Role of Drug Metabolism – Pharmacokinetics (DMPK) and Toxicity in Lead Optimisation
1/93
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
94 Terms
What are the main stages of drug discovery outlined in the DMPK document?
Exploration, Lead Selection, Lead Optimisation, Development"
What is the primary focus of Lead Optimisation in drug discovery?
Evaluate compound properties, including in vivo pharmacokinetics, and optimize activity, bioavailability, or other factors"
ADME
What was the biggest reason for compound failure in early drug discovery? Explain all reasons!
Poor pharmacokinetics (PK) and bioavailability
—
Clinical Safety: Toxicity or adverse effects in humans (e.g., liver damage).
Efficacy: Drug fails to work as intended in clinical trials.
Formulation: Challenges in creating stable, deliverable doses (e.g., solubility).
PK/Bioavailability:Poor absorption, distribution, metabolism, or excretion (ADME).
Commercial/Toxicology: Long-term toxicity or non-viable production costs.
Cost of Goods: Expensive to manufacture at scale. (Chemical Tractability)
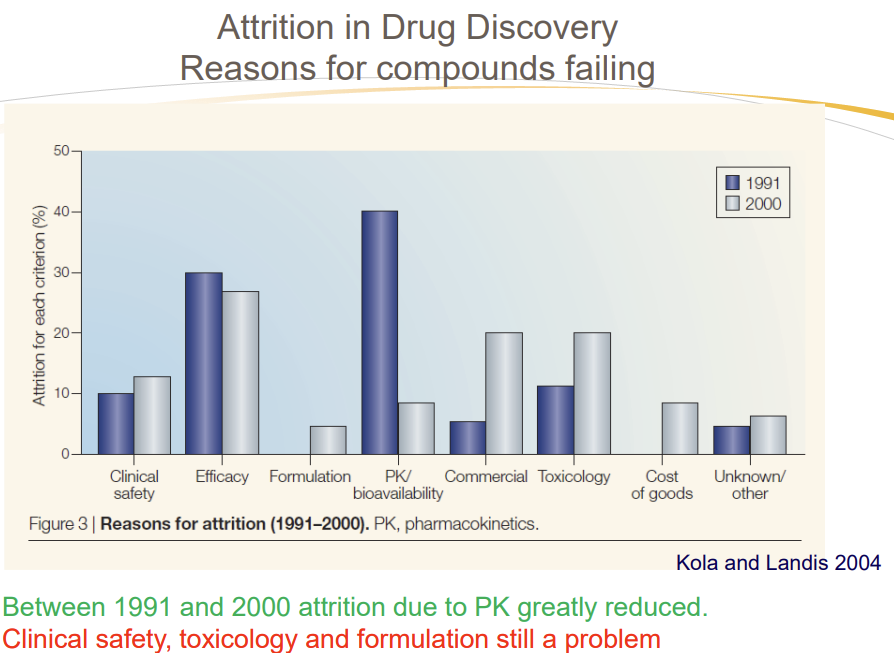
What was the attrition rate due to PK-related failures in early drug discovery?
Approximately 40%"
How did evaluating PK properties earlier impact failure rates by 2000?
Reduced PK-related failures to under 10%"
What does Pharmacokinetics (PK) study?
How the body interacts with the drug for the entire duration of exposure
How drug concentration changes as it moves through the body
What are the two main processes in PK?
Absorption and Distribution (In), Metabolism and Elimination (Out)"
What does ADME stand for in pharmacokinetics?
Absorption, Distribution, Metabolism, Elimination"
What is Absorption in the context of ADME?
The process a substance undergoes before entering systemic circulation"
What is Distribution in ADME?
The dispersion of a drug in body fluids and tissues"
What is Metabolism in ADME?
The biotransformation of a drug to a more polar entity for easier elimination"
What is Elimination in ADME?
The excretion of the drug from the body, e.g., in urine via the kidney"
What factors affect oral absorption and bioavailability?
Solubility, Permeability, Metabolism"
Why is the bioavailable dose lower for oral (PO) dosing compared to intravenous (IV) dosing?
Oral drugs face barriers like Dissolution, Solubility, Permeability
• The drug must dissolve in the stomach to be absorbed. Insoluble compounds form suspensions, pass through the intestines, and are eliminated in feces without absorption.
