yr 2 options A Craven medicinal chem
1/55
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
56 Terms
what is a drug?
a drug is a substance that has a physiological effect when introduced into the body of a living organism intended for use in medical diagnosis, cure, treatment or prevention of disease
pharmacokinetics vs pharmacodynamics
pharmacokinetics - what the body does to the drug
pharmacodynamics - what the drug does to the body
drug target interactions
biological targets are big compared to small molecule drugs
the key drug-target interactions usually happens in a local area of binding site
protein/biological target interacts with the drug in the binding site through binding interactions that are usually non covalent
there are several types of intermolecular bonding interactions which differ in their strengths - number and type present depends on the structure of the drug and the functional groups present
types of drug target interactions
electrostatic/ionic bonds
van der waals interactions
pi-pi interactions
hydrophobic interactions (the only one thats entropic not enthalpic)
hydrogen bonds
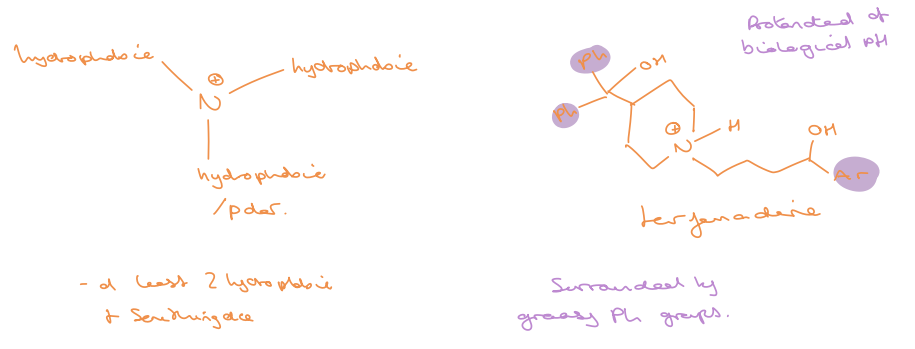
electrostatic/ionic drug target interactions
not very common but when present are usually the strongest interactions - almost always stronger than hydrogen bonds
eg. between NH3+ and COO- an ionic interaction (salt bridge) can be formed
An enthalpic effect
binding energy: 5-300 KJmol-1
van der waals drug target interactions
simple induced dipoles and their attractive forces
exist in all drug-protein interactions and can’t really be altered so mostly uninteresting
An enthalpic effect
rough binding energies: 0-10 KJmol-1
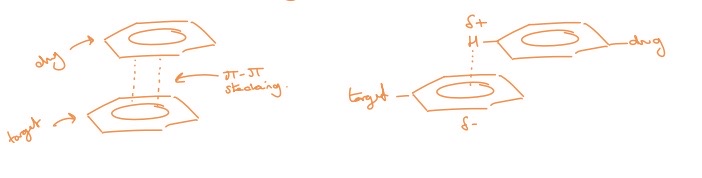
pi-pi drug target interactions
interactions between two aromatic rings (have clouds of electron density above and below the ring)
usually leads to regions of electron density building up in those areas but if you place and EWG or use electron deficient heteroaromatics, you can flip this and create a positive charged area that can stack onto another aromatic ring
pi-pi stacking
An enthalpic effect
binding energy: 1-5 KJmol-1
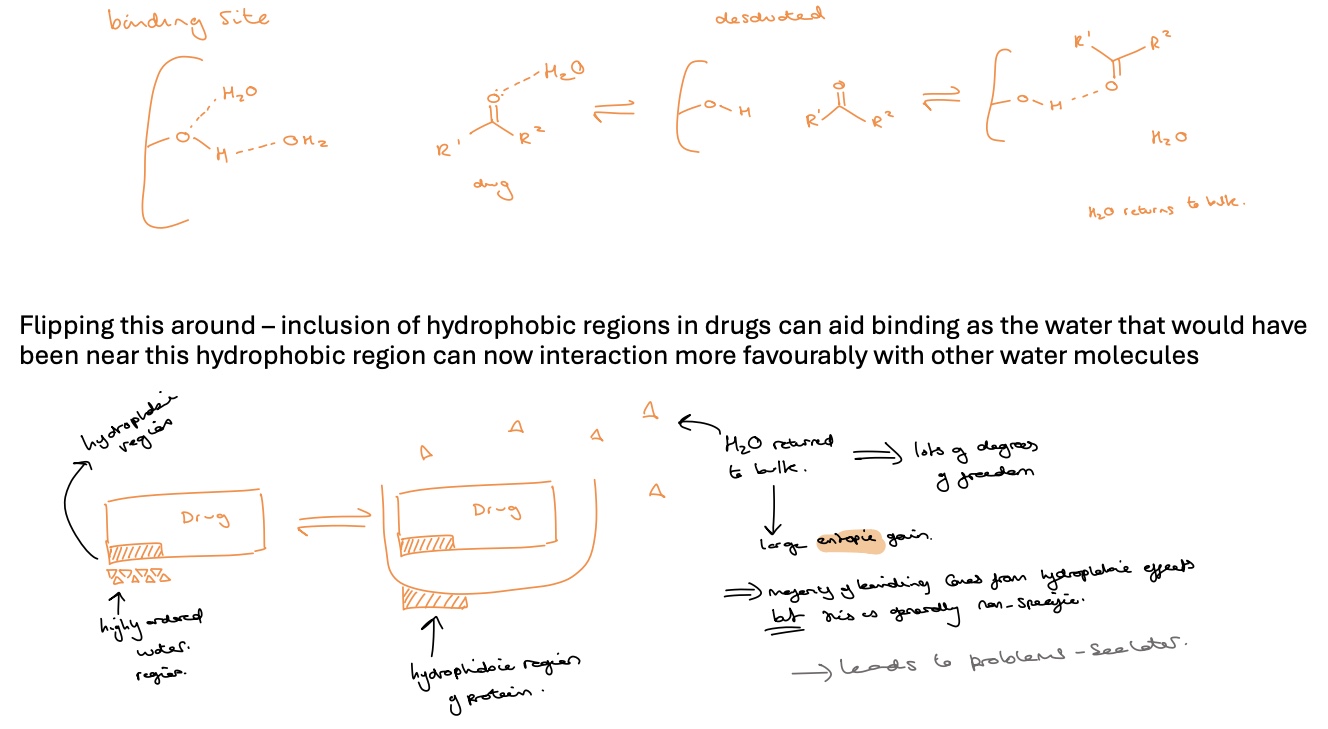
hydrophobic drug target interactions
an entropic effect
the water molecules surrounding the drug have to be stripped away before other interactions can take place
