Looks like no one added any tags here yet for you.
What is the PDH complex? Where is it located within the cell? What types of tissues are it prevalent in?
The pyruvate dehydrogenase complex is located on the inner mitochondrial membrane. It is distributed among the mitochondria of cells and is abundant in cells high in mitochondria, such as skeletal and heart muscle tissue.
How does pyruvate enter the PDH complex?
Through pyruvate channels called pyruvate translocate alongside H+ symport
What is the overall summarized constitution of the PDH complex?
This complex consists of 3 enzymes, 5 coenzymes, ATP coenzyme, and other proteins.
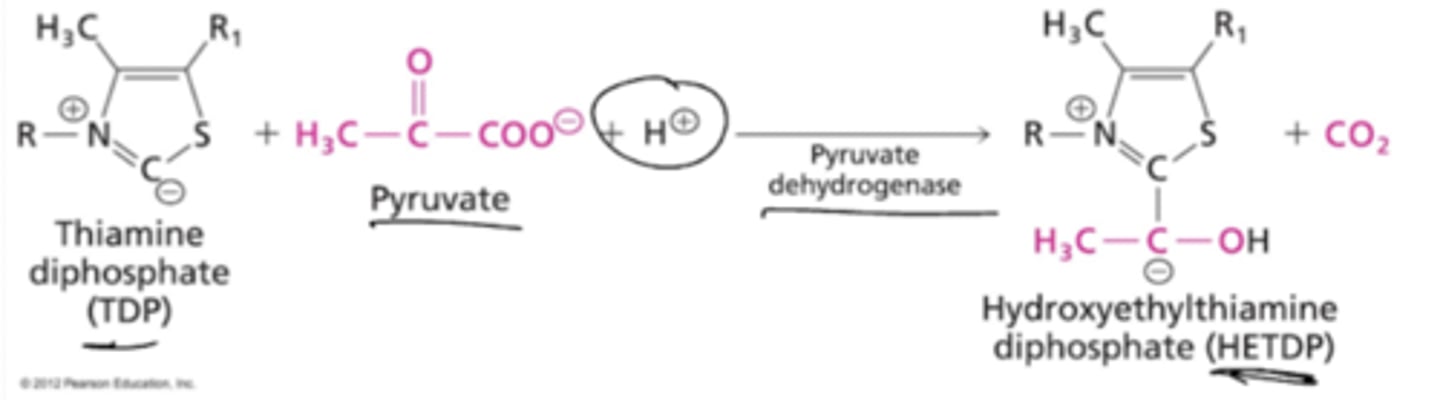
What is the first enzyme of the PDH complex? Describe its reaction.
Pyruvate Dehydrogenase (aka pyruvate decarboxylase) catalzes the reaction of thiamine diphosphate (TDP) and pyruvate with an H+ ion to form hydroxyethylamine diphoshate (HETDP) and CO2.