Solubility, influenced by the compound's ionization state.), absorption in the Intestines
• The dissolved drug must cross the intestinal membrane to enter the bloodstream.
Permeability, determined by the compound's lipophilicity.), permeability, and liver metabolism
• After absorption, the drug passes through the liver, where it may be metabolized by various enzymes (CYP450: CYP3A4 / CYP2D6 / CYP2C9 / CYP1A2). This reduces the amount of active drug reaching systemic circulation.

What is the first step for an orally administered drug to reach systemic circulation?
Dissolution in the stomach"
What key property influences dissolution in the stomach?
Solubility, influenced by the compound's ionization state"
What is the second step for oral drug absorption?
Absorption in the intestines"
What key property determines intestinal absorption?
Permeability, determined by the compound's lipophilicity"
What happens to a drug after intestinal absorption before reaching systemic circulation?
It passes through the liver, where it may be metabolized.
(CYP450: CYP3A4 / CYP2D6 / CYP2C9 / CYP1A2)
What is the Lipinski Rule of Five used for?
Predicting oral bioavailability and poor permeability/absorption"
What are the criteria of the Lipinski Rule of Five?
MW < 500, log P < 5, H-bond donors < 5, H-bond acceptors < 10"
What is an exception to the Lipinski Rule of Five?
Transporter substrates"
What is the significance of ≥30% bioavailability in preclinical studies?
High likelihood of good bioavailability in humans
Bioavailability is the fraction of an administered drug that reaches systemic circulation in its active form, becoming available to act at the target site.
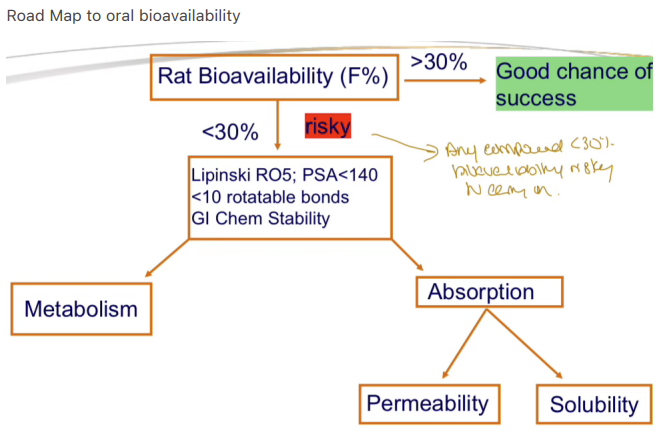
Why is testing every compound in vivo for PK studies impractical?
Time constraints and ethical concerns about excessive animal use"
What is Solubility in the context of drug development?
The maximum amount of a compound that can be dissolved in a certain volume of a solvent"
What factors influence solubility?
pH of the solvent:
↑ pH (alkaline) → ↑ Solubility (ionized COO⁻ form dominates).
↓ pH (acidic) → ↑ Solubility (ionized NH₃⁺ form dominates).
Temperature:
↑ Temperature → ↑ Solubility (endothermic dissolution, e.g., sugar in water).
Which form of a compound is more soluble: charged or neutral?
Charged (ionized) form"
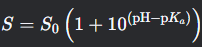
What is the Henderson-Hasselbalch equation for solubility of an acid?
S = S0 (1 + 10(pH - pKa))
What is the Henderson-Hasselbalch equation for solubility of a base?
S = S0 (1 + 10(pKa - pH))
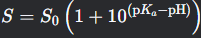
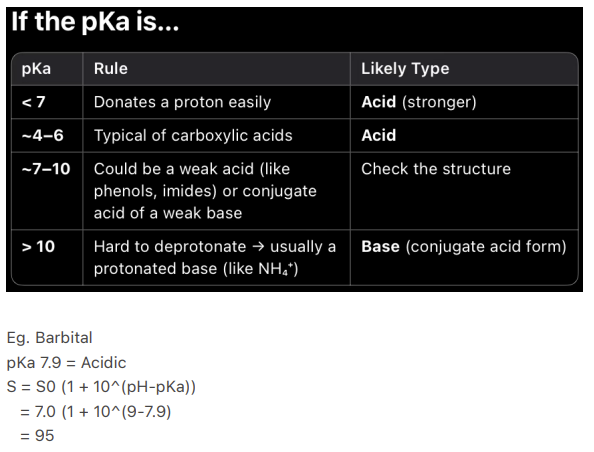
What does a pKa < 7 indicate about a compound?