this takes energy and if energy is required to desolvate both the drug and the binding site is greater than the stabilisation energy gained from the binding interactions then the drug may be ineffective
however inclusion of hydrophobic regions in drugs can aid binding as the water that would have been near this hydrophobic region can now interact more favourably with other water molecules
water returned to bulk (from hydrophobic region on drug- highly ordered water region) when drug and protein bind create lots of degrees of freedom in water therefore a large entropic gain
majority of binding comes from hydrophobic effects but this is generally non-specific
hydrogen bond drug target interactions
probably the most important interaction when it comes to specificity (binding to one target over another)
strength can vary from 16-60 KJ mol-1 (usually 12-20)
HBAs
anything with a partial negative charge
teh more negative the charge the stronger the H-bond
carboxylate > phosphate > carbonyls > alcohols/amines/ethers? > R2S/R-Cl
HBDs
generally an electron deficient hydrogen linked to O or N
the more electron deficient the stronger the bond formed
R-NH3+ > R-OH > R-NH2 or Ph-NH2 (not good HBAs)
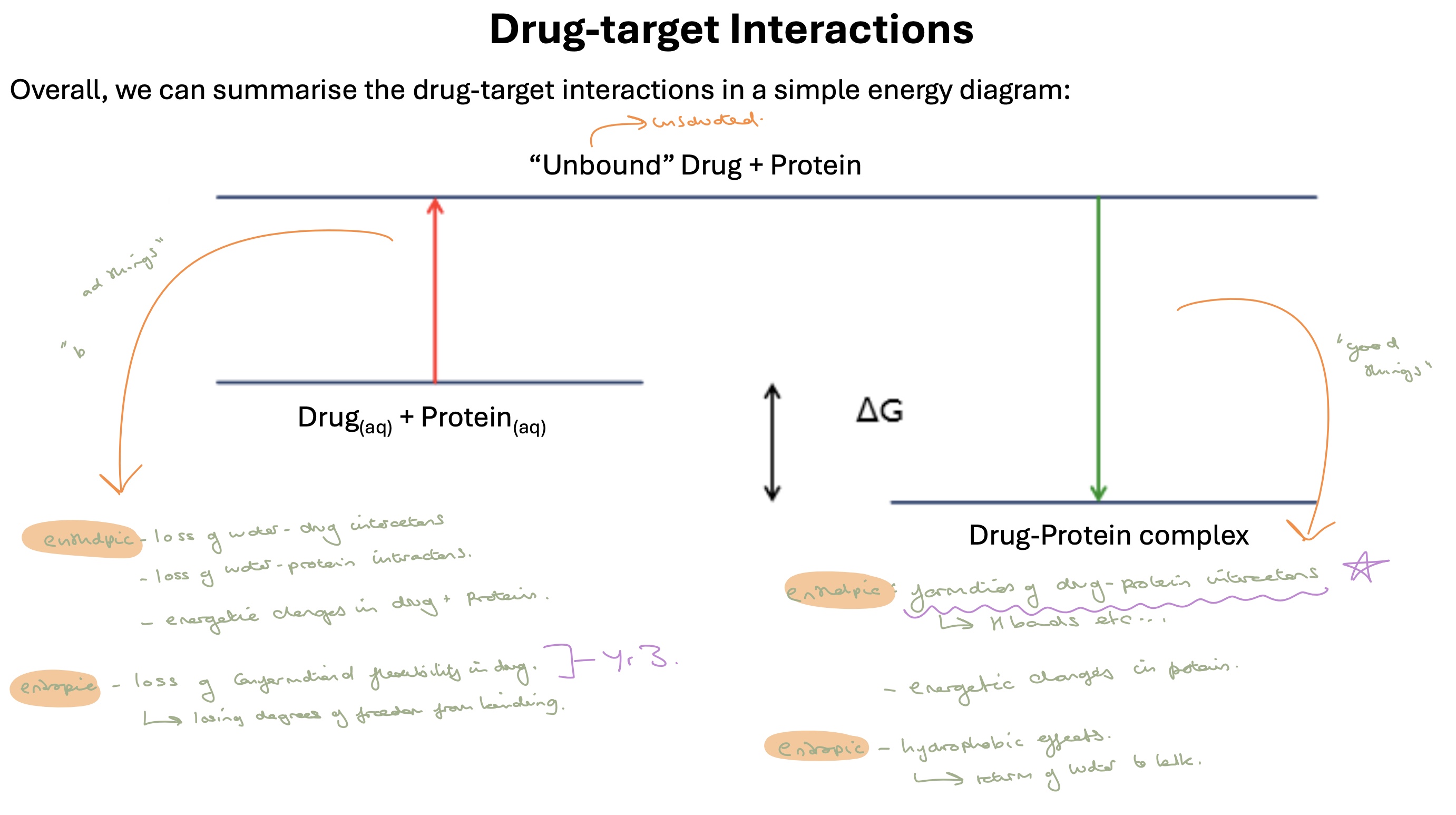
drug target interactons summarised in an energy diagram
quantifying interactions (IC50)
IC50 = inhibitory concentration - 50% effect
eg. the conc of a drug needs to kill 50% bacteria / have 50% effect - measure in M
look at photo for keq and Ki
∆G = -RT ln(eq) therefore ∆G = RTlnKi

pKa estimates - acids
acid- lower pka - stronger acid
ethanoic acid - 5
phenol - 10
tetrazol - 4.5
sulfonomide - 4
pka estimates - bases
base - higher pka - stronger base
NH3 - 9
MeNH2 , Me2NH - 11
Me3N - 10
imidazole - 8
pyridine - 5
alinine - 5
availabiliity of lone pair or stability of +ve charge dictates basicity
pka estimates - neutral
ketones
carboxomides
ethers
amines - trends in pKa
electron donating r groups tend to increase basicity
makes lone pair more available
stabilises resultant positive charge
electron withdrawing r tend to reduce basicity
makes lone pair less available
destabilises resultant positive charge
number of substituents
in aqueous solution need to consider teh solvation by water as weel - this stabilises the positive charge we make which increases basicity
