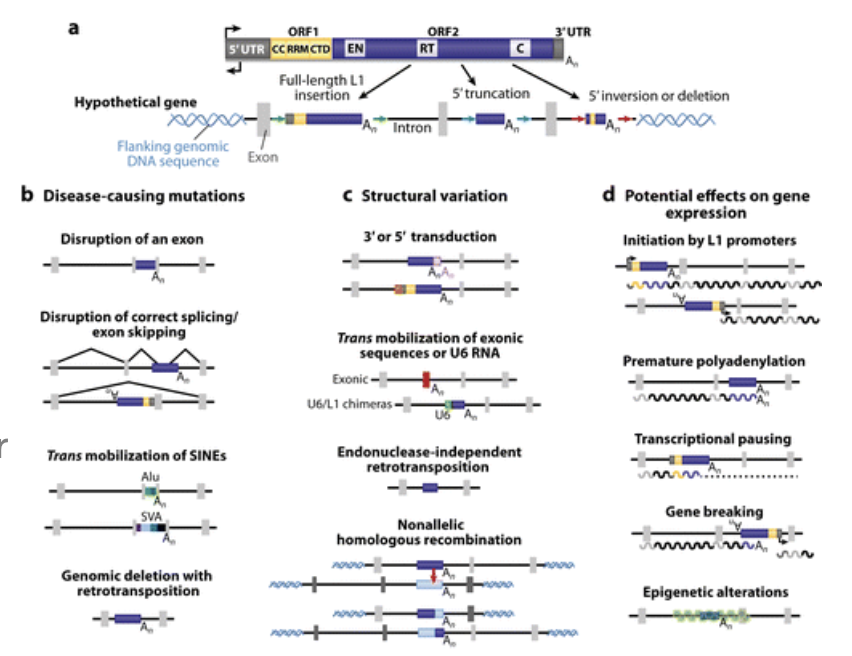
Transposons Insertions: Diseases
study of 240 hemophilia patients showed that 2 individuals had disease mutations caused by independent LINE1 insertions into Factor VIII
evidence of elevated LINE1 activity in cancers, is hypothesized to play a role in mutagenizing cancer genomes to promote cancer progression
estimated that 1 in 1,000 disease causing mutations is due to a novel LINE1 insertion
Transposons Insertions: Evolution
new transposons insertions help to drive variation and the evolution of adaptive traits
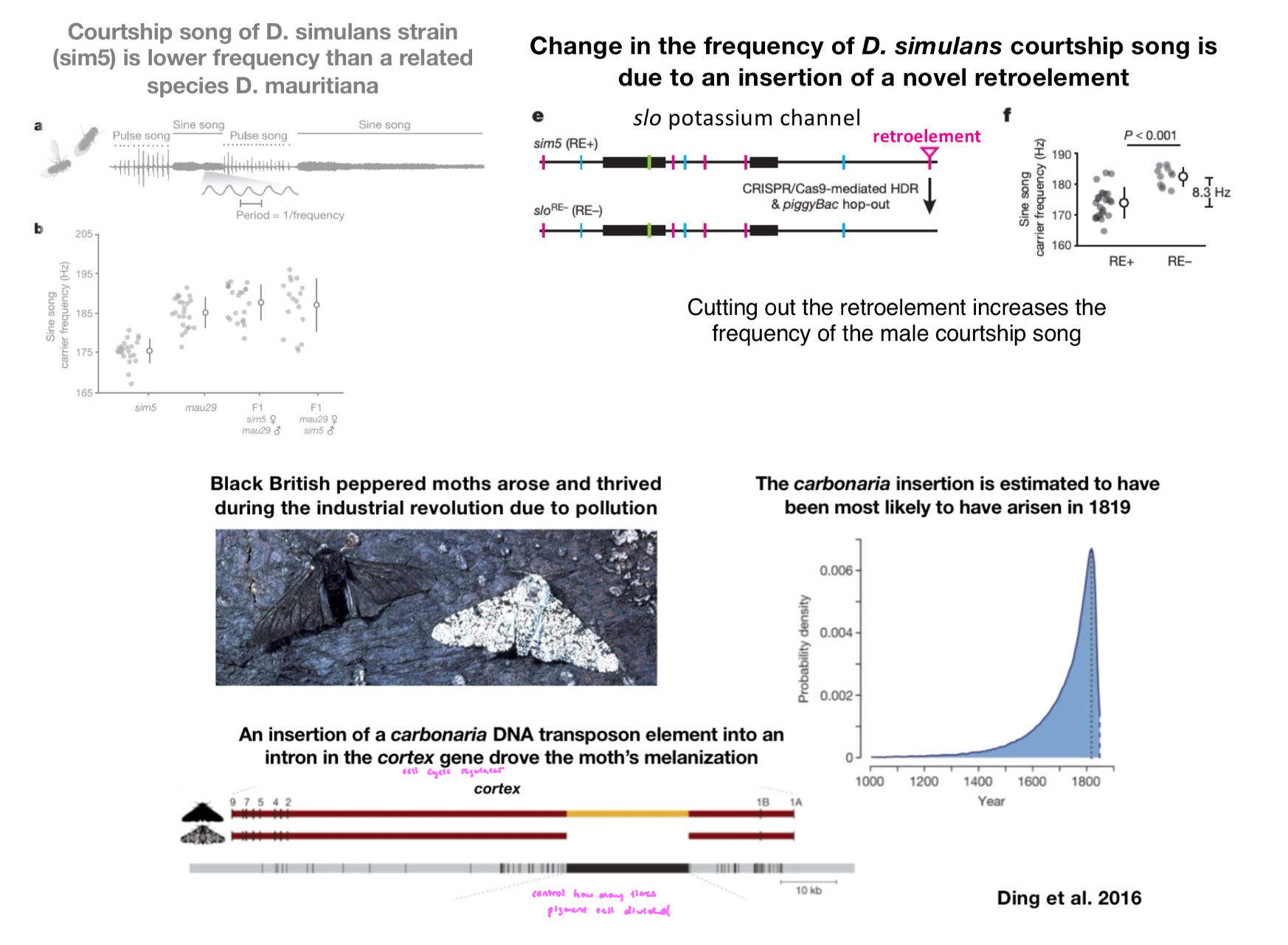
Arc Gene
important for many forms of neuronal plasticity (i.e memory formation)
Arc = activity regulated cytoskeleton
