BIOAVAILABILITY & BIOEQUIVALENCE, GENERIC & BIOSIMILAR DRUGs
1/41
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
42 Terms
What are branded or “innovator” medicines?
•Branded or innovative medicines are new medicines.
•A new medicine often comes about through the research and development of a new chemical molecule.
•The developer of that molecule gets regulatory approval for the medicine and a patent to protect their invention.
•They have sole exclusive rights to protect it which, if granted, last for about twenty years.
•The company has the exclusive rights to manufacture and market the medicine, allowing them to recoup their research and development costs.
•When a company develops a new medicine, it is known both by its generic name – the name of the active or key ingredient
– and by its brand name, which is the name which the developer sells the medicine under.
•The brand name is chosen by the developer and is usually something that’s easy for patients and healthcare professionals to recognise and pronounce, and the generic name will be a more scientific sounding name
Exclusivity period is normally around 20 years - given by regulatory bodies
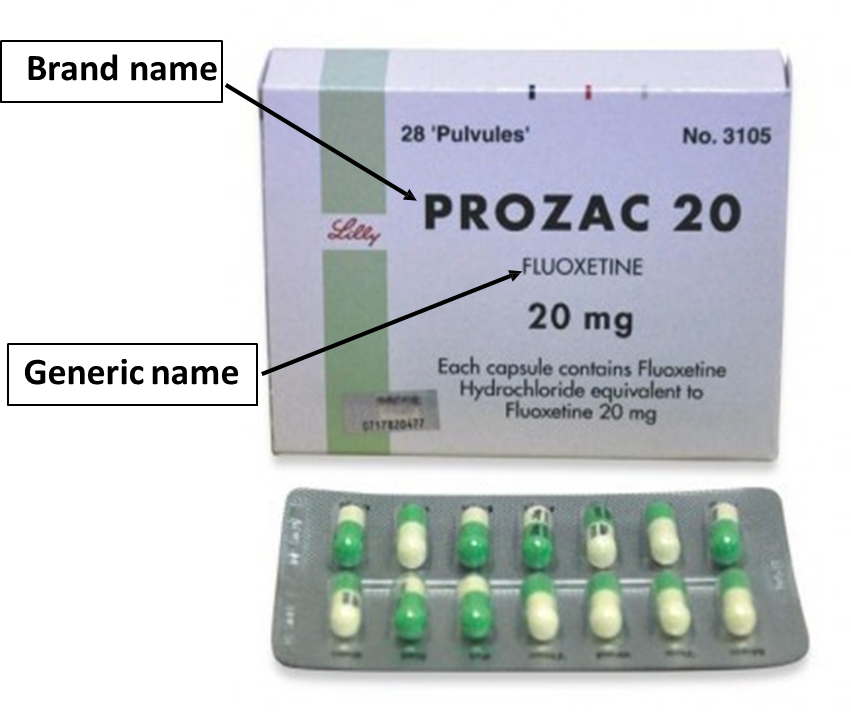
What is a generic medicine?
•Once the innovator patent expires, other companies can manufacture a copy of the medicine and either market the medicine using a different brand name (branded generic), or sell it as a generic product, with only the name of the active ingredient on the packaging.
•
•Generic products are defined as “medicinal product[s] [having] the same qualitative and quantitative composition in active substances and the same pharmaceutical form as the reference medicinal product, and whose bioequivalence with the reference medicinal product has been demonstrated by appropriate bioavailability studies.”
Bioequivalence studies ensure that the generic drug has a similar absorption rate as the original drug
In the EU, the Committee for Medicinal Products for Human Use (CHMP) guideline on bioequivalence (BE) requires that bioequivalence studies are carried out to show that the rate and extent of absorption of test product are equivalent to reference

What is a generic medicine?
A generic copy of a medicine
•Contains the same active ingredients as the original branded medicine
•Comparable in terms of its dosage form (tablets or capsules etc.)
•Comparable strength of API
•Same route of administration (how you take it).
•Must also be bioequivalent, which means that you can expect it to work in exactly the same way as the original product.
•Equally effective and safe as the branded one
•Brand-name and generic drug companies must do months-long "stability tests" to show that their versions last for a reasonable time.
•All manufacturing, packaging and testing sites must pass same quality standards as those of brand drugs

BIOEQUIVALENCE
Two medicinal products (a generic and a reference /Innnovator) containing the same active substance are considered bioequivalent, if:
§They are pharmaceutically equivalent - same API, same form and same dosage.
§Their bioavailabilities after administration in the same molar dose lie within acceptable predefined limits i.e. if their rates and extents of absorption are the same (not statistically different)
These limits are set to ensure comparable in vivo performance, i.e., similarity in terms of safety and efficacy
BIOAVAILABILITY
The extent and rate (concentration vs time) at which the active moiety is delivered from pharmaceutical form and becomes available in the systemic circulation
Bioavailability is measured using three main parameters:
•the area under the plasma drug concentration versus time curve (AUC) (rate and extent of absorption) - reflects total drug exposure over time
•
•the maximum plasma concentration (Cmax) (extent of absorption)
•the time to reach maximum concentration (Tmax) (rate of absorption)

The “true dose” is not the drug swallowed;
BUT is the drug available at the site of action to exert its effect (therefore, bioavailable)
Bioavailability depends on:
•Dissolution
•Absorption
•Survive metabolism??
May have a drug with very low bioavailability:
•Dosage form or drug may not dissolve readily
(issues with solubility & dissolution)
•Drug may not be readily pass across biological
membranes (i.e. issues with absorption)
•Drug may be extensively metabolized during
absorption process (first-pass, gut wall, liver
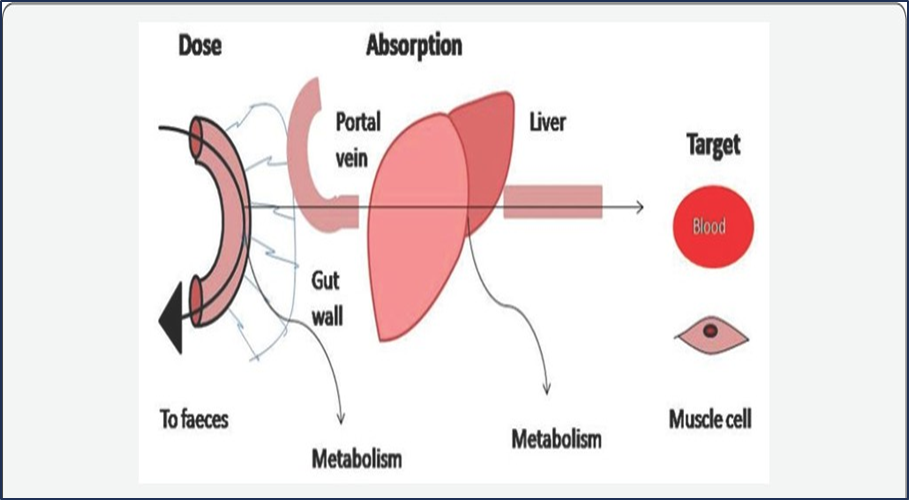

Variable bioavailability will lead to variable exposure to the drug (efficacy)
Factors Influencing Bioavailability
1. Pharmaceutical factors - drug physical and chemical properties
2. Patient related factors - sex, age, health status
3. Route of administration
1. Pharmaceutical factors
•Physicochemical properties of the drug
- Particle size
- Crystalline structure
-Salt form
•Formulation and manufacturing variables
- Disintegration and dissolution time
- Pharmaceutical ingredients
- Special coatings
- Nature and type of dosage form
2. Patient related factors
•Physiologic factors
-Variations in pH of GI fluids
-Gastric emptying rate - if its slower it will delay the emptying of drug into the intestine and will delay the dissolution and absorption of the drug
-Intestinal motility
-First-pass metabolism (how efficient is your liver detox?) - how much drug is metabolised before it reaches systemic circulation
-Age, sex
-Disease states
•Interactions with other substances
-Food
-Alcohol, smoking
-Other drugs
3. Route of administration
Affects pharmacokinetics: absorption, distribution, metabolism and excretion (ADME)
relevant to ALL drugs
large research/development area
frequent cause of failure of treatment
failure of compliance
failure to achieve effective level
produce toxic effects
can enhance patient satisfaction with treatment
Routes of administration
Two main routes:
1. Enteral (=gastrointestinal): medications enter body through digestive system (including rectum)
Oral or per os (PO)—by mouth
Per rectum (PR)—by rectum
Gastric or duodenal (e.g., feeding tube)
Sublingual
has challenges of 1st pass metabolism - metabolised in liver = loss of bioavailability.