solvation by H20 increases with more N-H bonds (more capability to h bond with H2O) - this works against EDGs (ankyl groups) so primary adn secondary are more basic that tertiary amines and NH3
acids - trends in pKa
electron donating r groups tend to reduce acidity
destabilise -ve charge
electron withdrawing r groups tend to increase acidity
stabilise -ve charge
determining extent of ionisation of compound
for acids
fraction unionised = 1/1+10^(pH-pKa)
pH = pKa : 50% unionised
pH = pKa -1 : 90% unionised
pH = pKa -2 : essentially completely unionised
for bases
fraction unionised = 1/1+10^(pKa-pH)
pH = pKa : 50% ionisedlipo
pH = pKa -1 : 90% ionised
pH = pKa -2 : essentially completely ionised
lipophilicity
a measure of how ‘greasy’ a compound is ie. how much it likes lipids (fats) and equivalent to hydrophobicity (how much water it hates)
lipophilic functionality includes:
aliphatics
aromatics
halogens (not all solvated by water despite lone pair)
polarity
a measure of how hydrophilic a compound is
polar functional groups include:
alcohols
amines
amides
ligand efficiency
a useful value for describing the efficiency of a drug’s binding to a target- some of the binding of a drug to a target is driven by hydrophobic interactions
hydrophobic interactions generally increase with the size of the molecule so we would expect larger molecules to bind better given all other binding interactions being equal
these interactions are key to specificity though so LE looks to remove hydrophobic interactions from the equation
same ∆G for and increasing N therefore less good binder and decreased LE
use heavy atom count as all atoms bar hydrogen typically contribute to hydrophobic effects
It is now widely used in drug discovery as it can identify high quality hits with a small binding energy which might get lost otherwise
through the drug discovery process LE will generally stay the same
equation for ligand efficiency
LE = -∆Gbinding / N
LE = 1.4 (-logKi)/N
LE = 1.4pKi/N
convert from J → cal Kcal = kj/4/184
n = number of heavy atoms ie. not hydrogen
lipophilic ligand efficiency (LipE)
instead of linking the binding to heavy atoms count this links it to logP as this links more directly to hydrophobic interactions
lipE = pKi -LogP
(pKi = pIC50)
high quality compounds that aren’t binding through hydrophobic effects have a high LipE eg above 6
quantifying lipophilicity and polarity