related to repetitive DNA b/c it evolved from a retrotransposon (type of repetitive DNA)
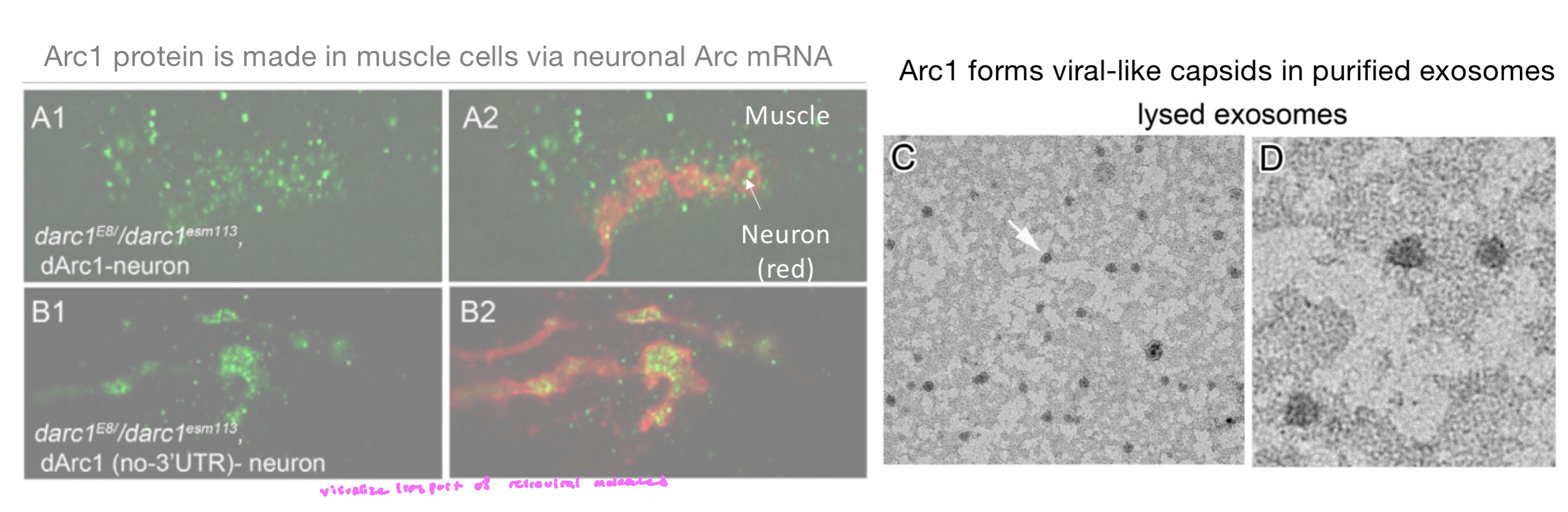
Arc1
Arc1 encodes a retroviral-like protein that traffics b/w neurons
independent domestication of retroelements happened many times in evolution (suggests strong selective advantage0
Arc1 protein is made in muscle cells via neuronal Arc mRNA
neurons produce Arc mRNA
Arc1 forms viral-like capsids in purified exosomes
these particles encapsulate Arc mRNA and are released in extracellular vesicles (exosomes)
our ability to form new memories requires a gene that evolved from a retro-element
loss of Arc → severe memory deficits
Why can identical repetitive DNA sequences have different biological effects?
