Role of Drug Metabolism – Pharmacokinetics (DMPK) and Toxicity in Lead Optimisation 2
1/76
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
77 Terms
What is Plasma Protein Binding (PPB)?
Drugs reversibly bind to plasma proteins, reducing the concentration of free drug in solution
Only the unbound (free) fraction of a drug is pharmacologically active, able to diffuse into tissues, and exert therapeutic effects.
What percentage of plasma is protein?
8%"
What are the three types of plasma binding proteins and their binding preferences?
Albumin (binds acidic drugs)
a1-acid glycoprotein (binds basic drugs)
Lipoproteins (binds lipophilic drugs)
What is the significance of unbound "free" drug in PPB?
Only unbound 'free' drug permeates membranes, bringing about efficacy.
How is the free fraction of a drug measured in vitro?
Using assays like equilibrium dialysis to measure drug concentration after mixing with plasma proteins
Low concentration = highly protein bound
What is a potential drawback of high PPB?
Can restrict exposure to the target, though it can be both positive and negative
—
1. Positive Effect: PPB Increases Half-Life (t½) by Restricting Clearance
Mechanism:
Drugs bound to plasma proteins (e.g., albumin) are too large to be filtered by the kidneys or efficiently metabolized by enzymes.
Only the free (unbound) fraction is available for elimination.
The bound fraction acts as a reservoir, slowly releasing the drug into circulation.
Outcome:
Longer t½ → Less frequent dosing needed [Half Life]
Warfarin (99% PPB) has a long t½ (~40 hours) because most of it stays protein-bound, slowly releasing the active form.
Diazepam (98% PPB) remains in the blood for days, allowing sustained effects.
2. Negative Effect: PPB Can Restrict Target Exposure
Mechanism:
Only the free drug can cross membranes and reach tissues (e.g., brain, tumors, intracellular targets).
If a drug is highly protein-bound, very little free drug is available to interact with the target.
Outcome:
Reduced efficacy if the free concentration is too low.
Saquinavir (HIV protease inhibitor, ~98% PPB) has poor tissue penetration, limiting antiviral efficacy.
Some antibiotics (e.g., ceftriaxone) may have reduced tissue penetration due to high PPB.
Why are compounds with 99.9% PPB considered poor starting points?
They have very low free drug concentration, limiting their effectiveness"
What is Drug Metabolism?
The biotransformation of pharmaceutical substances in the body to facilitate easier elimination"
Where do most metabolic processes involving drugs occur?
In the liver, where most enzymes are found
CYP450: CYP3A4 / CYP2D6
What are Phase I reactions in drug metabolism?
Modifications of drug structure to a metabolite:
Oxidation (e.g., cytochrome P450 enzymes like CYP3A4)
Reduction (e.g., nitro groups → amines)
Hydrolysis (e.g., ester/amide cleavage)
Hydration (adding water to double bonds)
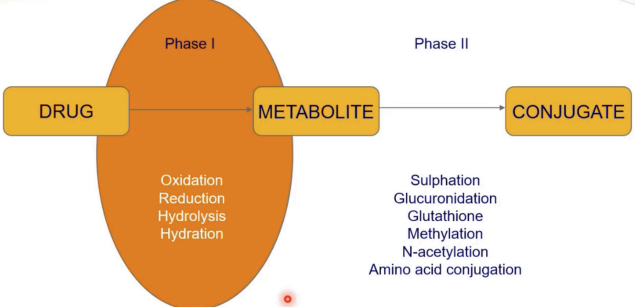
What are Phase II reactions in drug metabolism?
Additions (conjugations) of polar groups to the metabolite to form the conjugate
Glutathione conjugation (detoxifies reactive metabolites)
Methylation (e.g., COMT for catecholamines)
N-acetylation (e.g., NAT2 for isoniazid)
Amino acid conjugation (e.g., glycine conjugation of bile acids)
Glucuronidation (via UDP-glucuronosyltransferases, UGTs)
Sulfation (via sulfotransferases, SULTs)
What is the outcome of Phase I and Phase II metabolism?