2. Parenteral (par=around enteral=gastrointestinal): medications enter body by some other means
Intravenous (IV)
Intramuscular (IM)
Subcutaneous (SC or SQ) and transdermal routes
Inhalational
Topical
Intrathecal
1.Enteral:
Oral (PO)
The most frequently used
Simplicity and convenience (improve patient compliance)
Less desirable for drugs that are irritating to the GI tract
or when the patient is vomiting or unable to swallow.
Oral absorption requires….
Chemical stability of drug:
Þmust be acid stable or protected from gastric acid (e.g., by enteric coatings).
Stability of drug to enzymes
Þ Avoid undesired biotransformation
Tip: Biotransformation can occur wherever appropriate enzymes are available (Liver, plasma, kidney, lung, gut wall, etc.)
Factors affecting oral absorption
Gastric emptying time. For many drugs the greatest absorption occurs in the small intestine (large surface).
How gastric emptying time affects oral absorption?
For many drugs the greatest absorption occurs in the small intestine (large surface).
More rapid gastric emptying facilitates their absorption Þ the drug is delivered to the small intestine more quickly.
Conversely, factors that slow gastric emptying (e.g., food, anticholinergic drugs) generally slow absorption.
Intestinal motility
Increases in intestinal motility (e.g., diarrhea) may move drugs through the intestine too rapidly to permit effective absorption
Food (presence and type)
Affects gastric emptying time
Absorption of some drugs (e.g., clarithromycin) is improved by administration with food,, whereas with tetracycline it is the opposite.
May reduce the absorption of some drugs owing to physical interactions with the drug (e.g., chelation), affecting metabolism..
Examples:
•Calcium chelation for tetracycline. Avoid co-administration with dairy products as product will then be insoluble.
•Grape fruit may inhibit metabolism of some drugs, such as cilostazol (re-consider dose, activity vs toxicity)
Rectal absorption
Useful in situations in which the patient is unable to take medication orally (e.g., is unconscious, vomiting, convulsing).
Drugs are absorbed through the rectal mucosa.
Þ Because of the anatomy of the venous drainage of the rectum, approximately 50% of the dose bypasses the portal circulation
Þ an advantage if the drug has low oral bioavailability
However, drug absorption via this route is incomplete and erratic. In part because the variability in drug dissociation from the suppository
2. Parenteral:
Do not involve drug absorption via the GI tract
Reasons for choosing a parenteral route over the oral route:
- drugs with low oral bioavailability
-patients who are unable to take the drug by mouth (e.g., it irritates the GI tract, unconscious, nausea)
-need for immediate effect (e.g., emergency situations)
-control the rate of absorption and/or duration/extension of effect (e.g., unfractionated heparin - level of coagulation).
Which one is faster? Why?
Intravenous injections: 100% bioavailability as the drug is administered directly into the vascular space. Bypass absorption barriers & minimal exposure to metabolism.
Intramuscular injections are absorbed faster than subcutaneous injections. This is because:
Muscle tissue has a greater blood supply than the tissue just under the skin.
Muscle tissue can also hold a larger volume of medication than subcutaneous tissue.
PHARMACEUTICAL EQUIVALENT
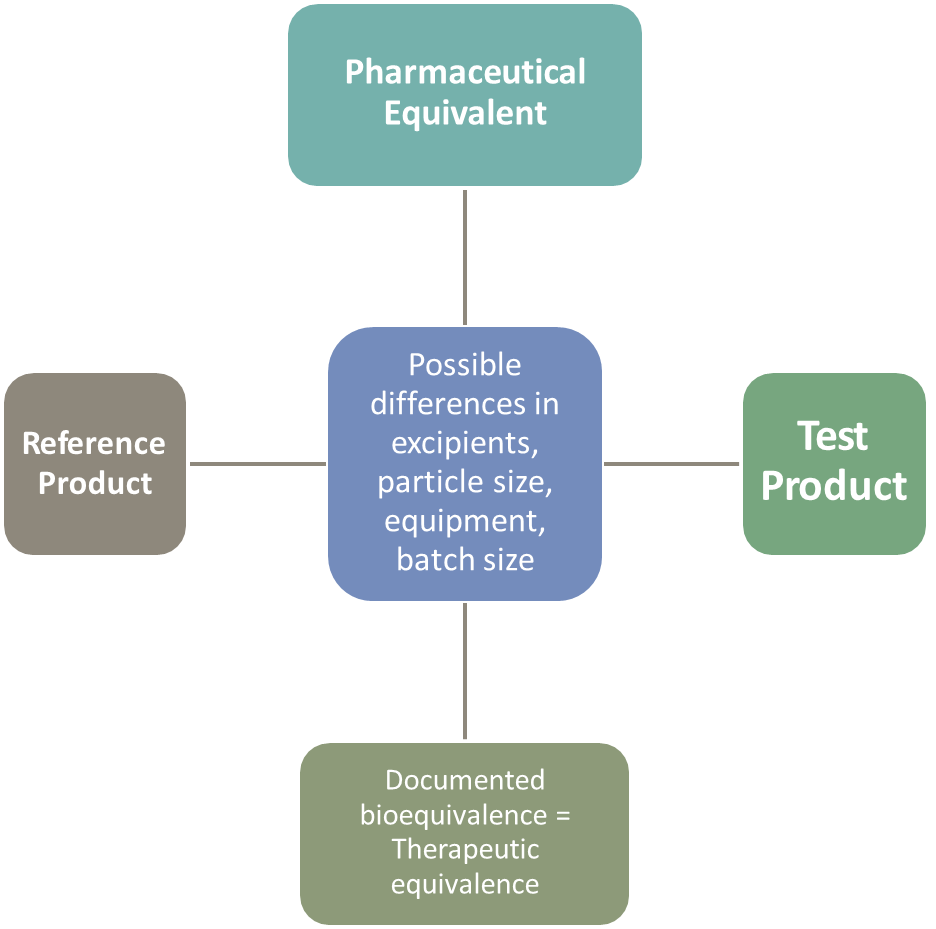
The generic drug needs to show that it is the same type of product (such as a tablet or an injectable) and uses the same time release technology (such as immediate-release, meaning for immediate effect of the drug, or extended-release, meaning one that is intended to slowly release the active ingredient over time). Differences such as the excipients used, particle size, manufacturing conditions etc may be different - still has to demonstrate bioequivalence. This is so that patients can switch between the two without experiencing differences in therapeutic outcome.