even if the 2 DNA sequences are identical at the nucleotide level, their biological effect can be completely different depending on where they are in the geome
sequence ≠ function by itself
eg. different contexts: different neighboring genes, regulatory elements, cell type
effects can be very different depending on where they land
eg. lands in promoter → alters transcription; exon → disrupts protein; intron → affects splicing
in Arc: one copy of the repetitive retrotransposon landed in the right genomic context and became essential for memory
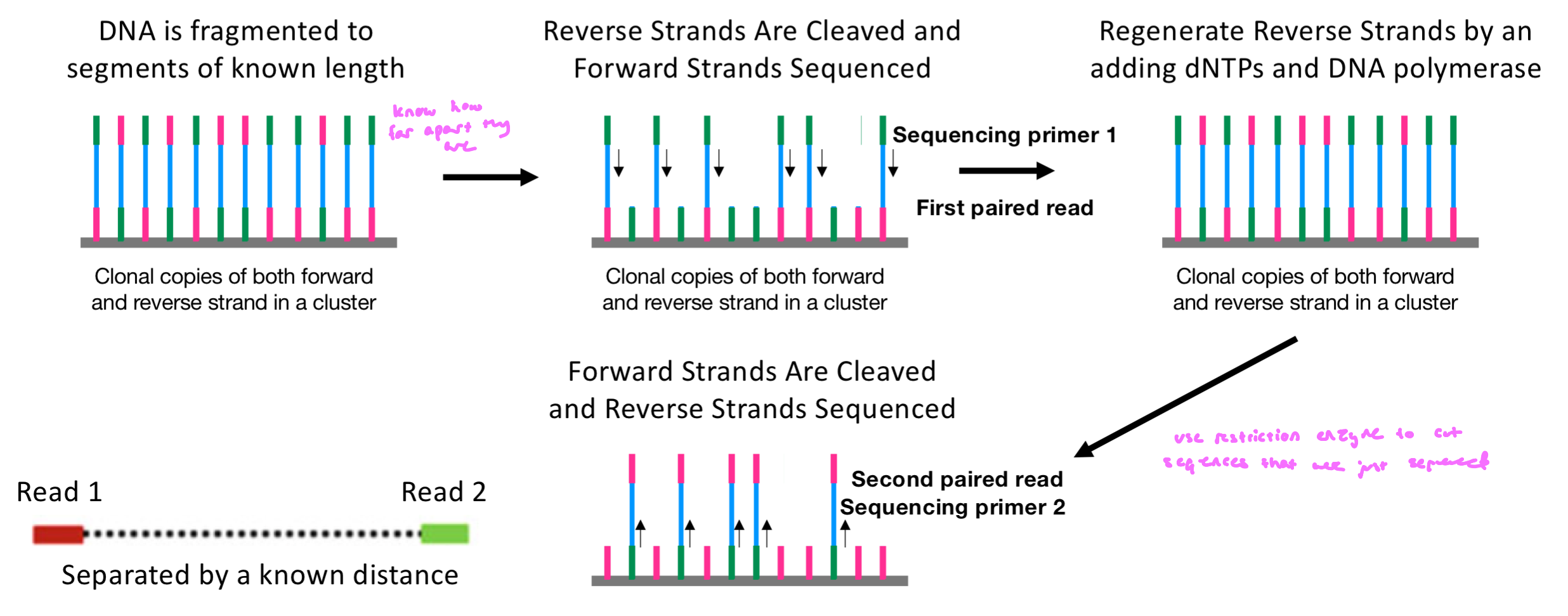
How does Paired-End Sequencing Generate Reads
Paired reads are produced thru sequential sequencing of forward and reverse strands
DNA is fragmented to segments of known length, adapters are ligated to both ends and contain sequencing primer binding sites
this defines the distnaces between reads
cluster generation (contain forward and reverse strands)
Read 1: reverse strands are cleaved, leaving single-stranded forward templates
forward strands are sequenced (first paired read; forward read sequenced)
sequencing primer binds to the adapter