Produces more polar products with higher aqueous solubility, readily excreted via bile and urine
How does metabolism affect drug exposure and bioavailability?
Increases clearance
Reduces exposure, causing LOW bioavailability
Exposure refers to the total amount of drug (AUC) that reaches systemic circulation. If metabolism is rapid, the drug is broken down before it can distribute or exert effects → lower AUC.
What is first-pass metabolism?
Drug metabolism that occurs before the drug reaches systemic circulation
Pre-systemic metabolism in the liver or gut wall after being absorbed from the gastrointestinal tract.
What are examples of Phase I metabolism reactions?
Oxidation, Reduction, Hydrolysis, Hydration"
What are examples of Phase II metabolism reactions?
Sulphation, Glucuronidation, Glutathione conjugation, Methylation, N-acetylation, Amino acid conjugation"
What are the most prominent enzyme families for Phase I metabolism?
Cytochrome P450 (CYP) and Flavine monooxygenase (FMO)
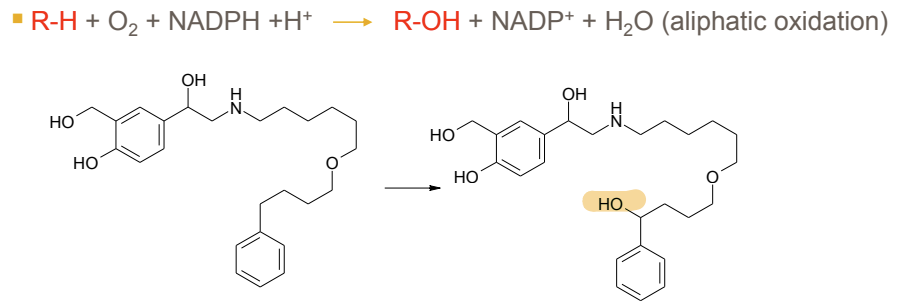
What is the general reaction for aliphatic oxidation in Phase I metabolism?
R-H + O2 + NADPH + H+ → R-OH + NADP+ + H2O
The drug (R-H) binds to CYP450’s heme iron (Fe³⁺), displacing a water molecule, where NADPH donates electrons (via NADPH-cytochrome P450 reductase) reducing the complex to complex to Fe³⁺–O₂⁻ which can be protonated highly reactive iron-oxo species which removes the inital R-H creating a carbon radical (R•) on the drug, quickly recombines with the Fe³⁺–OH oxygen, forming R-OH (the hydroxylated product).
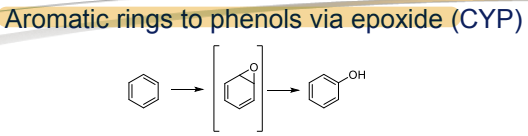
What is the Phase I metabolism reaction for aromatic rings?
Conversion to phenols via epoxide, catalyzed by CYP"
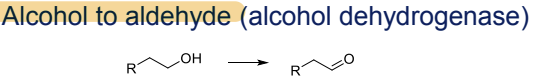
What is the Phase I metabolism reaction for alcohols?
Conversion to aldehydes, catalyzed by alcohol dehydrogenase
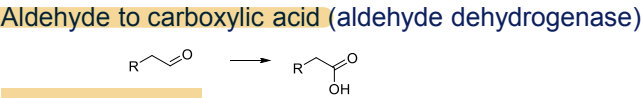
What is the Phase I metabolism reaction for aldehydes?
Conversion to carboxylic acids, catalyzed by aldehyde dehydrogenase
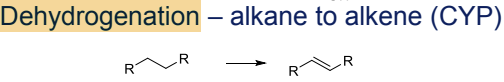
What is the Phase I metabolism reaction for alkanes?