It donates a proton easily, likely a stronger acid"
What pKa range is typical for carboxylic acids?
4-6"
What does a pKa of 7-10 suggest about a compound?
Could be a weak acid (e.g., phenols, imides) or conjugate acid of a weak base"
What does a pKa > 10 indicate?
Hard to deprotonate, usually a protonated base (e.g., NH4+)"
What is the pKa of Barbital, and is it acidic or basic?
7.9, Acidic"
What are strategies to improve solubility?
Incorporate ionizable/polar groups
Reduce lipophilicity
Optimize particle size
Use amorphous forms (Random, disordered structure with higher solubility due to lack of lattice energy)
Out-of-plane substitution (Adding functional groups to a rigid, planar (flat) drug molecule in a non-planar (3D) orientation to disrupt crystal packing and improve solubility)

How does adding an amino group affect solubility?
Adding ionizable groups increases solubility, e.g., from 8 µg/mL to 300 µg/mL"
How does adding an oxygen-containing group (e.g., hydroxyl) affect solubility?
Further increases solubility, e.g., to 500 µg/mL due to enhanced hydrogen bonding"
Why does reducing lipophilicity improve solubility?
Allows water molecules to form hydrogen bonds around less lipophilic groups.
Polar groups attract water’s oxygen atoms, forming a hydration shell that enhances dissolution.
How does reducing particle size enhance solubility?
Increases surface area, facilitating dissolution"
Why are amorphous compounds more soluble than crystalline ones?
Amorphous compounds lack ordered lattice structure, requiring less energy to dissolve"
What are negative effects of low-solubility compounds?
Poor absorption/bioavailability
Insufficient IV dosing
Low bioassay activity
Erratic assay results
Expensive formulations (Higher conc of drug needed to overcome poor bioavailability)
Frequent high-dose administration
What is Kinetic Solubility?
The concentration at which a compound starts precipitating from a supersaturated solution.
A supersaturated solution contains more dissolved solute than its equilibrium solubility limit under normal conditions, causing instability since It is a metastable state (not at true equilibrium).
How is Kinetic Solubility measured?
The concentration at which a compound starts precipitating from a supersaturated solution:
Dissolve in organic solvent (e.g., DMSO)
Add aqueous buffer until precipitation
Monitor with ChemiLuminescent Nitrogen Detection (HTS)
What is Thermodynamic Solubility?
Solubility measured by adding aqueous solvent to solid with long mixing times (>24 h) to achieve equilibrium
– Aqueous solvent added to solid directly
– Long mixing times (>24 h) to achieve equilibrium
– Solubility value varies with form of the solid (amorphous, crystalline, polymorph
- Different crystalline form, hydrate, solvate)
– Amorphous solid has higher solubility
– Used when a candidate is identified for development
Which is more accurate: Kinetic or Thermodynamic Solubility?
Thermodynamic Solubility"
What factors affect Thermodynamic Solubility values?
Form of the solid (amorphous, crystalline, polymorph, hydrate, solvate)"
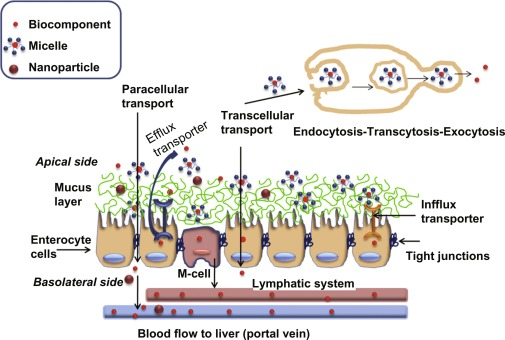
What is the primary mechanism of intestinal permeability?