Read 2: forward strand is removed, reverse strand is regenerated by adding dNTPs and DNA polymerase
sequencing primer binds on the other adapter
reverse read sequenced (2nd paired read)
final results: reads from opposite ends/orientation, but from the same fragment and separated by a known insert size
How does Paired-End Sequencing Generate Reads FIGURE
within the two adapters that are ligated onto either end of DNA molecules are sequencing primer binding sites
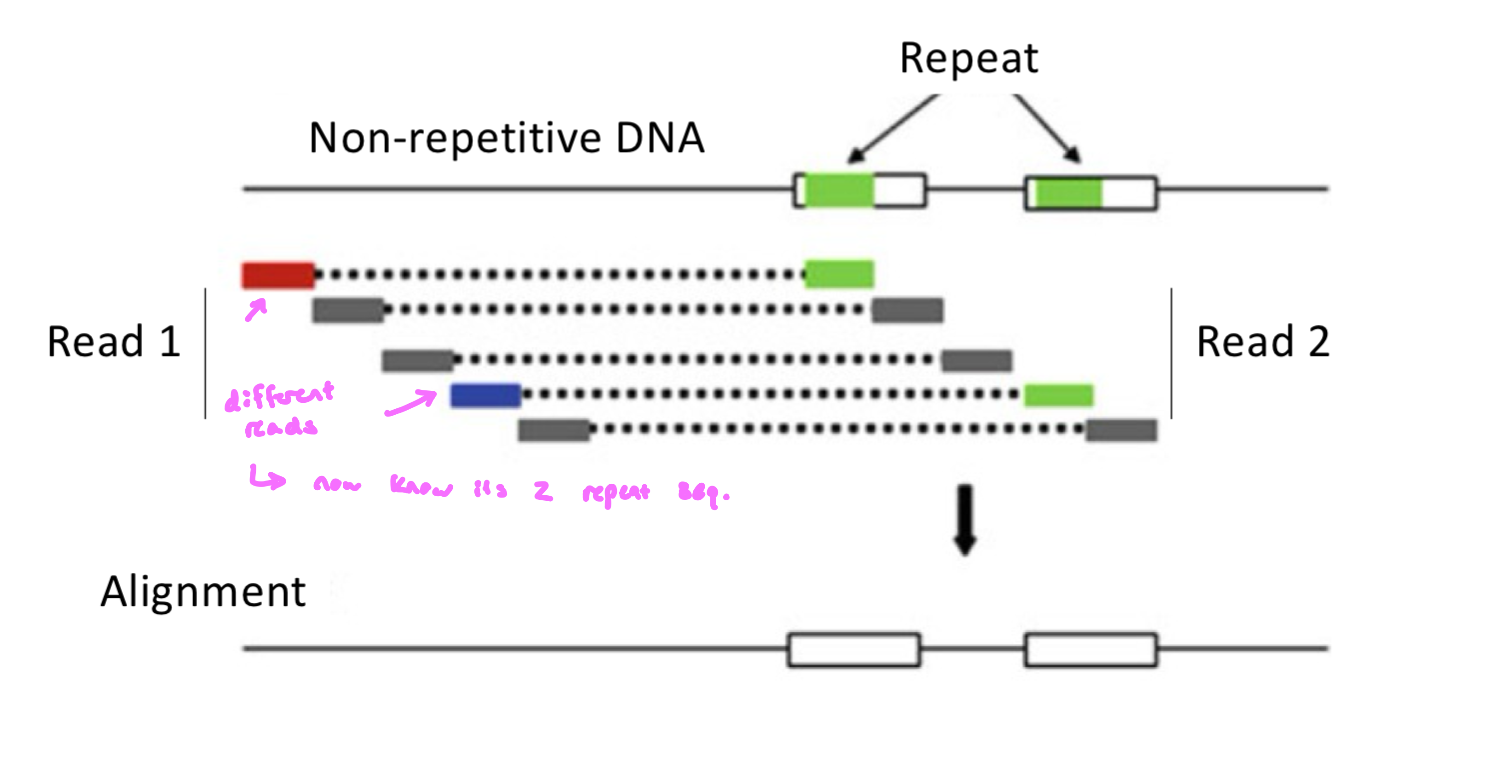
paired end sequencing can help us to map some repetitive sequences
Paired End Sequencing Mapping
Sanger Sequencing Elegant Approach
increase the read length: get sequencing reactions to approach the size of chromosomes
long read sequencing analyzes single DNA molecules
long read sequencing has higher error rate (0.2-3% vs 0.1%) and is more expensive than short read sequencing
short and long read sequencing using all platforms can be (and are frequently) combined
this approach led to the full sequencing of human chromosomes
PacBio Sequencing
Uses zero-mode waveguides (ZMWs) → tiny nanowells (~1 zeptoliter).