Dehydrogenation to alkenes, catalyzed by CYP
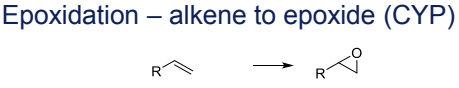
What is the Phase I metabolism reaction for alkenes? [Not too important]
Epoxidation to epoxides, catalyzed by CYP
—
The alkene (C=C) binds near CYP450’s heme iron (Fe³⁺). NADPH reduces Fe³⁺ to Fe²⁺, enabling O₂ binding → forms Fe³⁺–O₂⁻ (peroxo intermediate). Protonation cleaves O–O bond, generating Fe⁴⁺=O (Compound I). Compound I transfers an oxygen atom to the alkene’s π-bond, forming the epoxide ring. No radical intermediate (unlike aliphatic hydroxylation)
What is the Phase I metabolism reaction for tertiary amines?
N-dealkylation to secondary amines and a carbonyl, catalyzed by CYP
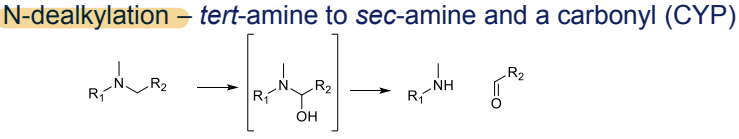
What is the Phase I metabolism reaction for ethers?
O-dealkylation to alcohols and a carbonyl, catalyzed by CYP
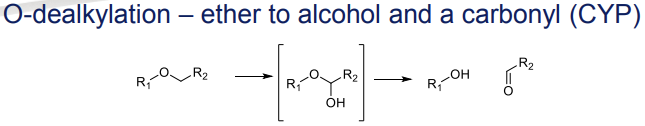
What is the Phase I metabolism reaction for sulphides?
S-dealkylation to thiols and a carbonyl, catalyzed by CYP"
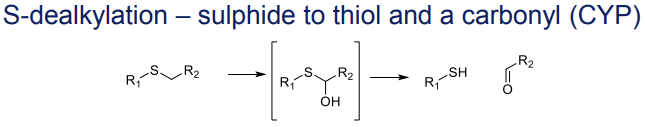
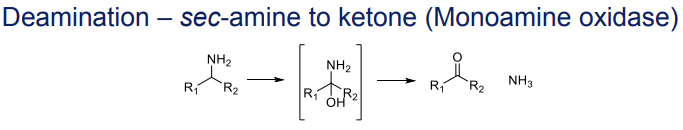
What is the Phase I metabolism reaction for secondary amines (deamination)?
Conversion to ketones, catalyzed by monoamine oxidase
What is the Phase I metabolism reaction for tertiary amines (N-oxidation)?
Conversion to amine N-oxides, catalyzed by CYP
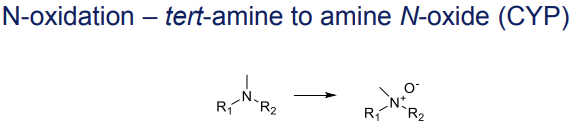
What is the Phase I metabolism reaction for secondary amines (N-hydroxylation)?
Conversion to hydroxylamines, catalyzed by CYP
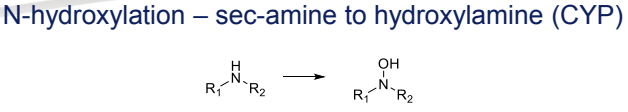
What is the Phase I metabolism reaction for sulphides (S-oxidation)?
Conversion to sulphoxides and sulphones, catalyzed by FMO (Flavin-containing monooxygenases)
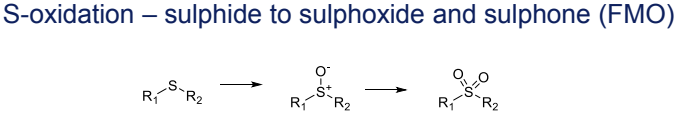
What is the Phase I metabolism reaction for cyclic amines?
Conversion to lactams, catalyzed by aldehyde oxidase
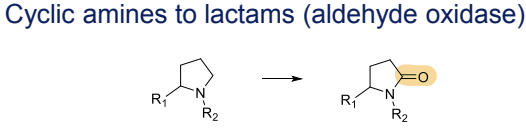
What is the Phase I metabolism reaction for ketones?