Passive diffusion (95%) from Gut Lumen → Apical Membrane of Enterocytes → Enterocyte (Cytoplasm) → Basolateral Membrane → Blood (Systemic Circulation)
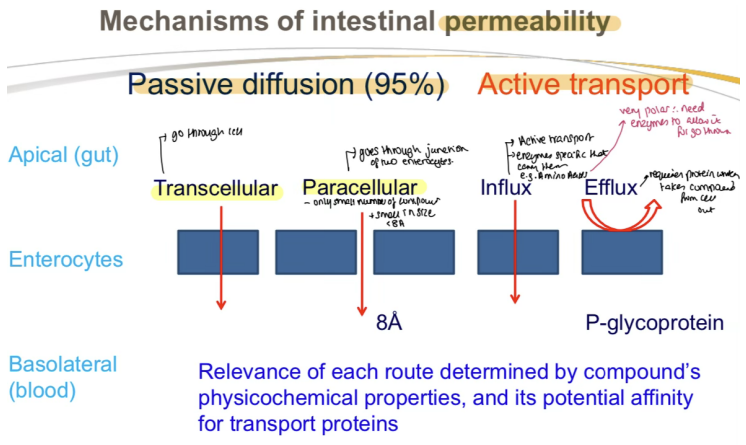
What are the two types of passive diffusion for intestinal permeability?
Transcellular and Paracellular
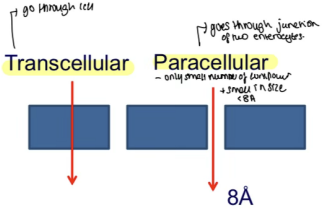
What is Transcellular Diffusion?
Drug crosses enterocytes by penetrating apical membrane, diffusing through cytoplasm, and exiting basolateral membrane"
What are the characteristics of drugs absorbed via Transcellular Diffusion?
MW > 300, log P > 0, unionized, soluble, HBA < 10, HBD < 5
—
Molecular Weight (MW) < 300–500 Da: Smaller molecules diffuse more easily through lipid membranes.
logP > 0 (Lipophilicity): Positive logP indicates higher lipid solubility (hydrophobic drugs cross membranes faster).
Unionised Form Dominates: Neutral molecules (non-ionized at physiological pH) bypass the polar barrier of cell membranes.
Governed by Henderson-Hasselbalch: Acids absorb best in acidic pH (stomach). / Bases absorb best in alkaline pH (intestines).
Adequate Solubility: Must dissolve in aqueous GI fluids to reach the membrane (balance between lipophilicity and solubility).
Hydrogen Bond Acceptors (HBA) < 10: Fewer HBA groups (e.g., carbonyl, ether) reduce polarity, enhancing membrane permeability. Desolvation penalty
Hydrogen Bond Donors (HBD) < 5: Fewer HBD groups (e.g., –OH, –NH₂) minimize strong interactions with water, favoring lipid partitioning. Desolvation penalty
What is Paracellular Diffusion?
Drug crosses around enterocytes through water-filled pores between cells
What are the characteristics of drugs absorbed via Paracellular Diffusion?
Small Size: MW <250–300 Da (must fit through tight junctions).
Hydrophilic: logP < 0 (polar/charged molecules bypass lipid membranes).
Charge Preference: Cationic (+) drugs favored due to negative charge of tight junction proteins (e.g., claudins).
Tight Junction Size:
Jejunum: ~8–10 Å (leakiest → better absorption).
Colon: <8 Å (tighter → reduced absorption).
Absorption Site: Primarily upper small intestine (jejunum).
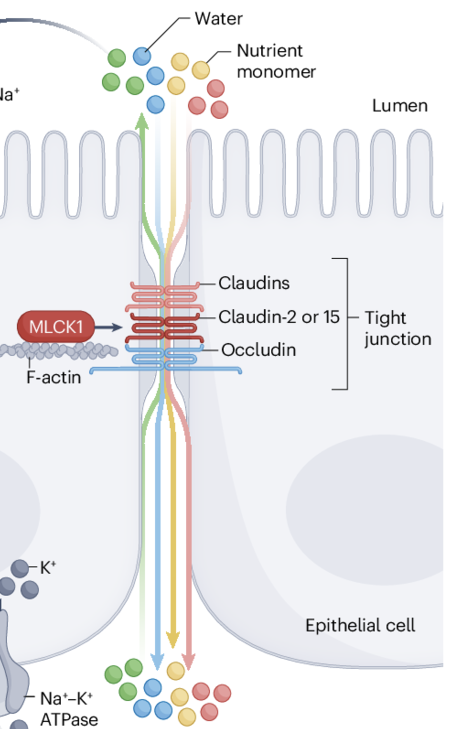
Why do positively charged molecules pass more easily through paracellular routes?