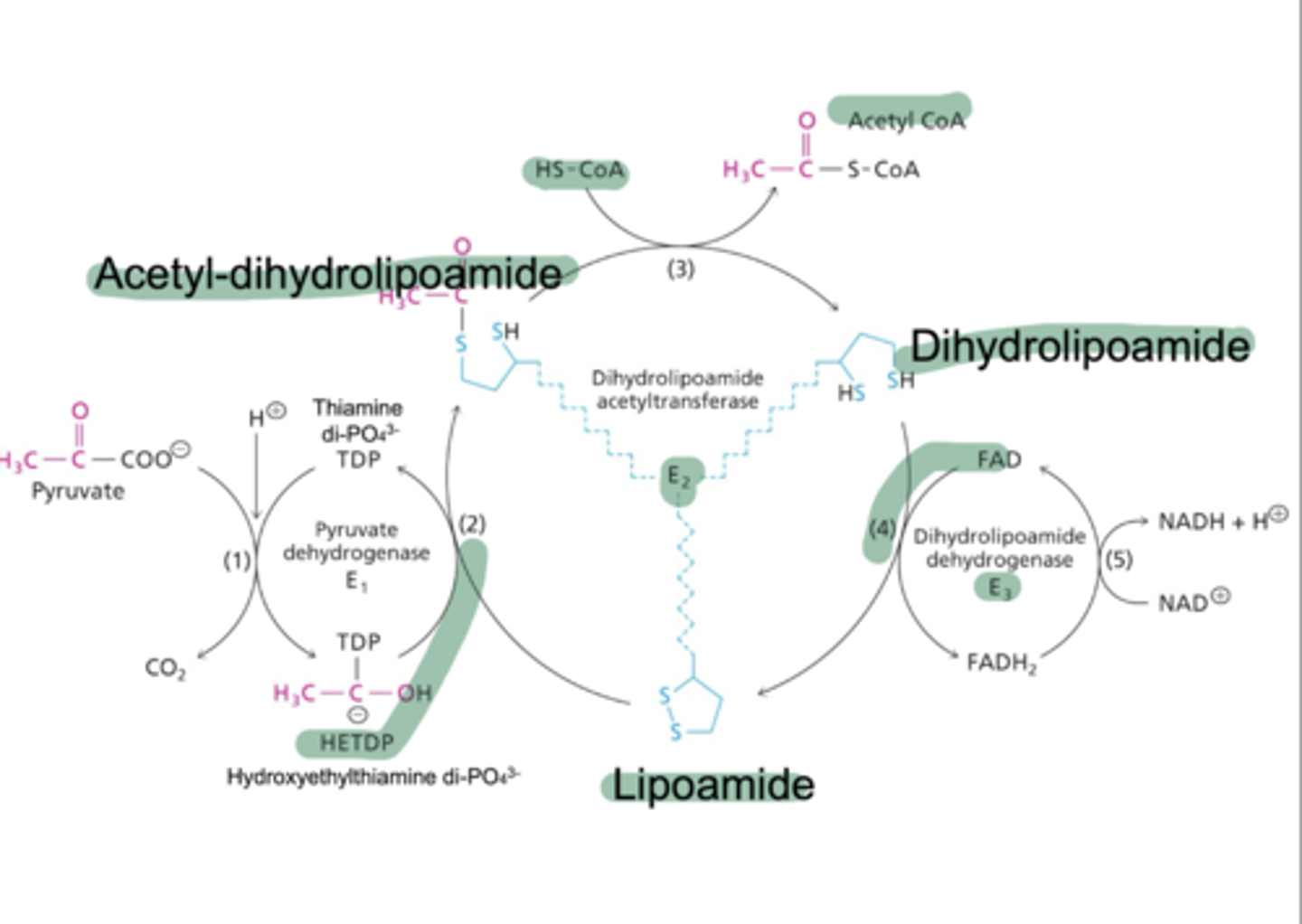
What is the second enzyme of the PDH complex? Describe its reaction.
Dihydroxylipoamide acetyltransferase catalyzes the reaction of HETDP with lipoamide to form a ylid and acetyl-dihydrolipoamide via acetylation. Acetyl-dihydroxylipoamide reacts with CoA to form dihydrolipoamide and acetyl CoA.
What is the third enzyme of the PDH complex? Describe its reaction.
Dihydroxylipoamide dehydrogenase catalyzes the reaction of dihydrolipoamide with an E3-FAD complex to re-form lipoamide via oxidation for reuse, and a reuced E3-FADH2 complex. This reduced complex with reacted with NAD+ to form an oxidized E3-FAD and a reduced NADH + H+.
What are the five cofactors involved in the PDH complex?
TLC For Nancy; Thiamine Pyrophosphate (B1), lipoic acid, CoA, FAD, and NAD+. NAD+ and CoA are cosubstrates used directly in reactions, TPP and lipoamide act as a swinging arm on E2, transfering the 2 carbon unit from the active site of E1 to E3 by substrate channeling. ATP is a regulator of the process.