logP/logD and pKa for acids
log D is ALWAYS smaller than logP - not just in acidic conditions
when unionised logP=logD
dip starts just before pKa = pH
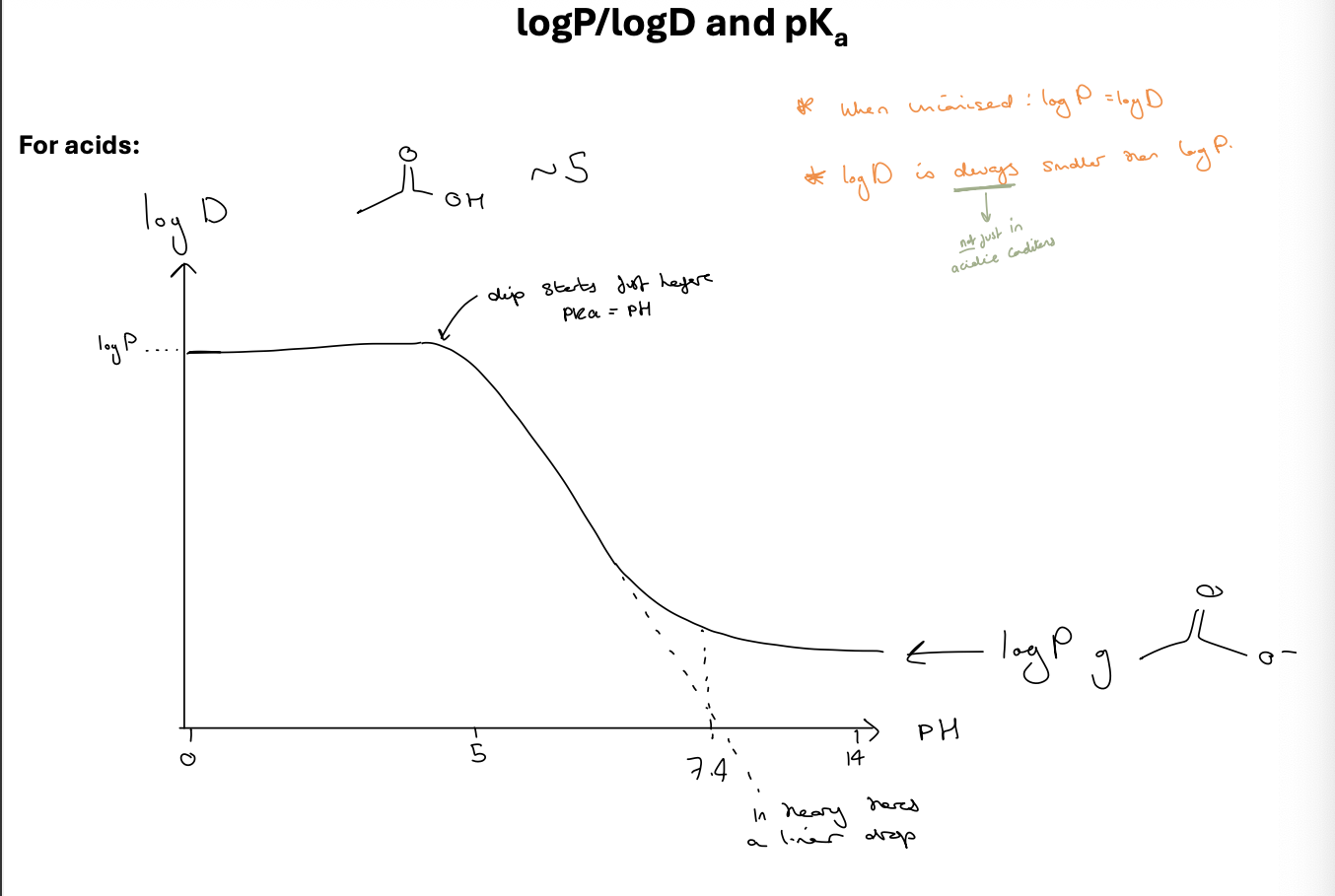
logP/logD and pKa
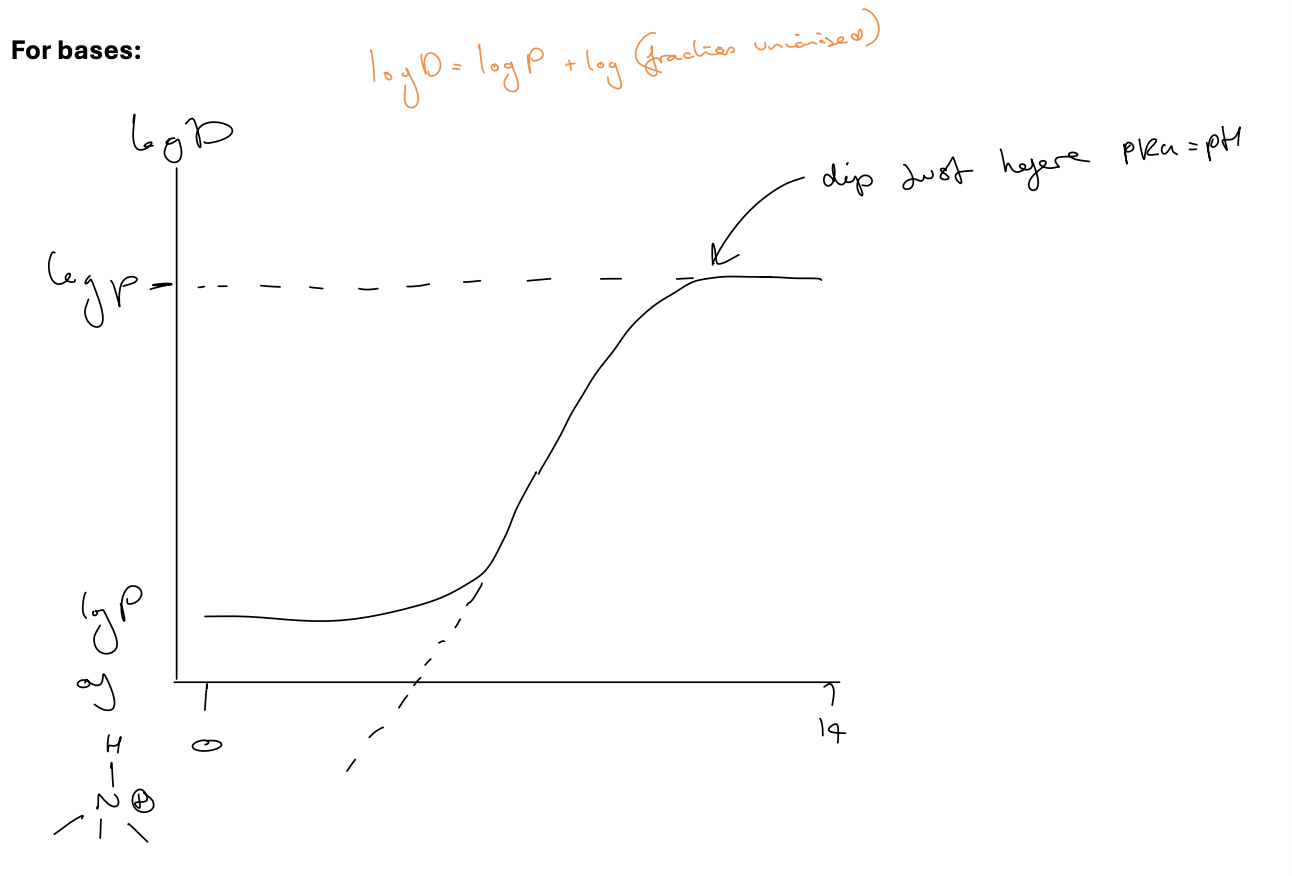
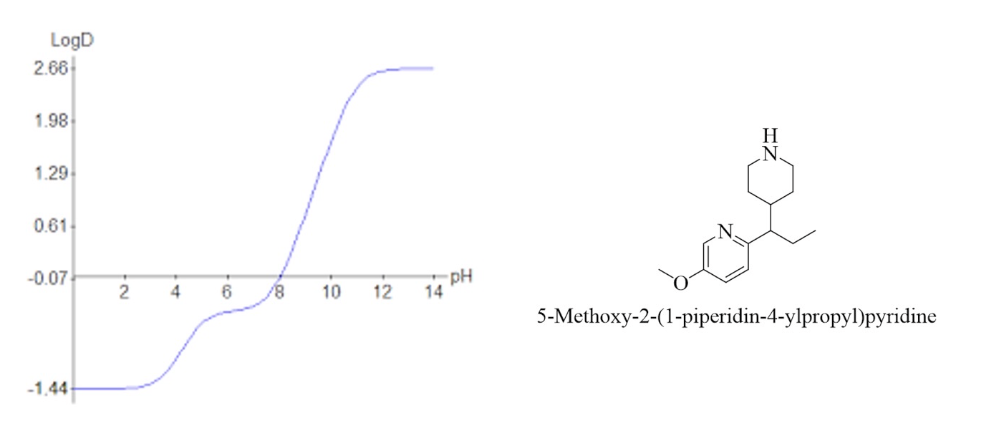
what does this graph tell you about this compound

difference between logP and logD
both a measure of lipophilicity / hydrophobicity
LogP treats the molecule as neutral
LogD looks at the ionisation of the molecule as well - dependant on the pH its measured at as this alters the ionisation of the drug
Calculating logP
often calculated using ‘fragmental calculators’
assume logP is an additive property ie. addition of a group changes logP by the same amount each time - falls down primarily when functionality changes
calculated logP = ClogP
can estimate logP based on fragments
H = 0
lipophilic substituents: pr > Me > Et > Cl > CF3 > Ph > F (positive values)
polar substituents: SO2Me > SO2NH2 > CONH2 > CN
pharmacodynamics
study of the pharmacological response to a drug
desired effects due to activity at the biological target ‘potency’
undesired effects due to activity at other targets ‘selectivity’
basically what the drug does to the body
pharmacokinetics
study of the movement of a drug through the body
absorption, distribution, metabolism, excretion (ADME)
gives an indication of the likely exposure of the drug in the body
basically what the body does to the drug
where does the drug go?
gut —(absorption)—> blood —(when drug can do its work)—> liver/kidneys —(elimination)—> excrete
brain - unique membrane between blood and brain called blood brain barrier - protects brain from things in blood
oral delivery - most common method
heart - very specific tissues/ion channels to worry about
stomach + intestine - GI tract, this is where we absorb the drug into blood
liver + kidneys - key organs in the excretion of drug
oral drug profiles
to monitor the absorption and elimination of a drug medicinal chemists will construct drug profiles of their drug in vivo
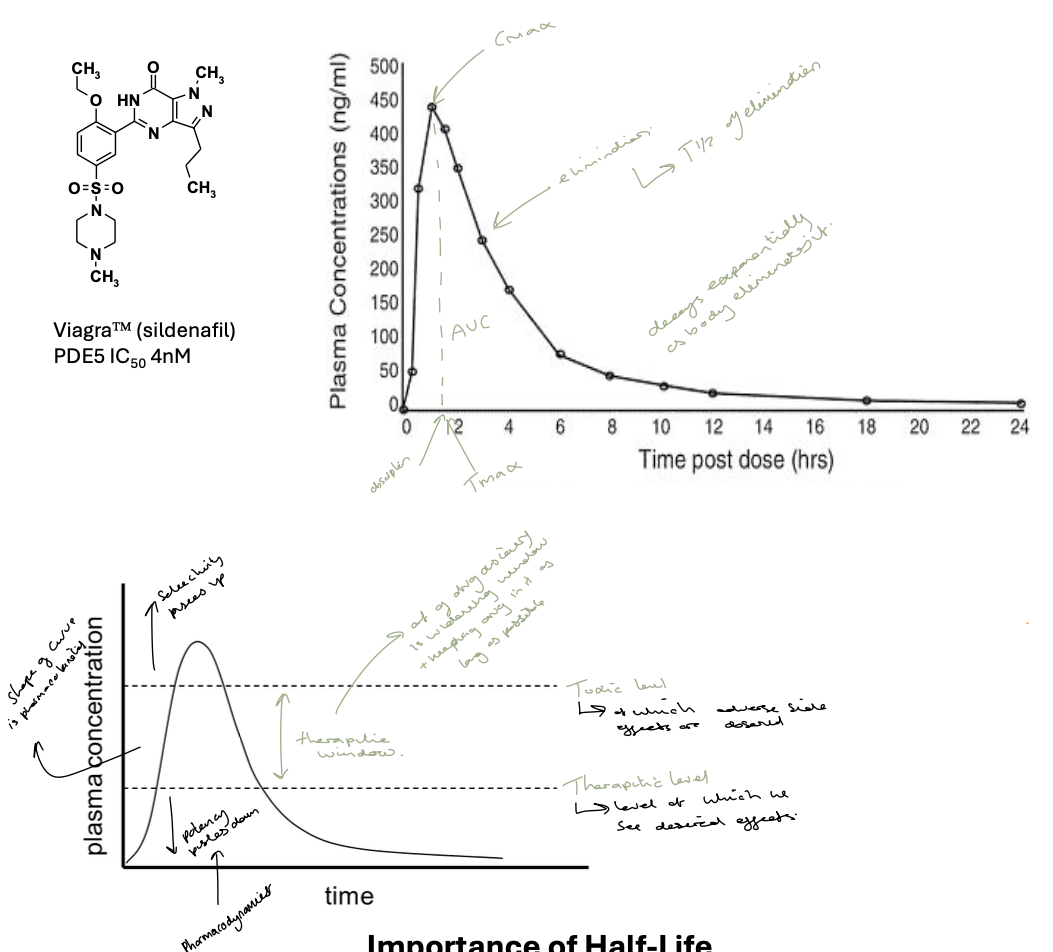
importance of half-life
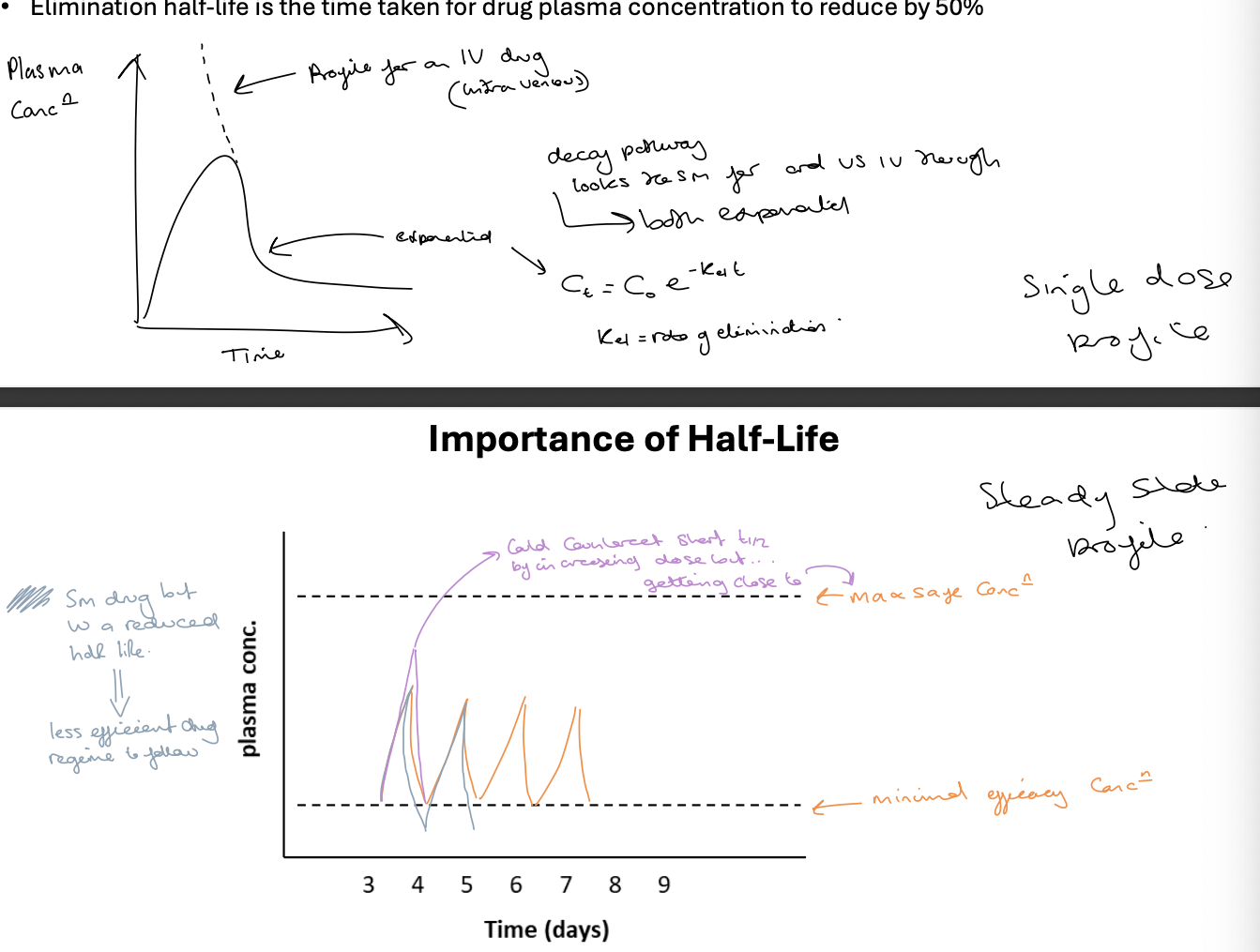
elimination half life is the time taken for drug plasma concentration to reduce by 50%
for ideal administration of oral drugs we want 24h doses