Junctional complex is negatively charged"
How do tight junctions affect paracellular absorption?
Become tighter (<8 Å) from jejunum to colon, making absorption more difficult"
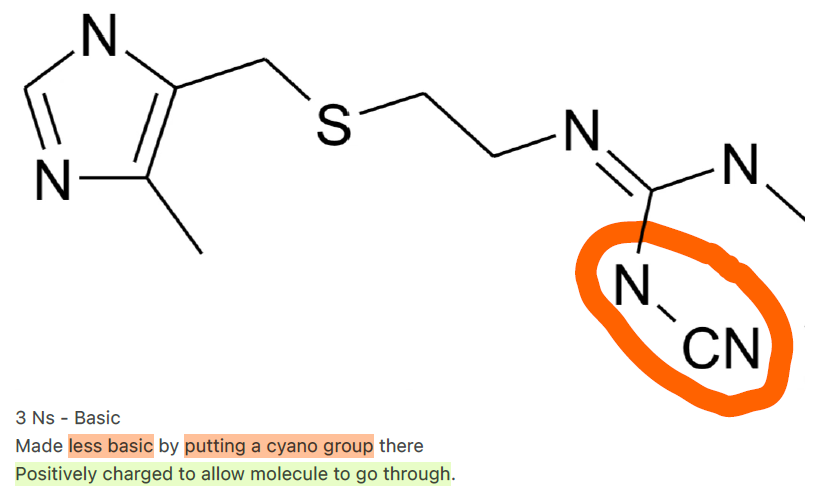
What is an example of a drug absorbed via Paracellular Diffusion?
Cimetidine"
What modification was made to cimetidine to enhance paracellular absorption?
Made less basic by adding a cyano group, allowing positive charge
Not too charged (avoids non-specific binding) with -ve claudin tight junction.
Overly cationic drugs (e.g., high pKa) may: Bind too tightly to junction proteins → slow diffusion. / Trigger steric hindrance if charge distorts the molecule’s shape.
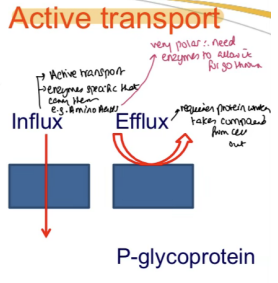
What is Active Transport in the context of intestinal absorption?
Extracellular / Intracellular:
Extracellular:
Transporters in the small intestinal mucosa facilitate absorption
Proteins (e.g., P-glycoprotein) actively transport compounds OUT of the enterocyte, back into the intestinal lumen, REDUCING absorption. This mechanism often limits bioavailability for certain drugs.

What is Active Transport in the context of intestinal absorption? (2)
Intracellular:
A large number of transporters are expressed by the small intestinal mucosa and play a major role in absorption
Substrates for these transporters exhibit absorption higher than expected from their diffusion across membranes
Influx subject to saturation of transporters
Enzyme specific carriers (Amino Acids)
Medicinal chemists have not exploited influx much in drug design – something for future
What is a characteristic of Active Transport substrates?
Exhibit higher absorption than expected from diffusion
What are examples of influx transporters?
Peptide Transporters (PEPT1, PEPT2) [Cephalexin]
Monocarboxylic acid transporters [Pravastatin]
Organic anion (OATP1, OAT1, OAT3) [Fexofenadine]
Organic cation (OCT1) [L - Carnitine]
![<ul><li><p>Peptide Transporters (PEPT1, PEPT2) [Cephalexin]</p></li><li><p>Monocarboxylic acid transporters [Pravastatin] </p></li><li><p>Organic anion (OATP1, OAT1, OAT3) [Fexofenadine] </p></li><li><p>Organic cation (OCT1) [L - Carnitine] </p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/4ca27ead-5452-4298-8ee3-dbb908bd0356.png)
What is Active Efflux Transport?
Transport of drugs from inside the cell back into the luminal space"
What are examples of efflux transporters?