Are these bioequivalent or not? Why?
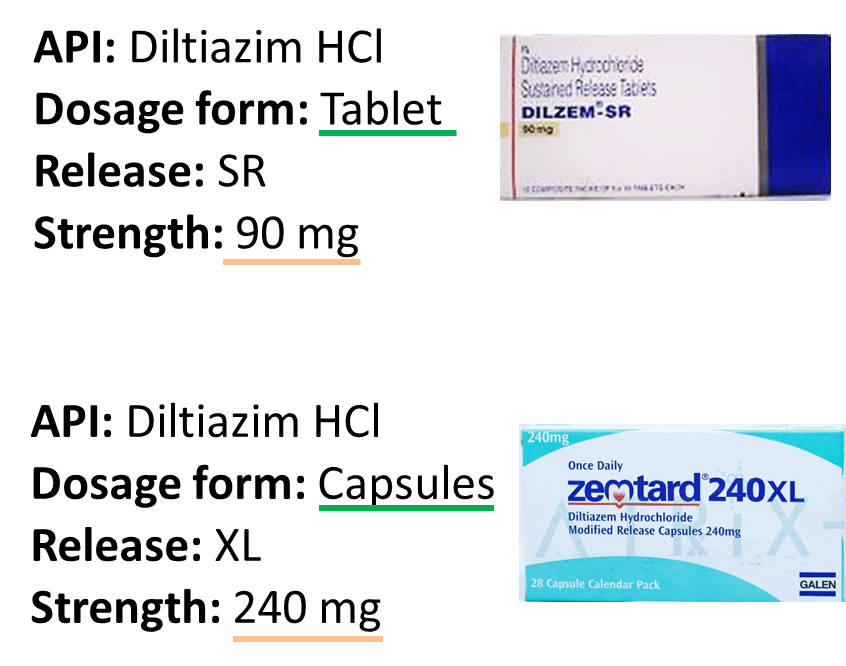
NOT bioequivalent
Are these bioequivalent or not? Why?

Bioequivalent
Are these bioequivalent or not? Why?

Generic medicines are strictly regulated
•Bioequivalence studies are the basis for the approval of generic medicinal products.
• Specific advice on how bioequivalence should be demonstrated to support scientific consistency in their conduct and assessment.
• European Medicines Agency’s (EMA) provides guidelines on generating relevant data and potentially improving the number of successful and well-conducted bioequivalence studies.
•The manufacturing process of new medicines and generic copies is strictly regulated. Regulatory bodies, such as the FDA and MHRA, perform stringent inspections on the manufacturing facilities and processes of pharmaceutical companies.
•The aim of the inspections is to verify the facilities are compliant with the strict quality control standards of the countries where the medicines are marketed.
Steps involved in development of generic product

•When an innovator drug is developed, evidence is required of its pharmacokinetics, efficacy and tolerability in volunteer study subjects as well as the target population - new drug so more trusting required
•However the development of a generic equivalent requires only evidence of its bioequivalence with the innovator drug in the study subjects.
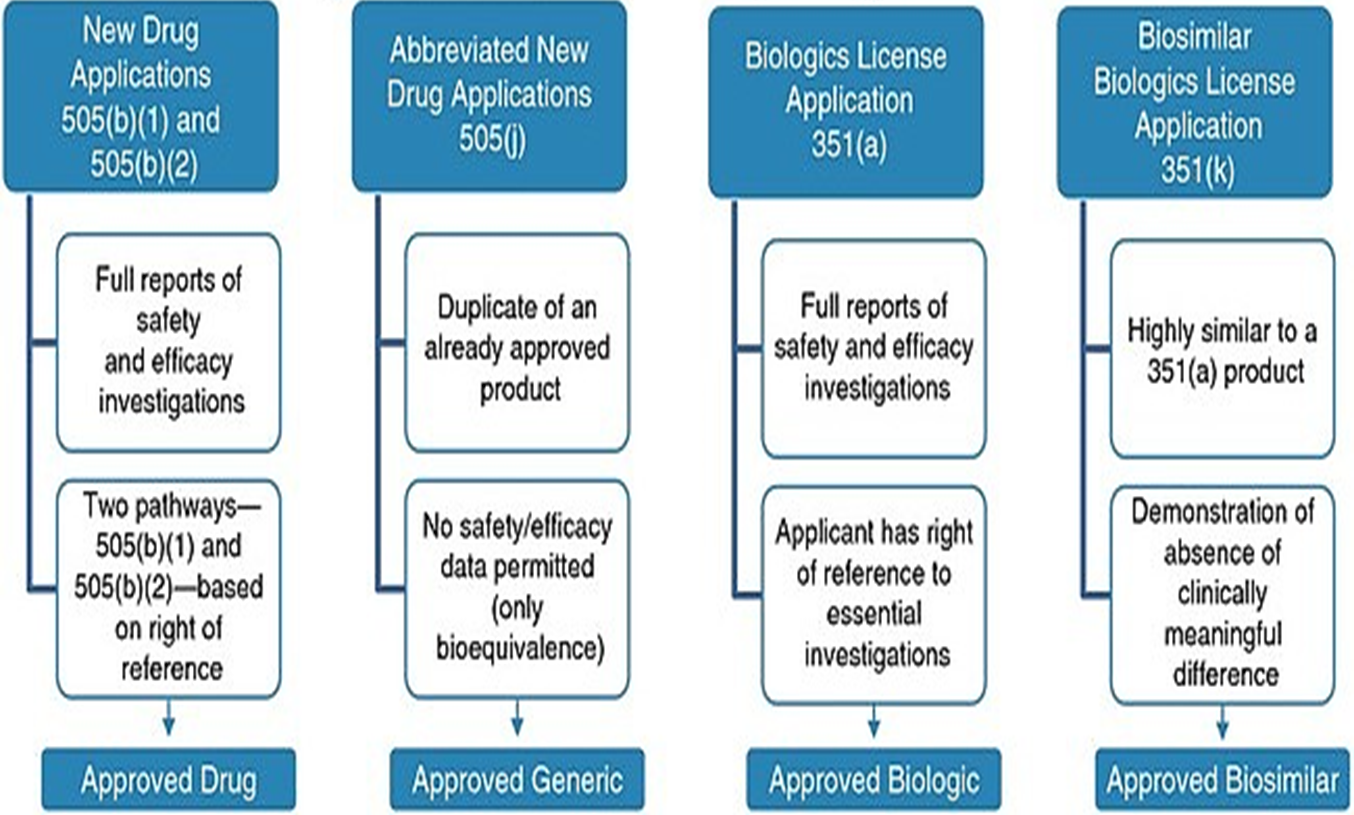
NDA (new drug application) vs. ANDA (abbreviated NDA) Review Process by FDA
Innovator Drug
NDA Requirements
1.Chemistry
2.Manufacturing
3.Controls
4.Labeling
5.Testing
6.Animal Studies
7.Clinical Studies
(Bioavailability)
Generic Drug
ANDA Requirements
1.Chemistry
2.Manufacturing
3.Controls
4.Labeling
5.Testing
6.Bioequivalence Study (in vivo, in vitro)
Note: Generic drug applications are termed "abbreviated" because they are generally
not required to include preclinical (animal) and clinical (human) data to establish safety and effectiveness.