Reduction to alcohols, catalyzed by alcohol dehydrogenase
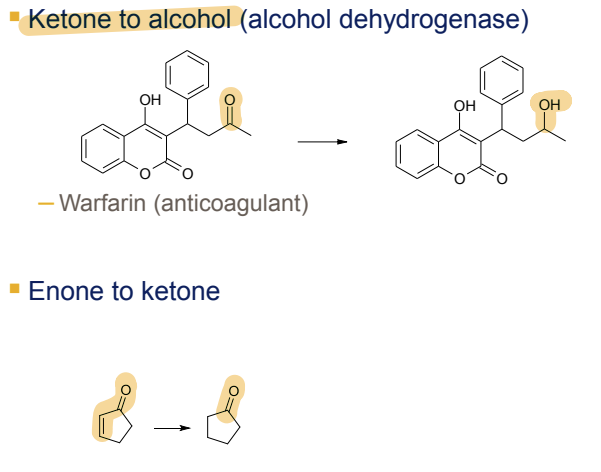
What is the Phase I metabolism reaction for aromatic nitro groups?
Reduction to anilines, catalyzed by NADPH-CYP450 reductase and nitroreductase
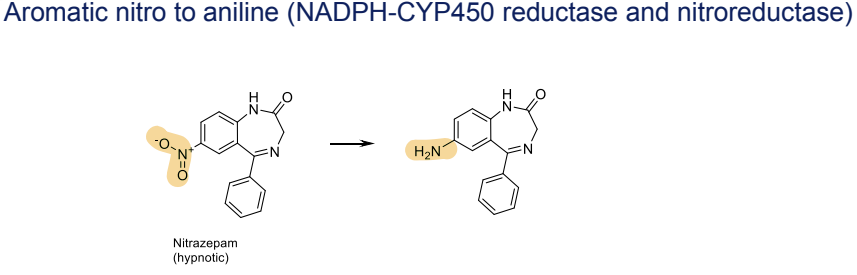
What are strategies to block Phase I metabolism of aromatic rings?
Block Hydroxylation of an Aromatic Ring by Adding Fluorine
Fluorine blocks the site of hydroxylation, reducing metabolism.
Reduces toxic metabolites (e.g., paracetamol becomes toxic after Phase I).
Slows Phase I, increasing drug half-life for longer therapeutic effects.
Example: Buspirone → 5-F-Buspirone
Buspirone undergoes CYP3A4 hydroxylation (t₁/₂ = 4.6 min). / Adding fluorine (5-F-Buspirone) increases t₁/₂ to 52.3 min.
Block Metabolism of a Methyl Group on an Aromatic Ring by Replacing with Chlorine
Methyl and chlorine are similar in size, but chlorination prevents metabolism at that site, slowing enzyme activity without altering the molecule's overall activity. Electron-withdrawing groups reduce electron density and stabilize the ring
Example: Tolbutamide → Chlorpropamide
Tolbutamide has a t₁/₂ of 5.9 hours. / Replacing the methyl group with chlorine (Chlorpropamide) increases t₁/₂ to 33 hours.
Block Metabolism of a Benzylic -CH₂- by Substituting with =CH₂ or =NOMe
Substituting with =CH₂ or =NOMe removes the hydrogen needed for metabolism, preventing the site from being metabolized.
General Insight: These modifications aim to slow Phase I metabolism, extending drug half-life and reducing toxic metabolites while maintaining therapeutic efficacy.