Each well contains one DNA polymerase bound to a single DNA molecule.
Only fluorophores at the polymerase active site are illuminated.
Fluorescently labeled nucleotides emit a signal when incorporated.
Signal detected in real time as bases are added.
Single-molecule sequencing → no PCR amplification required.
PacBio Sequencing

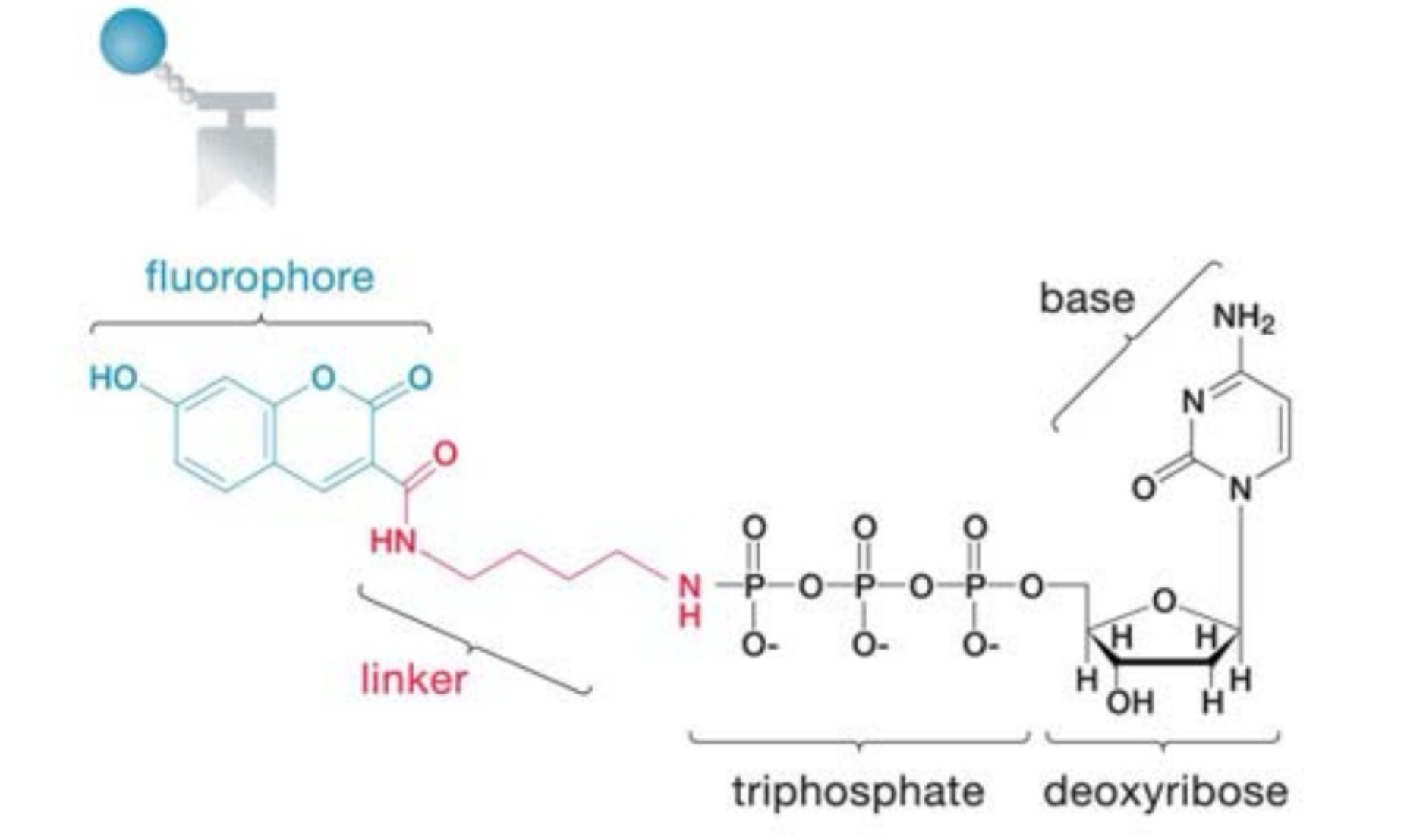
PacBio Hifi Sequencing
single molecule sequencing by synthesis (sequences one DNA molecule at a time)
no PCR amplification required
DNA polymerase incorporates nucleotides like normal replication and each base addition is observed in real time
bases are labeled with dyes thru the terminal phosphate group – incorporation will lead to cleavage of the dye
Real-time Detection of Base Incorporation
time for each base to be incorporated (miliseconds) is much faster than random diffusion (microseconds)