the shorter the half life the greater peak to trough ratio- need higher dose to maintain Cmin > efficacy
unless the drug is very safe once-a-day dosing requires a long half life eg. longer than 12 hours
half life is driven by two independent factors - volume of distribution and clearance
dosing must be carefully controlled to ensure that the plasma concentration remains in the therapeutic window
first graph is the single dose profile
second graph is the steady state profile - shows max safe and minimus efficacy concentrations
what are the 6 components of ADMET - key pharmacokinetic ideas
absorption
distribution
metabolism
excretion
elimination*
toxicity
absorption
process by which our body absorbs the drug
for oral drugs = GI tract - 4 main types
paracellular (molecule squeezing between cells in the gut wall) -rare
transcellular - passive diffusion (by far the most common)
transcellular - active transport (cell actively pulls drug through gut wall)
transcellular - efflex pumps - essentially the opposite of active transport - cells push drugs back into GI tract
factors to consider when looking at the absorption of a drug
logP
too polar - less able to desolvate and enter highly non-polar cell membranes
too lipophillic - may permeate cell membrane but fail to exit into aqueous media on the other side
ionisation
generally ionised compounds will not cross the cell membrane - do not line lipids
can use prodrugs to mask ionisable functionality until it reaches the target eg. masking carboxylic acids (carboxylates at body pH) as methyl esters which are non-ionisable so absorbed well but inactive - converts back to active form in vivo after absorption
altering basicity d a compound can lead to significant changes in permeability/absorption
altering pKa from 99% ionised to 90% ionised for example ensures enough drug is neutral for absorption to occur
follow lipinskis rules
Lipinski’s “rules” and the birth of cheminformatics
a drug has poor absorption if it obeys more than one of the following rules
ClogP < 5 (don’t want compound too greasy)
molecular weight < 500 (don’t want compound too greasy)
HBD <5 (not too polar or it wont want to leave water - too many H bonds therefore not absorbed)
HBA <10
this sparked a revolution in cheminformatics (computional) in drug discovery
distribution
transfer of a drug from the blood to the tissues eg. where site of action exists
metabolism