Instead, generic applicants must scientifically demonstrate that their product is bioequivalent
(i.e., performs in the same manner as the original drug).
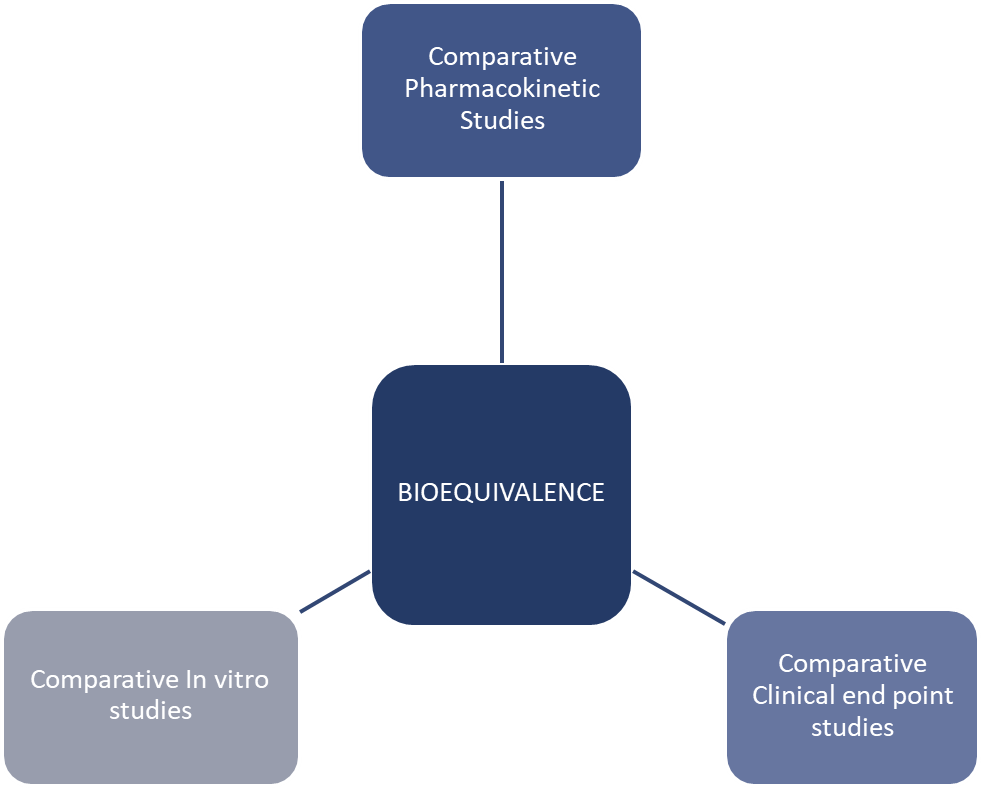
Bioequivalence Studies
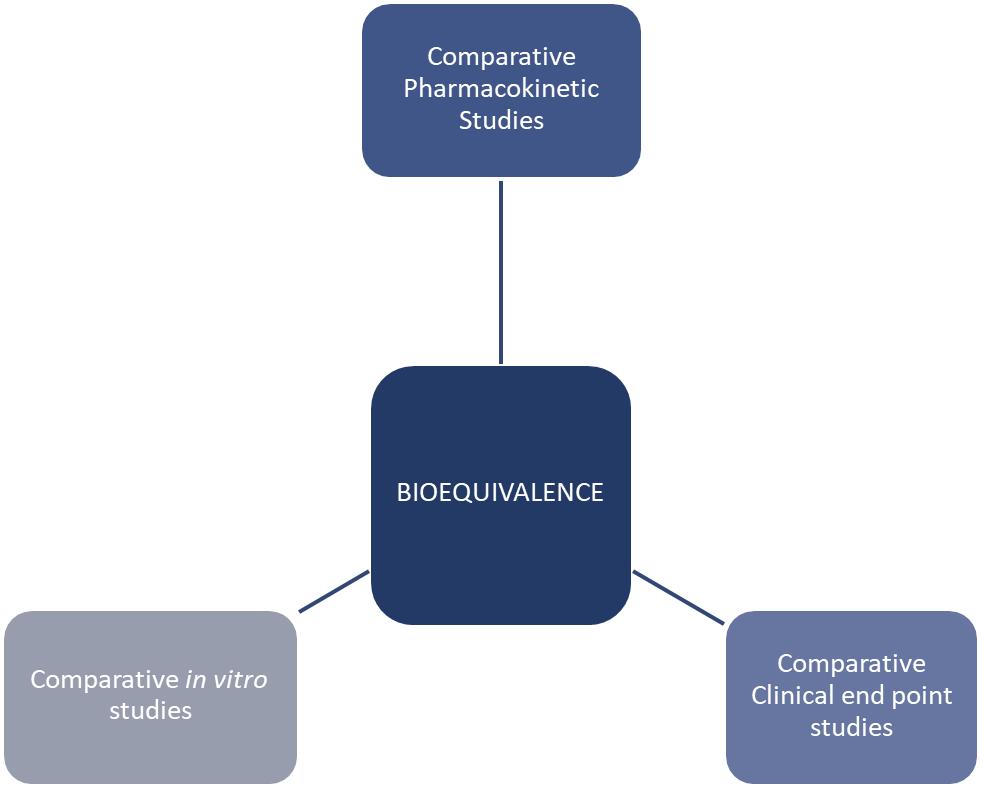
Pharmacokinetic - look into ADME
Comparative clinical end point studies - used when pharmacokinetic studies are not enough and focus on measuring therapeutic effect safety - look at clinical response and ADR
Comparative in vitro studies - these are lab based studies, look into dissolution profile, particle size, polymorphism, viscosity, pH and release characteristics
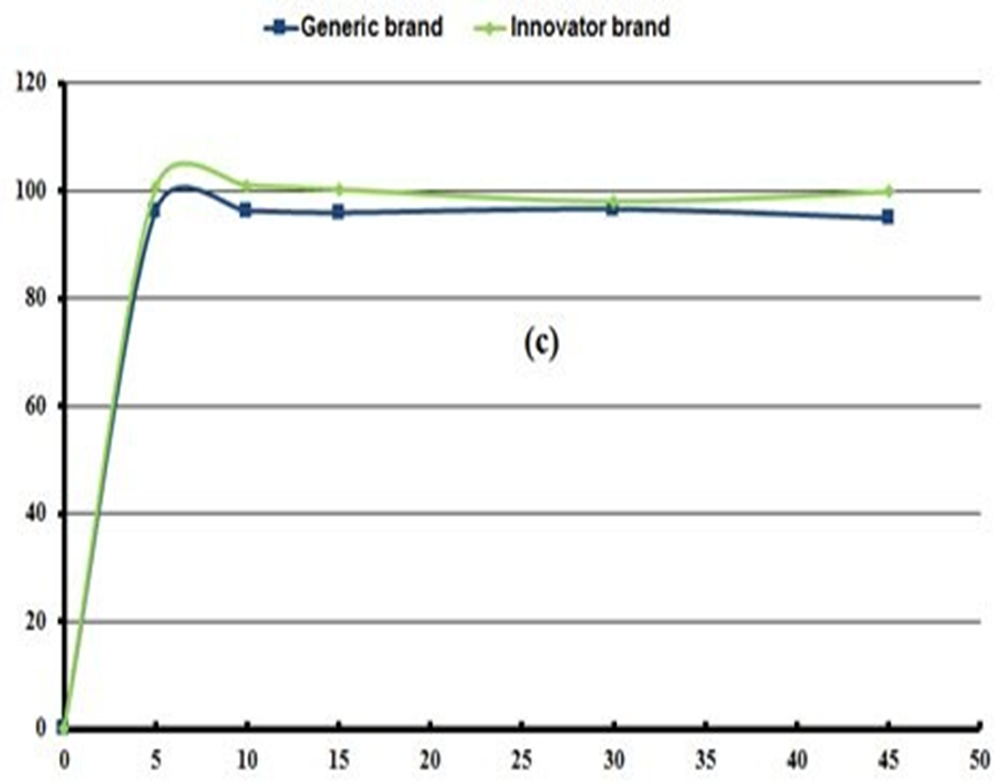
In vitro bioequivalence : Dissolution profile

In vivo bioequivalence study: Format and contents
Parameters | Specifications in EU |
Study design | Non-replicated, randomized, crossover studies |
Fasting/fed state studies | Fasting |
Volunteers | >12 (Min 80% power of acceptance criteria) |
Study dose | TEST: Made by the manufacturer |
REFERENCE: European reference product | |
Sampling points | At least 2 samples before expected Tmax, 3–4 terminal log–linear phase |
Analytical method validation parameters | Accuracy, precision, repeatability, intermediate precision specificity, detection. limit of quantitation, linearity range |
Moieties to be measured in plasma | Active drug/metabolites if applicable |
Pharmacokinetic parameters | Cmax, Tmax, AUC0–t and AUC0–∞, t1/2, λz |
Criteria for bioequivalence | 90% CI 80.00–125.00 for Cmax, AUCt, AUC0– ∞ (for highly variable drugs 75–133%) |
GCP requirements | ICH GCP guidelines |
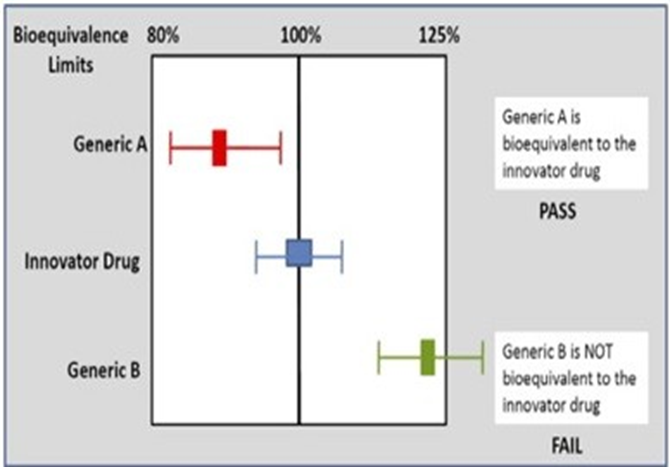
In vivo bioequivalence study : Are drugs A and B bioequivalent?
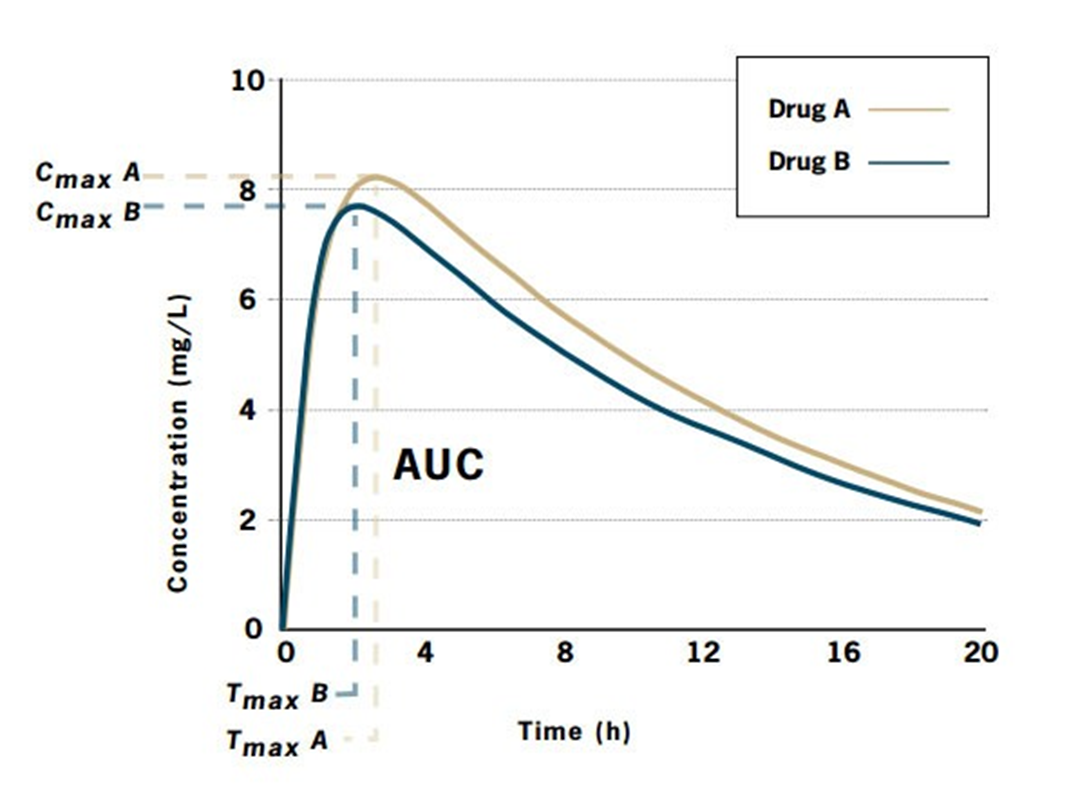
Drug A: | Cmax = 8.1 mg/L; Tmax = 2.6 h; AUC = 124.9 mg.h/L |
Drug B: | Cmax = 7.6 mg/L; Tmax = 2.1 h; AUC = 112.4 mg.h/L |