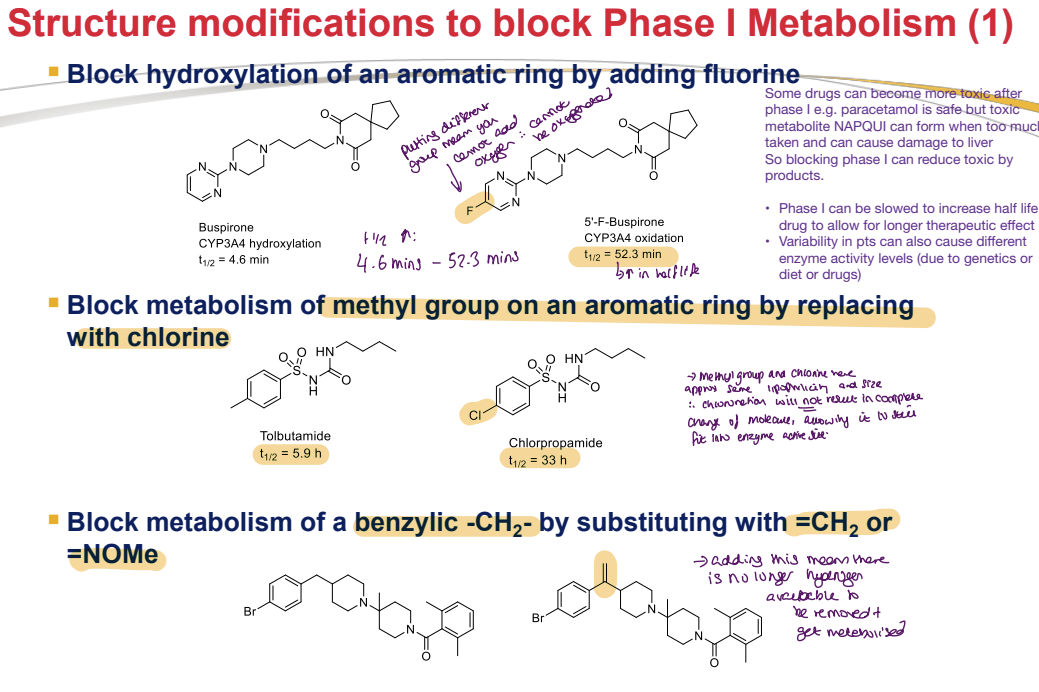
What are strategies to block Phase I metabolism of alkyl side chains?
Increase Bulkiness by Adding Aliphatic Groups Adjacent to the Labile Position
Aliphatic groups sterically hinder enzyme access, reducing Phase I metabolism.
Larger molecules are more likely to be protein-bound, slowing clearance from the body.
Extends drug half-life for prolonged therapeutic effect.
Example: Metoprolol → Betaxolol
Metoprolol (beta 1 receptor blocker for hypertension) has a human PK t₁/₂ of 4 hours. / Adding an aliphatic group (Betaxolol) increases t₁/₂ to 19 hours.
Remove Labile Functional Groups
Labile groups (e.g., -OH) are prone to reactions with other molecules, leading to degradation.
Removing these groups reduces degradation, increasing molecular stability.
Enhances shelf life (crucial for storage and transportation in varying environmental conditions).
Improves pharmacokinetic (PK) profile by increasing bioavailability.
Prolongs drug presence in the body by reducing rapid metabolism of labile groups.
Original stability: 1%. / After removing the labile -OH group: Stability increases to 60%.
What is a strategy to block Phase I metabolism of tertiary amines?
Convert to amides to prevent N-dealkylation or N-oxidation"
What are strategies to block Phase II metabolism ?
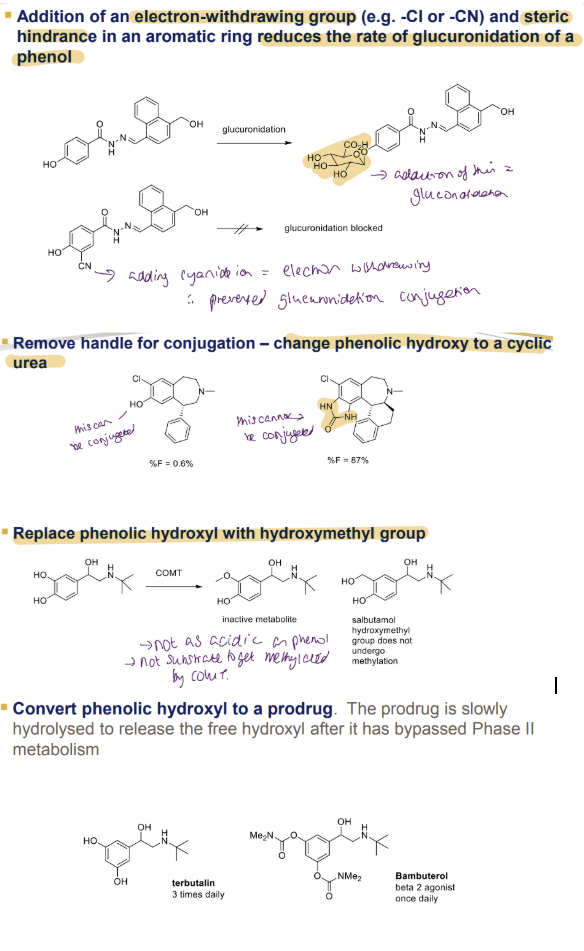
Effect of Electron-Withdrawing Groups and Steric Hindrance on Glucuronidation
Adding an electron-withdrawing group (e.g., -Cl or -CN) and steric hindrance to an aromatic ring reduces the rate of glucuronidation of phenol.