P-glycoprotein (Pgp)
Breast Cancer Resistance Protein (BCRP)
Where is efflux transport found?
Brain, Liver, Kidney"
How does efflux transport in the brain function?
Protects brain tissue by removing drugs"
How does efflux transport in the liver function?
Removes drug and metabolites to bile for elimination"
What is Permeability in in vitro assays?
The velocity of compound passage through a membrane barrier, e.g., Caco-2 cell line"
What does Papp (cm/s × 10^-6) correlate to?
Apparent Permeability - Human absorption and oral bioavailability
What is an alternative to the Caco-2 cell line for permeability assays?
MDCK cell line
What are strategies to improve permeability?
Change ionizable group to non-ionizable (e.g., acid to hydroxyl)"
1. Change Ionizable Group to Non-Ionizable (Acid → Hydroxyl): Neutral –OH avoids ionization, enhancing transcellular diffusion.
Modification: Convert carboxylic acid (–CO₂H) to a hydroxyl group (–OH).
2. Increase Lipophilicity (Add Methyl Group): Lipophilic groups improve membrane partitioning (logP ↑).
Modification: Add a methyl (–CH₃) or trifluoromethyl (–CF₃) group.
3. Replace Polar Group with Isostere (Acid → Tetrazole): Tetrazole mimics acid pKa but reduces H-bonding.
Modification: Swap –CO₂H with tetrazole (acidic but less polar).
4. Prodrug Approach (Ester Addition): Ester masks polar groups, improving absorption; hydrolyzed by esterases to active drug.
Modification: Add ester (e.g., pivaloyloxymethyl to ampicillin → pivampicillin).
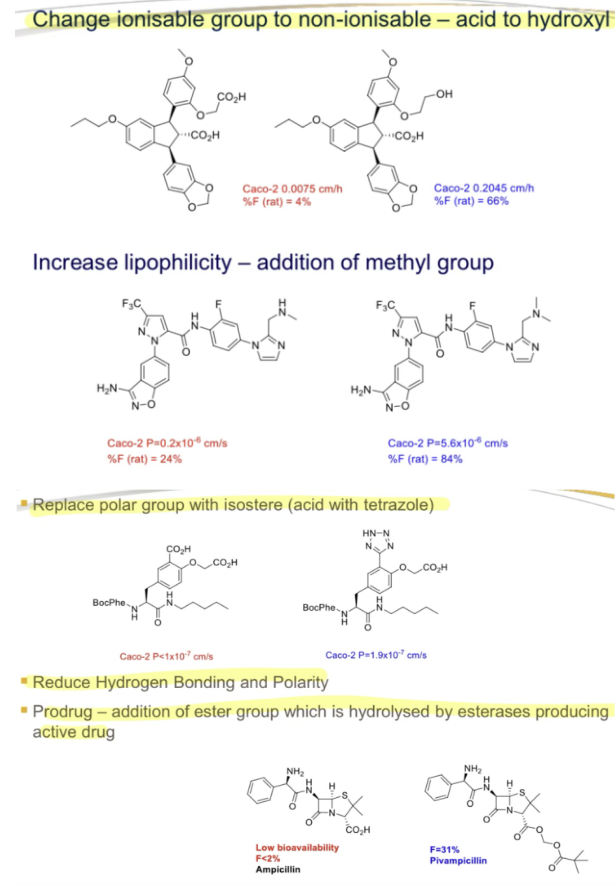
What is an example of a permeability structure modification?
Replace polar group with isostere (e.g., acid with tetrazole)"
What is a Prodrug strategy for permeability?
Add an ester group, hydrolyzed by esterases to produce the active drug"
What is an example of a drug with low bioavailability improved by a prodrug?
Ampicillin, bioavailability <2%"
What are formulation strategies for Class III compounds (high solubility, low permeability)?
Prodrug strategy to increase permeability"
What are the challenges with Class IV compounds (low solubility, low permeability)?
Very risky and costly, requiring many years in development"
What is the relationship between charge, ionization, hydrogen bonding, and solubility/permeability?
Increasing charge, ionization, or hydrogen bonding increases solubility but decreases permeability"
What is the relationship between lipophilicity, size, and solubility/permeability?