Drug A is the innovator product and Drug B is the generic product.
•The ratio of areas (generic: innovator), and therefore the relative bioavailability, is 0.9.
•To be accepted as bioequivalent, the 90% confidence intervals for the area ratio would need to fall thin the range 0.8-1.25.
Ongoing quality checks
•Once approved and licensed, each and every production batch of a medicinal product, branded or generic, must be certified by a Qualified person (QP) before being released for sale.
•The QP ensures that each batch has been manufactured in compliance with the ‘marketing authorisation’ and Good Manufacturing Practice (GMP).
•Checks include a full qualitative and quantitative analysis of all the active constituents
•The supply chain for medicines is also certified, monitored, and inspected from manufacturer to pharmacy (Good distribution practice).
•All medicines in use, original brands and generics, are subject to the same monitoring and reporting for adverse events.
•In the UK adverse events, such as side-effects, or previously unknown interactions with other medicines, should be reported to the MHRA (Medicines and Healthcare products Regulatory Agency). Yellow card scheme
Branded and Generic prescribing
• In primary care, if a medicine is prescribed by brand name, the pharmacist may dispense only the specified brand and is reimbursed for doing so.
• If a medicine is prescribed by generic name, the pharmacist may dispense any suitable generic or branded product and is reimbursed at a set price, listed in the Drug Tariff.
•
• Increasing the level of generic prescribing in the UK has long been encouraged.
Advantages of Generic prescribing
Generic medicines are significantly cheaper than the original innovator products
•It takes a lot less time and money to develop a generic medicine than it does to develop an innovative medicine.