Example: Phenol undergoes glucuronidation, but when a -CN group is added, glucuronidation is blocked due to electron withdrawal, preventing glucuronide conjugation.
Removing Handle for Conjugation
Change the phenolic hydroxyl (-OH) to a cyclic urea to prevent conjugation.
Example: With phenolic -OH: %F (bioavailability) = 0%. / With cyclic urea: %F = 87%, indicating no conjugation occurs.
Replacing Phenolic Hydroxyl with Hydroxymethyl Group
Replace the phenolic -OH with a hydroxymethyl (-CH2OH) group.
Results: Hydroxymethyl derivative is an inactive metabolite. / Salbutamol (with hydroxymethyl) is an active drug, not susceptible to methylation by COMT (Catechol-O-Methyltransferase), unlike phenolic -OH.
Converting Phenolic Hydroxyl to a Prodrug
Convert the phenolic -OH into a prodrug that is slowly hydrolysed to release the free hydroxyl after Phase II metabolism.
Examples: Terbutaline: Administered 3 times daily. / Bambuterol: A beta-2 agonist, administered once daily, demonstrating a slower release profile.
What is a strategy to block Phase II metabolism of phenols (2)?
Change phenolic hydroxyl to a cyclic urea to remove the conjugation handle"
What is a strategy to block Phase II metabolism of phenols (3)?
Convert phenolic hydroxyl to a prodrug, which is slowly hydrolyzed to release the hydroxyl after bypassing Phase II metabolism"
What is the purpose of in vitro solution stability assays?
To determine compound stability at different pH values (1-12) and reject unstable compounds early"
What are the pH ranges in the digestive tract for solution stability assays?
Stomach: 1-2, beginning of small intestine: 4.5, further down intestine: 6.6, colon: 6-9"
How is solution stability determined in vitro?
By HPLC over time at 37°C in a range of pH buffers (1-12)"
What is the purpose of in vitro plasma stability assays?
In vitro plasma stability assay:
To determine susceptibility to hydrolysis by plasma proteins and support structure modification studies"
How is plasma stability determined in vitro?
By LCMS over a 6-hour period at 37°C in plasma buffered to pH 7.4"
Which plasma is commonly used in generic plasma stability assays?
Sprague-Dawley male rat plasma"
What is recommended for plasma stability assays to align with efficacy studies?
Use the same species plasma as used in efficacy pharmacology experiments"
What is the purpose of liver microsomal stability assays with NADPH?
To assess Phase I oxidations by CYP and FMO, providing clearance (mL/min/g liver)"
Describe Phase II Metabolism
Metabolising enzymes attach polar endogenous molecules (e.g., glucuronic acid, sulfate) to drugs/metabolites to enhance water solubility for excretion.
Example: Glucuronic acid via glucuronosyl transferases / Sulphate via sulphotransferases / Glutathione via glutathione transferases
Prerequisite: The drug must have a "handle" (functional attachment group like –OH, –NH₂, –COOH) for conjugation.
Phase I often creates this handle (e.g., CYP450 adds –OH).
Phase I: CYP2D6 hydroxylates codeine → morphine (–OH added).