free nucleotides diffuse too quickly to produce a signal
DNA polymerase highly processive (can synthesize >70 kilobases w/o falling off)
enables very long reads
get a fluorescent spike (strong signal that lasts) for correct bases
DNA polymerase holds each nucleotide long enough for a detectable fluorescent signal, allowing extremely long single-molecule reads
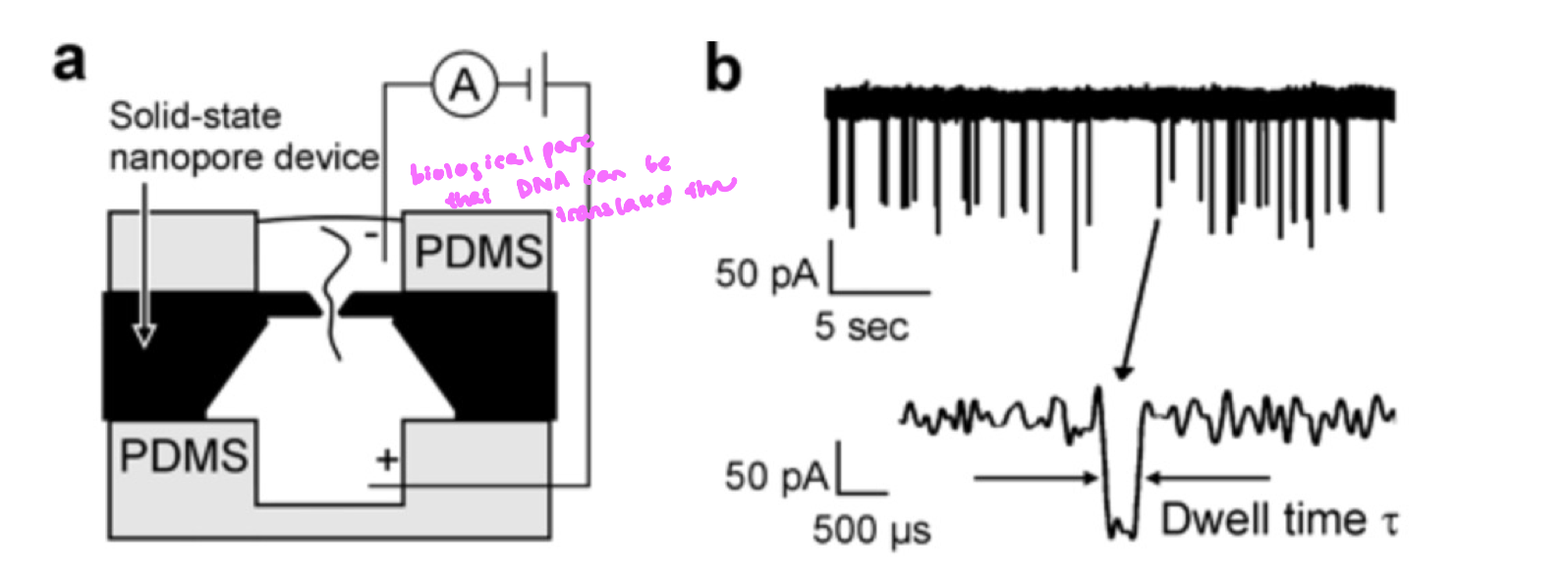
Oxford Nanopore Sequencing
directly measures DNA translocation
similar to gel electrophoresis, applying a voltage gradient leads negatively charged DNA to travel towards the anode, only route is thru a pore
translocation of DNA blocks the natural current of ions thru a pore
as each nucleotide passes thru the pore, the DNA partially blocks the ionic current
different bases cause distinct current changes