•
•Generic manufacturers do not incur the significant risks and costs associated with the research and development of innovative product
•
•Generic manufacturers can afford to sell their medicines for a lower price than those who sell equivalent branded medicines.
Advantages of Generic prescribing
•The generic medicines industry helps save the NHS over
£13 billion per annum.
•The average cost to the NHS of a generic medicine is
£3.80, the average cost of a branded medicine is around
£20.95.
•Without generic medicines, the NHS medicines bill would be approximately twice as much as it is now.
•
•Competition from companies who produce generic medicines also encourages researchers within the pharmaceutical industry to develop new medicines.
Medicines should be prescribed by brand name in the following situations:
•Where there is a difference in bioavailability between brands of the same medicine, particularly if the medicine has a narrow therapeutic index.
•Modified-release (MR) preparations are not interchangeable, particularly if the medicine has a narrow therapeutic index.
•Where products contain more than one ingredient and brand-name prescribing aids identification.
•Where there are important differences in formulation between brands of the same medicine. For example, fentanyl patches, which are available as matrix formulations (Fencino®)and reservoir formulations (Fentalis®). Although studies* showed that the release of fentanyl from these two types of patches is similar, it is recommended the patient remains on the same type of patch.
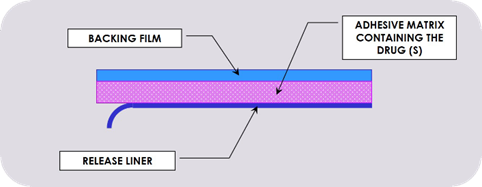
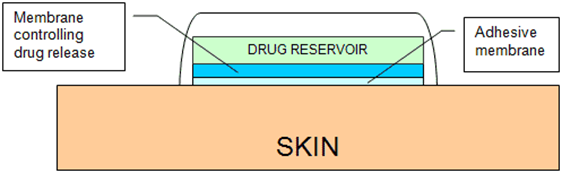
•Where administration devices have different instructions for use and patient familiarity with one product is important. Examples include inhaler devices from different companies
•
•Where branded and generic preparations have different licensed indications.
•
•For some patients, where differences in product name, presentation, appearance or taste may lead to anxiety, confusion, dosing errors and reduced adherence. Difficulties for patients with autism, learning difficulties or mental health problems should be considered