Phase II: UGT2B7 glucuronidates morphine → morphine-3-glucuronide (inactive) or morphine-6-glucuronide (more potent).
—
Conjugation Process: Once the hydroxyl group is added, Phase II enzymes like UDP-glucuronosyltransferase (UGT) or sulfotransferase can attach a large, water-soluble molecule to the hydroxyl group, forming a conjugate. This conjugated drug is generally more water-soluble, which enhances its excretion in the urine or bile.

What is the purpose of Phase II stability assays?
To assess conjugation reactions (e.g., glucuronidation) for compounds with susceptible groups like phenols and alcohols"
What cofactors are used in Phase II stability assays?
Appropriate cofactors, e.g., uridine diphosphate glucuronic acid for glucuronidation"
What is the purpose of hepatocyte assays in metabolic stability?
To provide broader metabolic stability data using fresh or cryopreserved hepatocytes without cofactors"
How is metabolite structure elucidation performed?
By LCMS/MS or LC-NMR using microsomes, S9, or hepatocytes, requiring scale-up for characterization"
What are in silico metabolic stability methods used for?
To predict metabolic stability and sites of instability at the design stage before synthesis"
What are examples of in silico metabolic stability tools?
Metasite, Meteor"
What is Volume of Distribution (Vd)?
A mathematical expression of the apparent volume into which a drug is dissolved, indicating how widely it is distributed in the body (L/kg)"
What does a low Vd (<0.07 L/kg) indicate?
Compound stays in blood, characteristic of hydrophilic compounds or those highly bound to plasma proteins"
What does a high Vd (>200 L/kg) indicate?
Compound binds to tissues with little in blood, characteristic of highly lipophilic compounds"
What does a moderate Vd (~0.7 L/kg) indicate?
Compound is moderately lipophilic, moderately bound to plasma proteins and tissues, distributing evenly"
How is Vd calculated?
Vd = Dose / C0, where Dose is mg/kg body weight and C0 is initial blood concentration after IV dose"
What is Area Under the Curve (AUC)?
The integrated area under a plot of drug plasma concentration vs. time"
How is bioavailability (%F) calculated?
%F = (AUCpo / AUCiv) × (Doseiv / Dosepo) × 100"
What does an AUC >500 ng h/mL for a 10 mg/kg oral dose over 0-6 h indicate?
An 80% chance of bioavailability >20%"
What is Clearance (Cl)?
Indicates how rapidly a drug is removed from blood and eliminated (mL/min/kg)"
What are the two main clearance pathways?
Renal clearance (Clr) via kidney to urine, and hepatic clearance (Clh) via liver to bile and feces"
How is Clearance (Cl) calculated?
Cl = Dose / AUCiv"
What is the significance of blood flow in PK studies?
Clearance is sometimes expressed as a percentage of total blood flow to liver and kidney"
What are the hepatic and renal blood flow rates in rats?
Hepatic: 55 mL/min/kg, Renal: 5 mL/min/kg"
What are the hepatic and renal blood flow rates in humans?
Hepatic: 20 mL/min/kg, Renal: 2 mL/min/kg"
What is Half-life (t1/2)?
The time for drug concentration in systemic circulation to reduce by 50%"
How is Half-life (t1/2) calculated?
t1/2 = 0.693 Vd / Clh"
How is Half-life (t1/2) used in dosing?
To calculate how frequently a dose must be administered to maintain therapeutic concentration, typically every 1-3 half-lives"
What is the desired Vd range for lead optimization?
Low: <1 L/kg, High: >10 L/kg"
What is the desired Clearance (Cl) range in rats for lead optimization?
Low: <10 mL/min/kg, High: >45 mL/min/kg"
What is the desired Half-life (t1/2) range in rats for lead optimization?
Low: <1 h, High: >3 h"
What is the desired Bioavailability (%F) range for lead optimization?
Low: <20%, High: >50%"
What is the desired AUC range in rats for lead optimization?
Low: <500 h ng/mL, High: >2000 h ng/mL"
What is the desired tmax range for lead optimization?
Low: <1 h, High: >3 h"