detects DNA bases by measuring changes in ionic current as DNA molecule passes thru biological pore under applied voltage
can detect modified bases and structural variants
Oxford Nanopore Sequencing FIGURE
Oxford Nanopore Sequencing: Shelf Life
because nanopore sequencers use biological channels, there is a limited shelf life
1 month at room temp, 3 months at 4ºC
accessibility: data acquired in the lab via a laptop w/ a USB connection

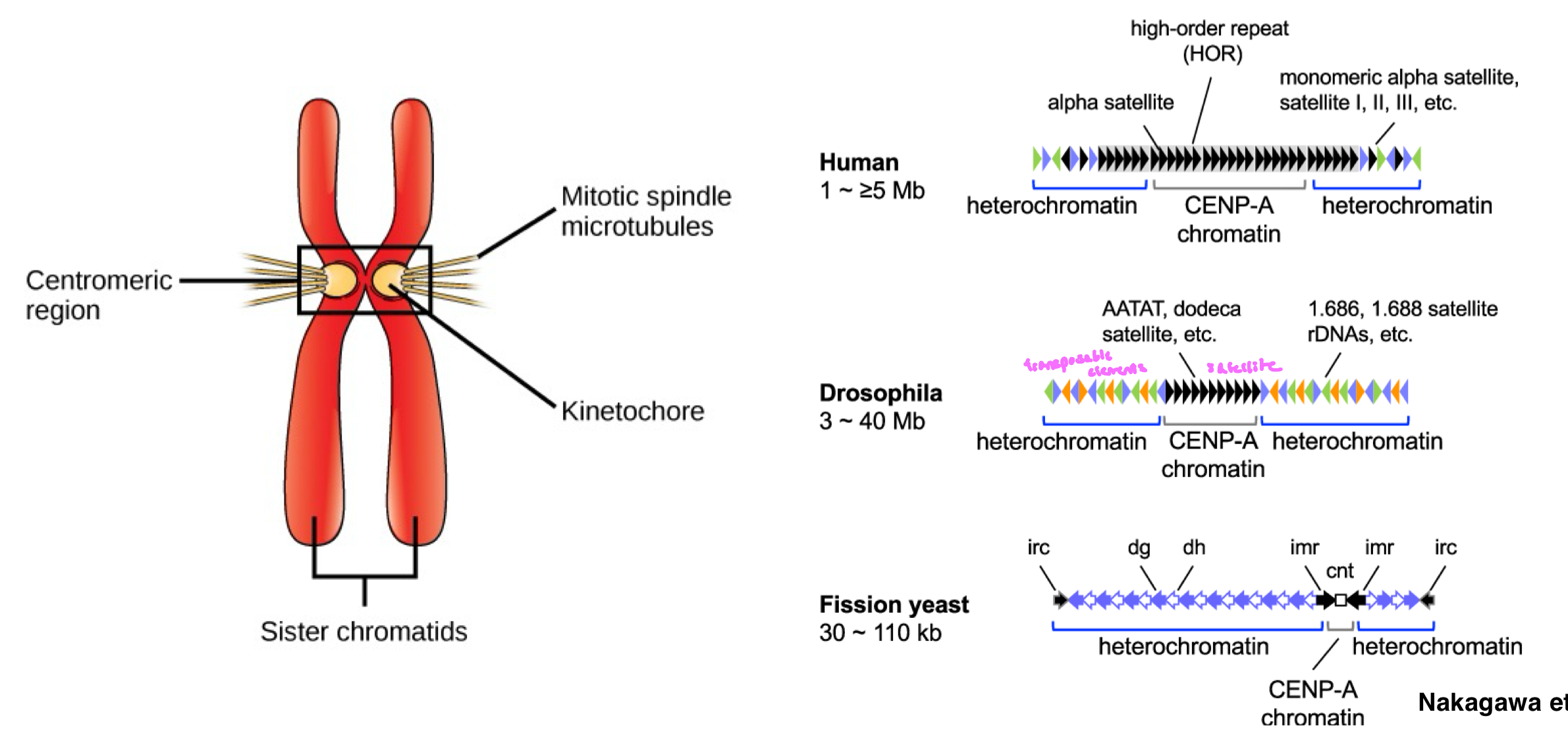
What is the sequence of centromeric DNA?
Composed of highly repetitive DNA → difficult to sequence (“genomic dark matter”).
Essential for chromosome segregation.
Flanked by heterochromatin rich in transposable elements.
In humans: main repeat is alpha satellite DNA.
Organized as monomer repeats → higher-order repeats (repeated blocks).
Other species (e.g., Drosophila) also have repetitive centromeres, but sequences differ.