modification of the drug by the body to help it be excreted
excretion
transfer of drug out of the body eg. urine, faeces
elimination
either the same as excretion (what phil uses) or combination of metabolism and excretion
toxicity
assessing the side effects - fundamentally caused by drug binding to other targets
unique to a drug class - some common liabilities eg. hERG ion channel - spans most small drugs
blocking hERG channels leads to prolongation of QT interval leading to fatal cardiac arrhythmias
common motif (toxicoplane) that causes hERG liability show in photo
Volume of Distribution (Vd)
volume required to hold all drug added to the system (body) at the same concentration to that measured in the circulating compartment (blood) - a theoretical parameter
a measure of how well the drug distributes out of the blood into the tissues- units L or l/kg - small values suggest a drug is closely confined to plasma and doesn’t distribute well to tissues
x mg of drug in one dose - if all absorbed and all staying in blood then plasma concentration = x/volume of blood in the body (3.5 L)
Vd = dose / drug concentration in plasma
small values = minimal distribution
large values = large distribution
neutral and acidic compounds tend to have limited distribution into tissues and have low volume of distribution (0.2-2 L/kg)
basic compounds display a higher volume of around 0.5-30 L/kg
as a general rule, Vd tends to increase with increasing lipophilicity
clearance
part of the elimination pathways
the rate at which a compound is removed from systemic circulation (via any method)
usually described in mL/min/kg
primarily through kidneys or liver or both
CL = Q x Er
Q = blood flow - for liver = 20 mL/min/kg
Er = extraction ratio = increased Er = increased clearance
[X]a → liver→[X]b (blood with drug) or → clean blood
half life relationship to Vd and CL
C = Cmax e -Ket
Ct = C0 e-kel t
Kel = CL / Vd
t1/2 = 0.693/Kel = (Vd x 0.693)/CL
an increased Vd leads to an increase in half life and an increase in CL leads to a decrease in half life
Vd more widely distributed means less drug moving through the liver
metabolism phase 1
initial alteration of functionality to something more polar
OXIDATION
eg. hydroxylation- usually happens on aromatics or adjacent to heteroatoms
N,S-oxidation → amine → N-oxide (R2N-O-) or R2S → R2SO2
N, O, S - dealkylation