How is the PDH complex regulated through molecularly?
Reactants and coeznymes (pyruvate, NAD+, CoA) are activators of the complex (NAD+ activates E3, CoA activates E2). Products such as NADH, acetyl CoA, and CO2 inhibt the complex (NADH inhibits E3, and acetyl CoA inhibits E2).
How is the PDH complex regulated through enzymatically?
Pyruvate dehydrogenase kinase and phosphatase (PDK and PDP) are enzymatic regulators of the complex. PDK phosphorylates PDH complex, rendering it inactivate, which PDP dephosphorylates it, activating it. PDP activity is also stimulated by Ca2+, which activates the PDH complex.
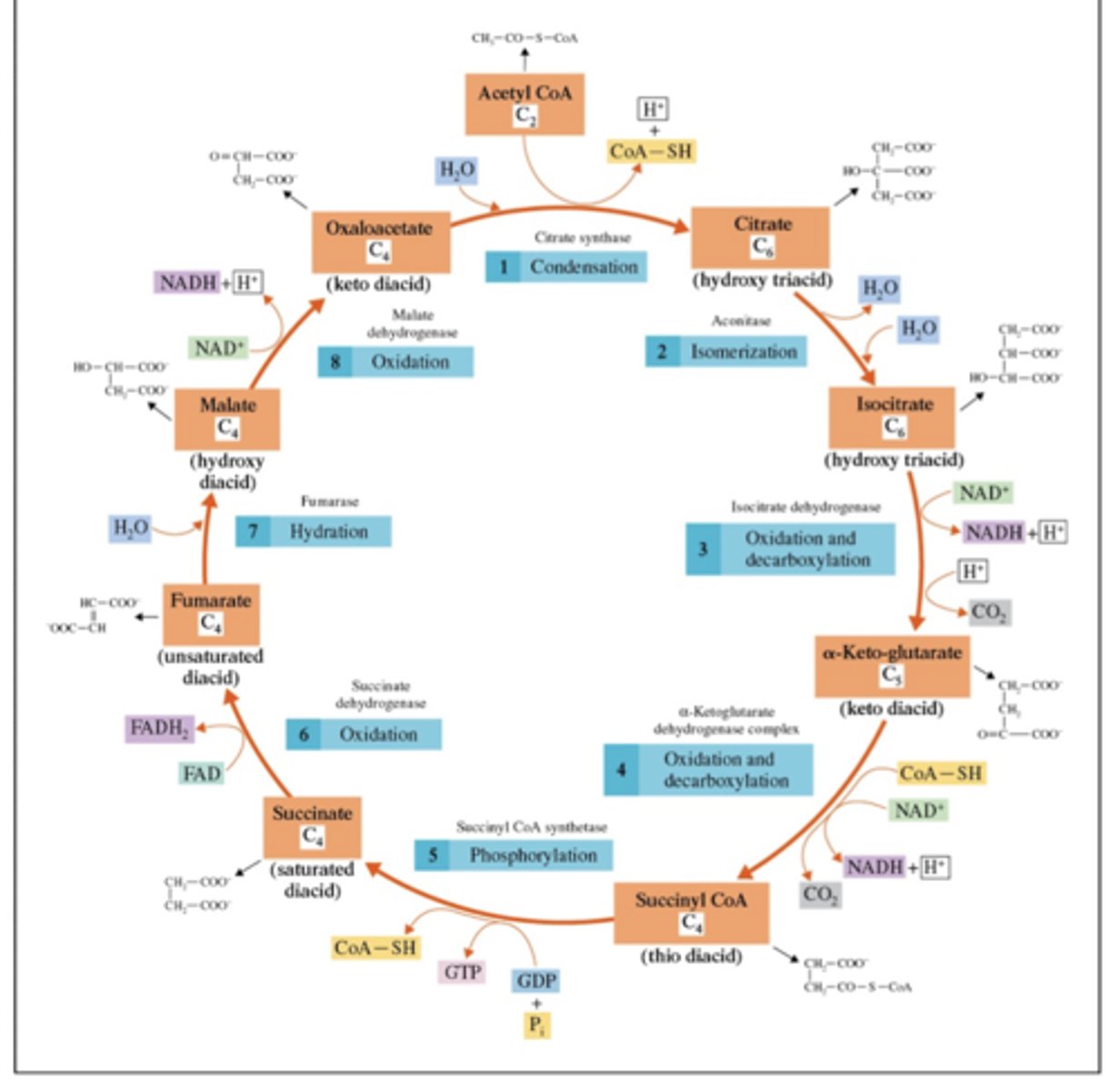
Draw out the TCA cycle, including reactants, products, and intermediates.