•
•Where the product is a biological rather than chemical entity. Many such agents are licensed as biosimilar medicines.
Biological medicines are more complex than small molecules
•Biological medicines are derived from living cells or organisms and consist of large, highly complex molecular entities which may be difficult to characterise.
•Due to the variability of the biological system and the manufacturing process, biological medicines may show a certain degree of variation, even between batches of the same product - have to be in a more controlled environment as a result of this
•
•Biological medicines are used to treat various diseases such as cancer, rheumatoid arthritis, psoriasis, Crohn’s disease, renal anaemia and diabetes.

What is Biosimilar Product
•A biosimilar is a biological medicinal product that contains a version of the active substance of an already authorized original biological medicinal product (reference medicinal product) in the EEA (European Economic Area).
•Similarity to the reference medicinal product in terms of quality characteristics, biological activity, safety and efficacy based on a comprehensive comparability exercise needs to be established - important as even minor changes can affect how it would interact
Biosimilars are not an exact replica due to the complexity of biological molecules, so unlike generics which are identical to their small molecule counterparts
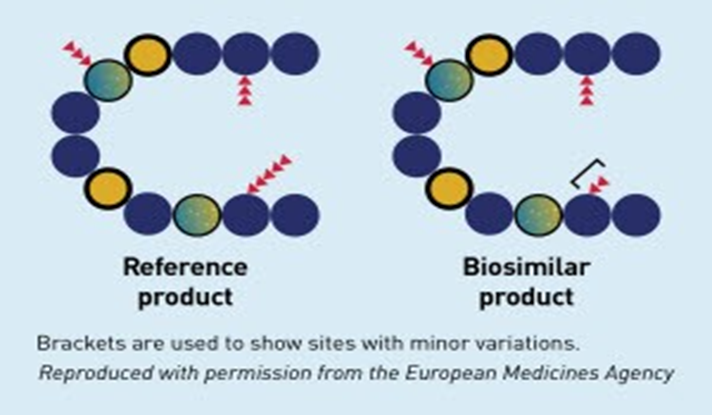

Biosimilar Scenario
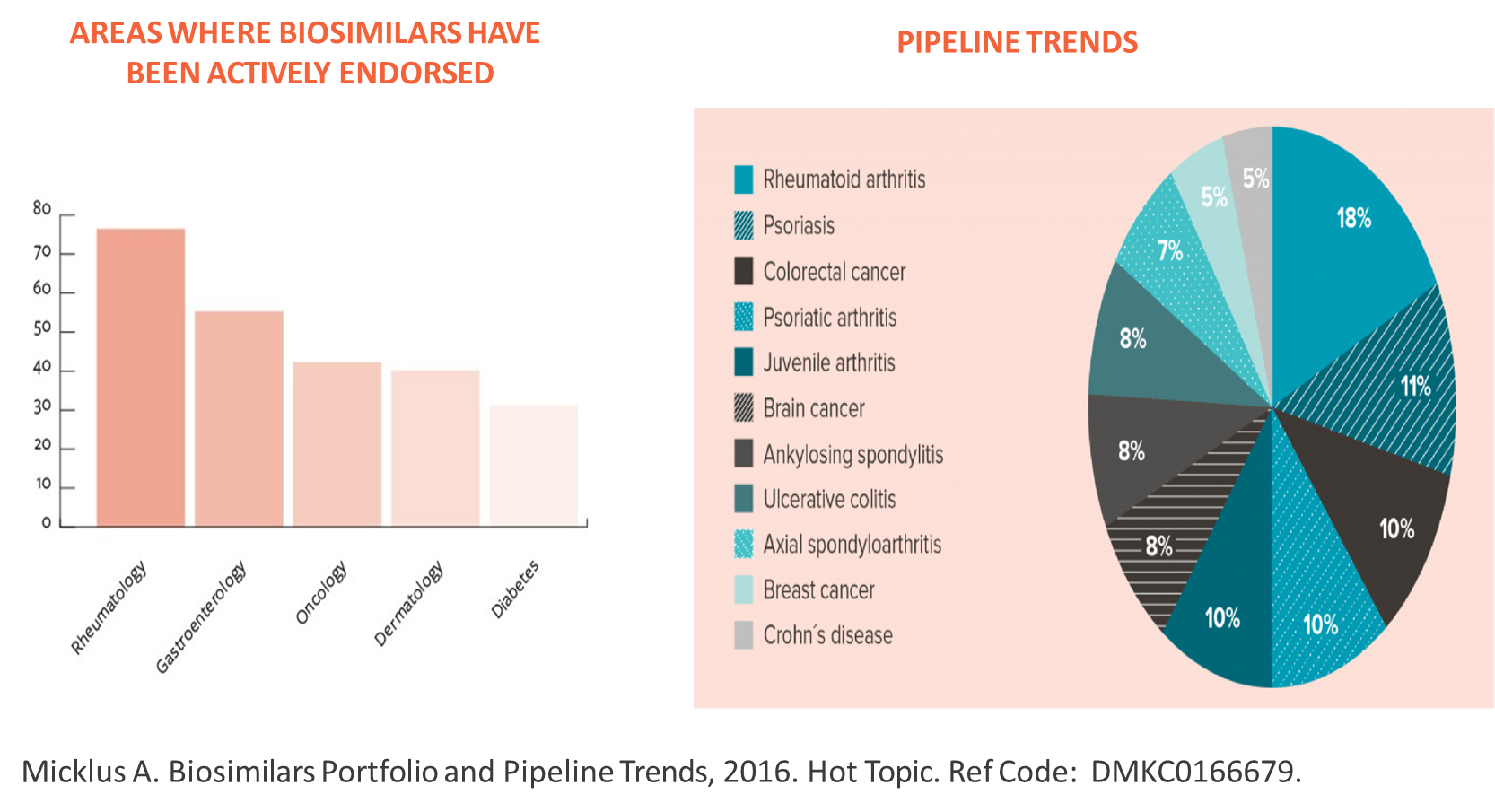
Drugs tend to be expensive to make as they tend to be used for conditions which require long term treatment and overall used for conditions which requires more expensive drugs as it is
Biosimilar Vs Generic
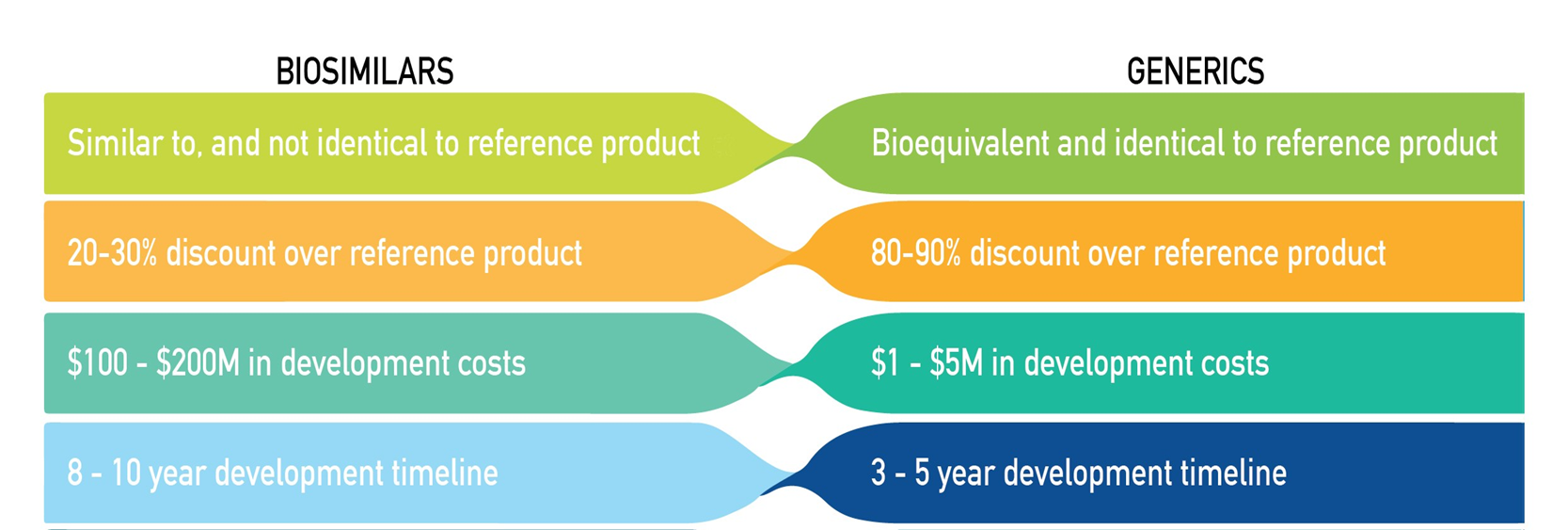
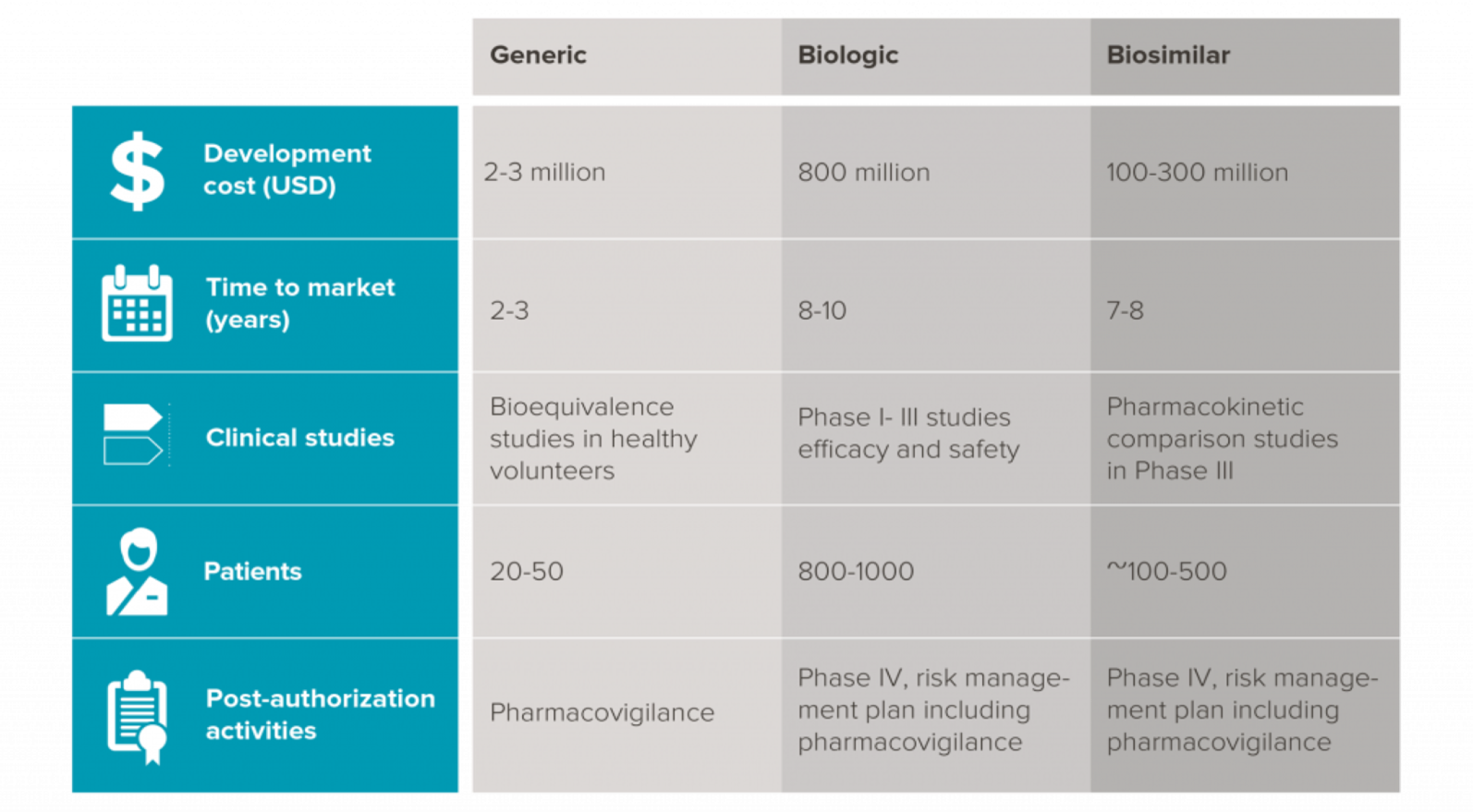
Biosimilars require phase 3 authorisation to ensure it performs similarly to the original drug
So the regulatory agencies would require thorough, comparisons to ensure that the biosimilar will work as intended and, without any unexpected side effects or without, reduced effectiveness.