What is the sequence of centromeric DNA? FIGURE
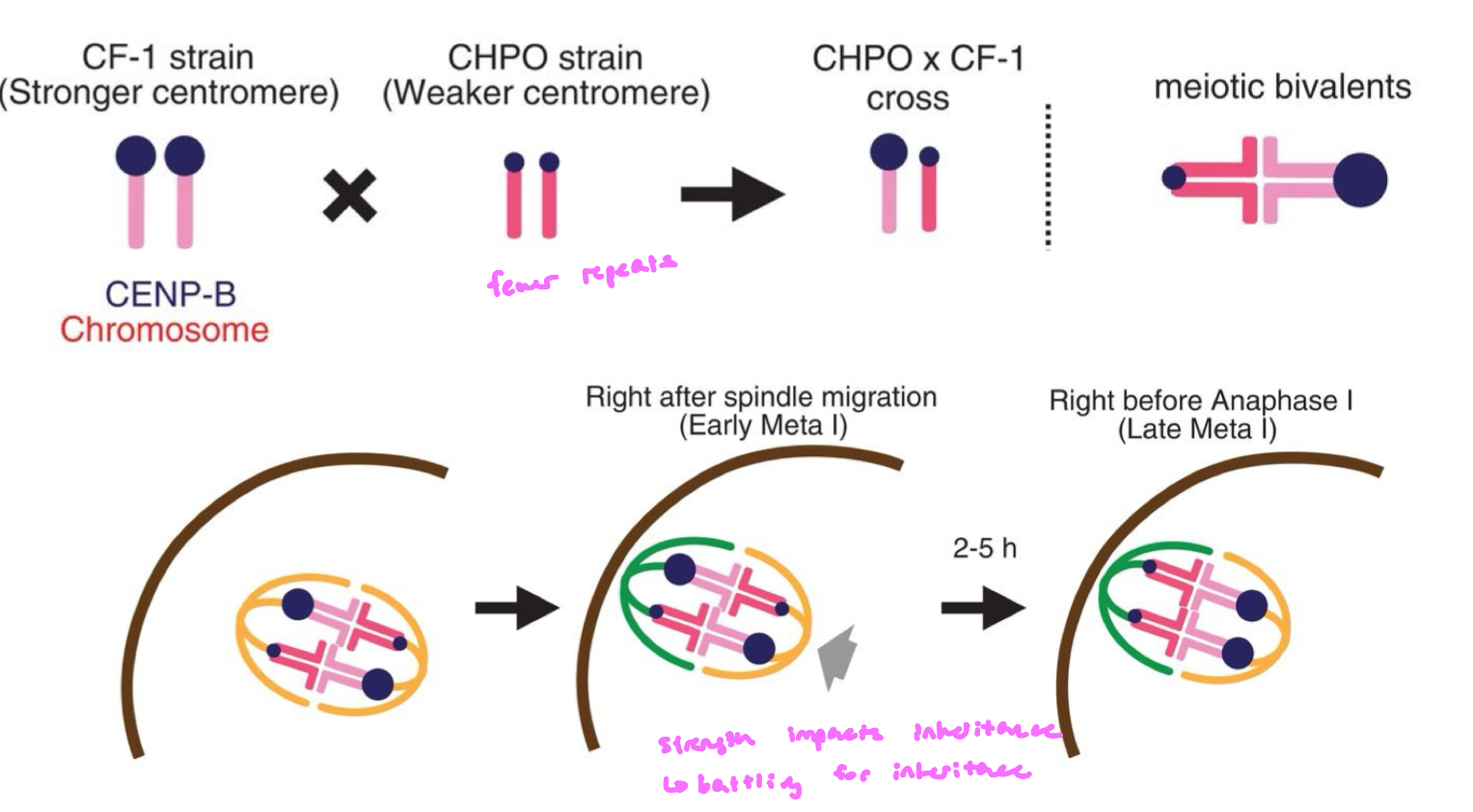
Satellite Repeats in Centromeres
give an evolutionary advantage
chromosomes w/ more repeats will end up inherited at a 60:40 ratio vs chromosomes w/ fewer repeats
more repeats = stronger centromere = higher chance of being inherited
during female meiosis, ¼ meiotic products becomes the egg, and chromosomes w/ stronger centromeres (more repeats) are more likely to attach to the egg pole
centromere repeats aren’t evolving b/c they make the organism more fit, which may lead to species that are less fit (b/c centromeres are cheating in meiosis)
Satellite Repeats in Centromeres FIGURE
What did long-read sequencing reveal about fly centromeres, and how are transposable elements involved?
long read sequencing led to the discovery that fly centromeres are filled w/ islands of retroelements (not just homogenous satellite repeats)
centromeres are sites of evolutionary warfare since they compete for inheritance during female meiosis
transposons (“soldiers”) can contribute to centromere function and possibly increasing strength for centromere drive
What does comparison of human centromeres reveal about their variability and evolution?
gapless assemblies: genome assemblies have gaps in repetitive regions
humans are highly variable: ~50% of centromeric sequences can’t even be aligned b/w two individuals due to higher order repeat sequences
the positions of centromeres can differ by >500 kilobases
the size of centromeres can differ by up to 3 fold
further evidence that human chromosome evolution is shaped by complex and often non-Mendelian forces
human chromosome evolution is dynamic and influence by repetitive DNA and not just coding sequences
Conclusions
repetitive DNA has important functions in biology and disease
the full sequencing of genomes necessitates the incorporation of long read sequencing approaches
several sequencing approaches are combined tgt to assemble an accurate genome
short read sequencing gives reads of the highest accuracy whereas long read sequencing enables analysis of repetitive regions