How does the concentration of NADH affect the TCA cycle?
NADH acts as a feedback inhibitor of the enzymes that produce it, signifying that there is a sufficient quantity of reduced electron carriers produced to further be oxidized. It is a feedback inhibitor of a-ketoglutarate dehydrogenase and isocitrate dehydrogenase
How does the concentration of succinyl-CoA affect the TCA cycle?
Succinyl CoA acts as competitive inhibitor of citrate synthase, and a feedback inhibitor of a-ketoglutarate dehydrogenase
How does the concentration of citrate affect the TCA cycle?
Citrate acts as a feedback inhibitor for many enzymes in the glucose to ATP chain. It acts as a feedback inhibitor for citrate synthase, as well as an inhibitor for PFK-1. It also promotes fatty acid synthesis - also promotes gluconeogenesis
How does the concentration of ATP and ADP affect the TCA cycle?
ATP is an inhibitor for citrate synthase, isocitrate dehydrogenase, and a-ketoglutarate dehydrogenase
ADP activates isocitrate dehydrogenase, leading to increased TCA speed
How is citrate synthase a regulatory enzyme for the TCA cycle?
It regulates the TCA cycle through allosteric regulation.
Inhibitors include ATP, NADH, succinyl CoA, and citrate (redirects to fatty acid synthesis).
Activators include ADP and oxaloacetate/acetyl-CoA.
How does isocitrate dehydrogenase regulate the TCA cycle?
Through allosteric regulation.
Activated by ADP, NAD+, and calcium ions.
Inhibited by ATP, NADH, and a-ketoglutarate formation.
How does a-ketoglutarate regulate the TCA cycle?
Regulated by allosteric regulation.
Activators include Calcium ions, NAD+.
Inhibitors include ATP, NADH, and succinyl CoA (product inhibitor).
How is a-ketoglutarate dehydrogenase similar to the PDH complex? What enzymes are present in the aKGDH complex?
These two coplexes require the same coenzymes: thiamine pyrophosphate, lipoamide/lipoic acid, CoA, FAD, and NAD+. The enzymes are a-ketoglutarate dehydrogenase (E1 with TPP), succinyltransferase (E2 with lipoamide prosthetic group), dihydrolipoamide dehydrogenase (E3 with FAD).
What are the main characteristics of the succinate dehydrogenase complex?
It is located on the inner mitochondrial membrane, only the trans isomer of the dehydrated substrate is formed, and the substrate analog malonate is a competitive inhibitor of the SDH complex.
What are some of the inputs/substrate of the TCA cycle?
Carbohydrates (they are turned into pyruvate), fatty acids (turned into acetyl CoA), alanine (turned into pyruvate), glutamine (into a-ketoglutarate), odd chain fatty acids (to propionyl CoA into succinyl CoA), and some amino acids (into succinyl CoA or fumarate)
What are some of the outputs/intermediate products of the TCA cycle?
Oxaloacetate (turned into amino acids, urea, pyrimidine nucleotides or carbohydrates), fatty acids and steroids (from citrate), a-ketoglutarate (glutamate to amino acids and nucleotides), and succinyl CoA (into porphyrins)
What is the energy yield of NADH, FADH2/QH2? What does this indicate about the complete oxidation of acetyl CoA?