REDUCTION
carbonyls to alcohols
-NO2 → -NH2
HYDROLYSIS
of esters or amides
metabolism phase 2
appendage of natural functionality to produce highly polar substrates
GLUCRONIDATION
add a sugar (glucose) to an alcohol
SULFONATION
adding sulfate (SO2OH) to alcohol
SILDENAFIL’S route of metabolism (on separate flashcard)
SILDENAFIL’S route of metabolism
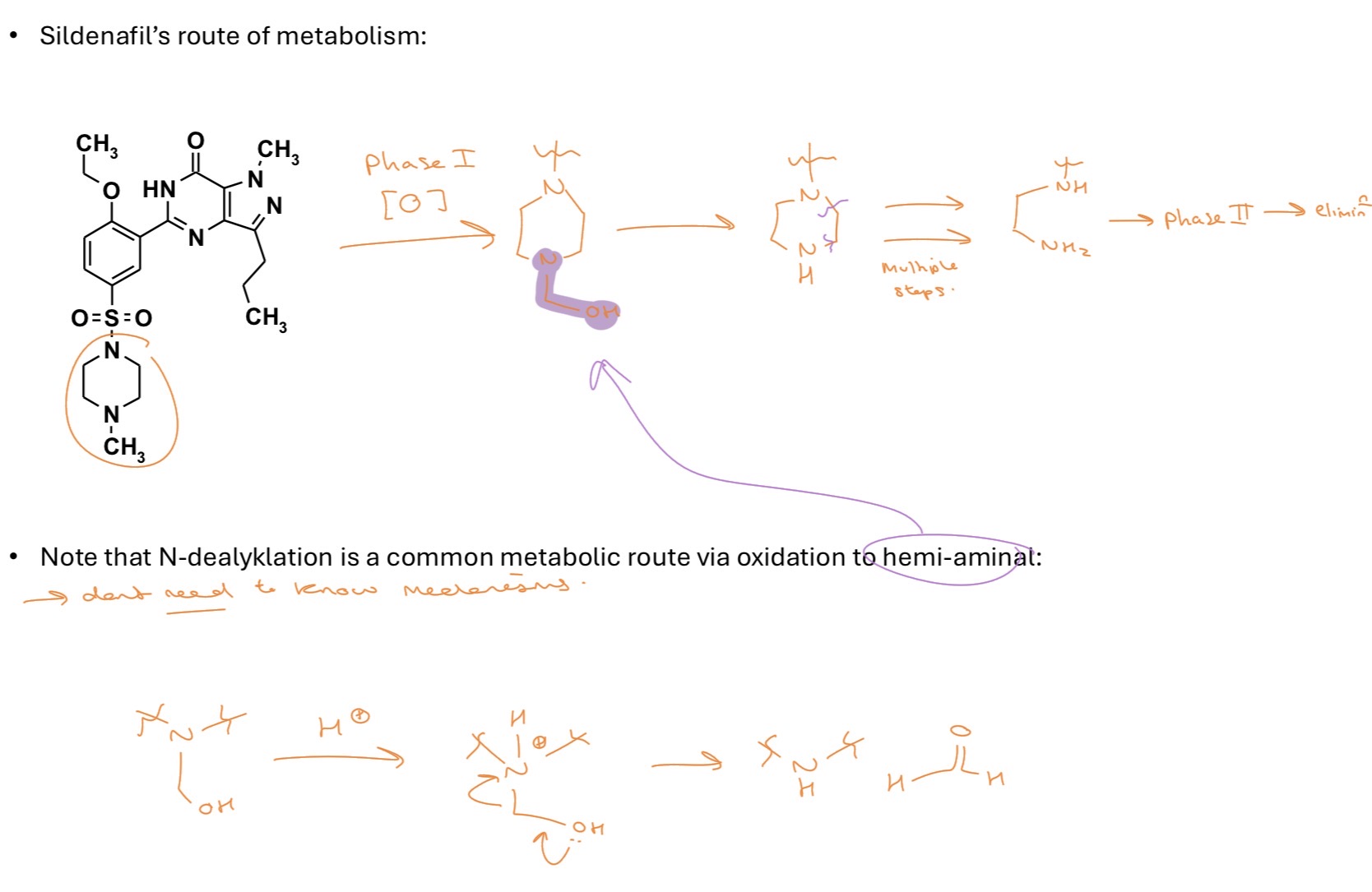
reducing metabolism
primarily done through ‘blocking’ the site - generally a C-H we’re trying to get rid of
eg. adding Fs is a classic approach as its nor much bigger than H but the C-F bond in strong
or altering an ether to an sulfoxide ie. R2O → SR2O2
need to be aware of admet changes though
not always that simple though, the metabolic enzymes involved tend to interact more strongly with more lipophilic substrates
often a correlation between logD and elimination and for compounds with logD >3 then reducing logD is the best tactic
for compounds with logD >3 then blocking the metabolic site often just moves the site of metabolism to somewhere else.
bioavailability (F)
%F = % of administered dose reaching systemic circulation (bloodstream)
a key pharmacokinetic parameter - determines the relationship between the administered dose and the concentration of drug in the body
AUC (area under curve) is proportional to %F
increased absorption - increased F
increased distribution - increased F
decreased metabolism - increased F
decreased elimination - increased F
natural products
natural products have evolved to interact with biological systems - why would an organism waste energy making something that couldn’t interact with biological systems
63% of small drugs are derived/inspired by natural products
eg. penicillin, lovastatin
fast follower approach
sometimes a new drug that comes along is described as first-in-class or blockbuster - drugs that do something new or different either treating a previously untreatable disease or targeting a new biological target within the same disease area
more often new small molecule drugs are described as fast-follower or ‘me too’ drugs - where a company takes an already developed drug compound and adapts it to make it better whilst avoiding patent infringements
high-throughput screening (HTS)
the vast majority of compounds synthesised don’t reach the market and are instead stored un libraries and are routinely tested against new biological targets