Increasing lipophilicity and size increases permeability but decreases solubility"
How does the range of permeability compare to solubility?
Permeability varies over a narrower range than solubility
Permeability: ~0 to >10 × 10⁻⁶ cm/s (Papp)
Solubility: ~ng/mL to >100 mg/mL
What is the fold difference between a high- and low-permeability compound?
Up to 50-fold
50 times more permeable!
What is the fold difference between a high- and low-solubility compound?
Up to one million-fold"
What is the net effect of a structural modification that improves solubility by 1000-fold but reduces permeability by 10-fold?
A 100-fold improvement in absorption
Important to consider both solubility and permeability determine extent of absorption.
What is the role of medicinal chemists in managing solubility and permeability?
Balance the two properties to optimize absorption"
Physicochemical properties for Solubility and Permeability are often opposed:
1. Trade-offs Between Solubility & Permeability
Increasing solubility (but reducing permeability):
↑ Charge/Ionization (e.g., –COO⁻, –NH₃⁺) → Better water interaction.
↑ Hydrogen Bonding (e.g., –OH, –NH₂) → More polar, less lipid-friendly.
Increasing permeability (but reducing solubility):
↑ Lipophilicity (e.g., –CH₃, –CF₃) → Better membrane crossing.
↑ Molecular Size (e.g., aromatic rings) → May hinder dissolution.
2. Magnitude of Variability
Permeability Range: ~50-fold difference between high/low.
Solubility Range: Up to 1,000,000-fold difference (e.g., 1 µg/mL vs. 1 g/mL).
3. Net Impact of Structural Changes
Example: A 1000× solubility boost with a 10× permeability drop still yields a 100× net gain in absorption.
4. Balancing Act in Drug Design
Goal: Optimize both properties (e.g., prodrugs, moderate logP, strategic H-bonding).
Tool: Lipinski’s Rule of 5 (guideline for oral drug-like balance).
5. Practical Implications
Low solubility? Use salt forms, nanosizing, or co-solvents.
Low permeability? Reduce H-bond donors/acceptors or target transporters.
What is the Biopharmaceutics Classification System (BCS) used for?
Classifying development candidates based on solubility and permeability for oral absorption"
What is the Biopharmaceutics Classification System for Development Candidates
1. Class I: High Solubility + High Permeability
Ideal for oral absorption – Minimal formulation challenges.
Example drugs: Metoprolol, Propranolol.
2. Class II: Low Solubility + High Permeability
Formulation Strategies:
Nanoparticles (increase surface area).
Salt forms (e.g., sodium or hydrochloride salts).
Lipid-based formulations (e.g., self-emulsifying drug delivery systems).
Example drugs: Ketoconazole, Naproxen.
3. Class III: High Solubility + Low Permeability
Strategies:
Prodrugs (e.g., Valacyclovir → Acyclovir).
Permeation enhancers (e.g., chitosan, SNAC).
Example drugs: Atenolol, Cimetidine.
4. Class IV: Low Solubility + Low Permeability
Implications:
High-risk development – Often requires alternative routes (IV, inhalation).
Example drugs: Paclitaxel (oral form), Furosemide.
Overview of Biopharmaceutics Classification System for Development Candidates
Class I/II: Focus on solubility enhancement for Class II.
Class III: Target permeability barriers (prodrugs, transporters).
Class IV: Rarely pursued orally due to high cost/complexity.
What are the characteristics of BCS Class I compounds?
High solubility and high permeability, ideal for oral absorption"
What are the characteristics of BCS Class II compounds?
Low solubility and high permeability"
What formulation strategies are used for BCS Class II compounds?
Nanoparticles, salts, lipid-based formulations to enhance solubility"
What is the challenge for BCS Class II compounds?
Drug can cross membrane easily but dissolves slowly in the GI tract"
What are the characteristics of BCS Class III compounds?
High solubility and low permeability"
What strategy is typically required for BCS Class III compounds?
Prodrug strategy to increase permeability"
What is the challenge for BCS Class III compounds?
Drug dissolves easily but struggles through membranes"
What are the characteristics of BCS Class IV compounds?
Low solubility and low permeability"
What are the development challenges for BCS Class IV compounds?
Very risky and costly, requiring many years in development"