NADH yields 2.5 ATP, FADH2/QH2 yields 1.5 ATP, and the total oxidation of one acetyl CoA (in which 2 total come from glucose) yields 10 ATP (each individual yields 3 NADH, 1 QH2/FADH2, and 1 ATP/GTP
What are the two shuttles for NADH? What are their respective yields?
The Malate-Aspartate shuttle NADH yields 2.5 ATP (leading to 32 ATP per glucose), and the Glycerol Phosphate shuttle NADH yields 1.5 ATP (leading to 30 ATP per glucose)
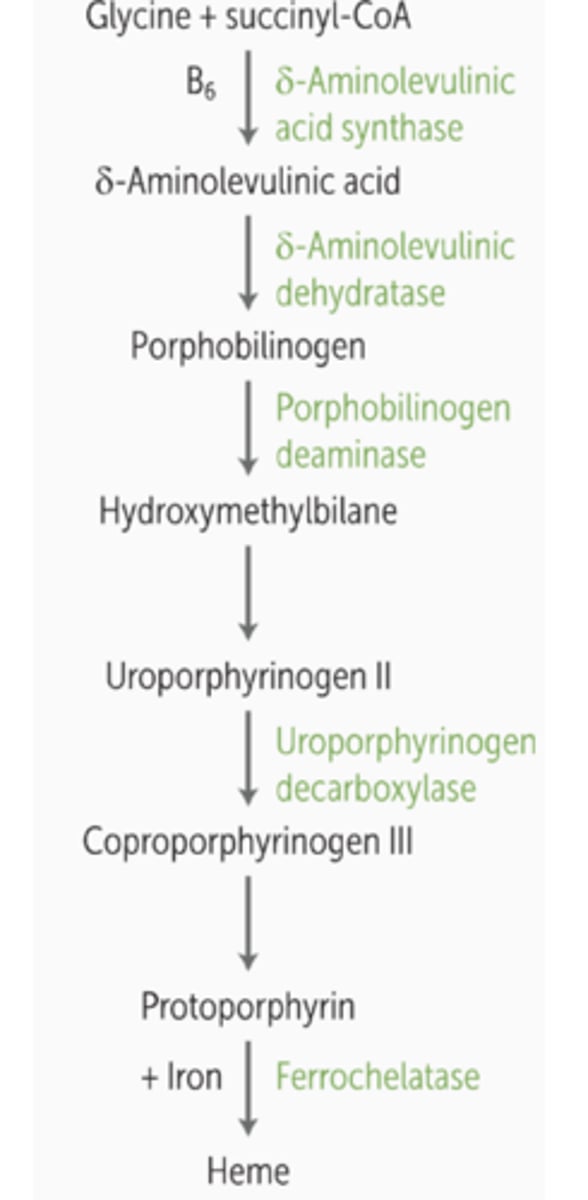
Name the pathway and intermediates for the conversion of succinyl CoA to heme synthesis.
Name the characteristics for the conversion of oxaloacetate to gluconeogenesis.
When glycolysis is "reversed", oxaloacetate is used to convert back to glucose
Name the characteristics for the conversion of a-ketoglutarate and oxaloacetate.
A-ketoglutarate is used to synthesize amino acids such as glutamate, glutamine, proline, and arginine by enzymes glutamate dehydrogenase, glutamine synthetase. Furthermore, a-KG combines with a free ammonia with NADH to form glutamate, which can be transaminated. Oxaloacetate can be used to synthesize aspartate, asparagine, methionine, lysine, and threonine by enzymes aspartate aminotransferase, and asparagine synthetase.
OAA is transaminated with glutamate to form aspartate, which is further converted into others
Name the characteristics for the conversion of citrate and fatty acids.