How are biosimilar medicines authorised for use?
•Marketing authorisation applications for biotechnology-derived medicines, including biosimilar medicines, are by law reviewed centrally by the European Medicines Agency (EMA).
•The resulting marketing authorisation, issued via a decision by the European Commission, is valid in all EU Member States.
•EU adopted a specific regulatory pathway that provides a robust regulatory process through overarching, quality, non-clinical and clinical, and product class-specific scientific guidelines for biosimilar medicines.
•The main part of the evaluation is a detailed head-to-head comparison of the biosimilar medicine with its reference medicine to show that there are no clinically significant differences between them.
•Biosimilar medicines require distinct regulatory pathways from those applied to generic medicines as they are not exact replicates of the originator (reference) medicine.
•The shortened and simplified regulatory approval process used for generic medicines is not sufficient to demonstrate similarity
Why should biosimilar medicines be used ?
Biosimilars are expected to be a cost-effective alternative to high-priced branded biologics, resulting in a positive impact for the main stakeholders:
üOpportunities to treat more patients with appropriate therapies
(physicians).
üCost savings and financial sustainability of healthcare systems (payers).
üImproved access to medicines (patients).
üReasonable return of investment with the continued attractiveness of R&D investment in new medicines development (industry).
•According to NHS’s recent savings data, NHS England saved £324 million in the last financial year by switching from using 10 higher priced medicines to better value biosimilars and generics.
•Switching patients to biosimilars of these biologics led to significant cost savings
•Infliximab biosimilars delivered £99.4 million in savings
•Etanercept biosimilars delivered £60.3 million
•Rituximab biosimilars delivered £50.4 million
•Cumulative savings of over £220 million in cancer and autoimmune diseases such as rheumatoid arthritis and inflammatory bowel diseases.
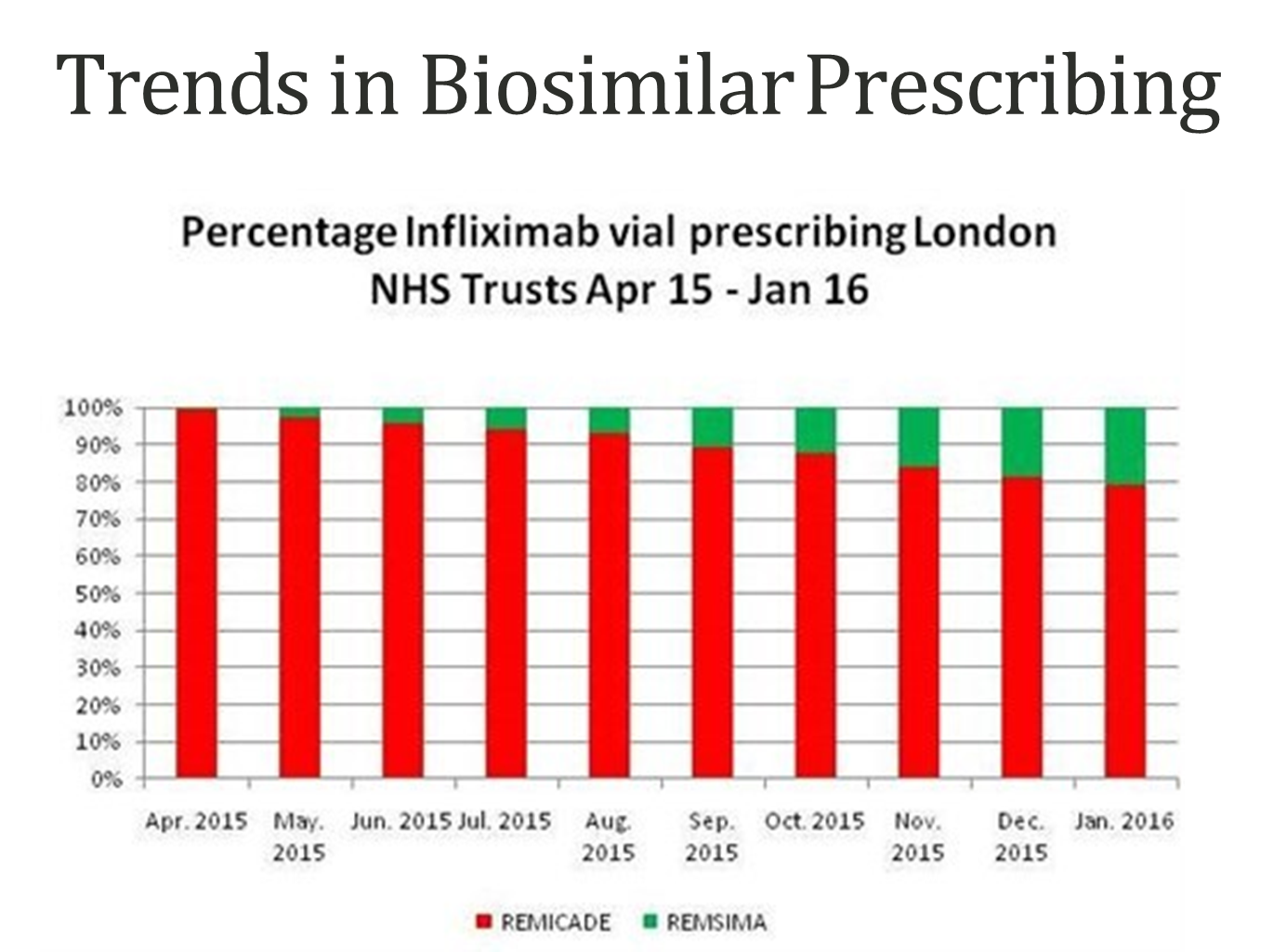
Trends in Biosimilar Prescribing
Is automatic substitution permitted?
•Automatic substitution, defined as the practice of dispensing one medicine instead of another equivalent and interchangeable medicine at the pharmacy level without consulting the prescriber, is not appropriate for biological medicines, including biosimilar medicines and is not permitted at this time.
•
•As biosimilar medicines often use the same international non-proprietary name (INN) as their reference product, so the main way to ensure automatic substitution does not take place, is through prescribing brand name.
•
• Brand name prescribing should be adhered to by all prescribers for biological medicines, including biosimilar medicines, and is in line with recommendations from the MHRA and NICE, as well as being enshrined in EU law
•
•UK prescribers are taught to prescribe using the INN, or ‘generic’ name, and this is established practice within the NHS for most products.
•It is important to ensure that prescribers are aware of the different requirements associated with biological medicines, including biosimilar medicines.
Can a patient already established on an originator biological medicine be switched to a
biosimilar medicine?
•Prescribers, of course, are always able to switch treatments for a given patient, provided it is safe to do so and there are appropriate monitoring arrangements in place
•There is growing practical NHS experience that demonstrates the safety and efficacy of biosimilars in clinical practice.
•The evidence regarding interchangeability is still developing.
•Switching between a reference product and its biosimilar should be managed at the discretion of the individual prescriber in partnership with the patient, with appropriate monitoring in place.
•Evolving evidence and treatment guidance should be made available to patients and prescribers to support them in their decision-making.