Citate is used as a carrier molecule for transporting acetyl-COA out of the mitochondria and into the cytoplasm - then citrate is converted back to acetyl COA and oxaloacetate by citrate lyase (which is activated by insulin), which the acetyl COA is used for fatty acid synthesis
Acetyl CoA is converted to malonyl COA by acetyl-CoA carboxylase (activated by citrate), and malonyl COA enters fatty acid synthesis complex (FAS) where it is elongated to form palmiate (16:0), a key fatty acid. Therefore, FAS is regualted by citrate as a precursor and regulator. ACC is activated by citrate, high citrate = nrg excess, and ATP demand increases, leading to citrate redirection into the TCA for energy prod
What are the causes of arsenic poisoning?
This occurs due to exposure of arsenic containing compounds, which inhibits PDC (by binding to lipoic acid, a-ketoglutarate dehydrogenase, and branched-chain ketoacid dehydrogenase, disrupting TCA, leading to impaired ATP production and lactic acidosis). This disrupts OXPHOS (my mimicing phosphoate and replacing it in ATP synthesis, forming arsenate esters that hydrolyze easily, reducing ATP prod and leading to cell energy failure)
Furthermore, this leads to oxidative stress, generating ROS (leads to mitochondrial damage, lipid peroxidation, and apoptosis)
What are the clinical manifestions of acute and chronic arsenic exposure?
Acute poisoning appears as severe gastrointestinal symptoms (vomiting, rice-water diarrhea, and pain), cardioascular collapse (hypotension , arrhythmias, shock), and neurological issues (such as seizures, delirium, and coma). Chronic poisoning appears as skin changes (hypigmentation, palmar and planter keratosis, Mee's lines), neuropathy (stockingh-glove distribution parasthesia, weakness), cancer risk (high risk skin, lung, bladder, and liver), and bone marrow suppression (anemia, and leukopenia)
How is arsenic poisoning diagnosed?
Blood and urine levels of arsenic in acute cases, hair and nail analysis in chronic hcases, and metabolic acidosis with high lactate (PDH inhibition)
How is arsenic poisoning treated?
Immediate decontamination, chelation hterapy (dimercaprol - BAL, or DMSA - succimer binds arsenic and promotes excretion). Also, penicillamine may be used for chronic cases. Supportive care includes IV fluids, correction of electrolyte imbalance, and symptom management
What are the causes of beriberi?
Also known as vitamin B1 deficiency. It is caused by poor intake of B1 (such as through polished rice, and malnutrition), increased demand for B1 (pregnancy, lactation, hyperthyroidism, fever, and sepsis), and malabsorption (chronic diarrhea, bariatric surgery), and alcoholism (which impairs absorption and utilization)
What are the symptoms of dry beriberi?
Dry berberi (neurological manifestations): include peripheral neuropathy (symmetrical affecting the legs more than arms), parasthesia (numbness, muscle wekaness, foot drop), and Wernicke-Korsakoff syndrome (in alcoholics). Wernicke encephalopathy leads to confusion, ataxia, and opthalmopegia. Korsakoff leads to amnesia, confabulation, and psychosis
What are the symptoms of wet beriberi?
high-output heart failure due to vasodilation. edema , tachycardia, dyspnea, cardiomegaly - can lead to shock and death of untreated.
Infantile (in breastfed infants of thiamine-deficient mothers): Heart failure, aphonia, vomiting, convulsions fatal if not immediately treated
What is PDH complex deficiency and what are some of its common features?
PDH complex deficiency is the impaired conversion of pyruvate to acetyl-CoA. Some features include neurological symptoms (developmental delays, hypotonia, and ataxia), lactic acidosis, and progressive neurodegeneration
How is PDH complex deficiency diagnosed?
Analyzing elevated lactate/pyruvate levels in blood, genetic testing for PDH complex gene mutations, and enzyme activity assays.
How is PDH complex deficiency treated?
Keto diet, DCA (dichloroacetate) to stimulate PDP activity, and thiamine supplementation.