advantages
cheap if you have the equipment bc you already have compounds
get lots of info very quickly
head start on synthesis
disadvantages
using ‘bad’ compounds eg. may be effective but safety concerns
not exploring chemical space - focused on areas already explored
hit or miss - what happens if none hit?
phenotypic screening vs target-based drug discovery (TBDD)
drug discovery was historically achieved by testing patients/animals with drugs and seeing what happened - this is phenotypic screening as the phenotype is the treatment of the disease - care about how it works not why (only learn why they work as chemical/biological techniques advance)
TBDD - more recent - identify a biological target that we think should treat a disease then test molecules against that target - but even if you find a compound capable of initiating a target there’s no guarantee it can do so in vivo and even if it can there may be many other complication in the biological system
so phenotypic screening has returned to the forefront
advantages of phenotypic screening
studies have shown that phenotypic screening is more effective at getting first-in-class
more confidence in in vivo activity - pharmakokinetics
disadvantages of phenotypic screening
what is the mechanism of action? net target which is lots of effort to find
TBDD is shown to give better best-in-class drugs
virtual screening
what if you don’t have compounds or are unable to store them?
solution is to run screening in silico - virtually
need crystal structure of protein of interest
then ‘dock’ compounds with the protein
gives starting point
serendipity
lead compounds are often found as a result of serendipity ie. chance
but their discovery required someone with an inquisitive nature to recognise the significance of chance discoveries and take advantage of these events
the discovery of cisplatin and peniciliin are the two most famous
eg. chlormethine and cyclophosphamide - survivors of mustard gas attacks were more susceptible to to infections - shown white blood cell counts were down - treatment for leukemia that causes excess white blood cells
glyceryl trinitrate - explosive industry - workers often suffered headaches - resulted from dilution of blood vessels in the brain - oral tablet used to treat angina
disulfiram - rubber industry - acquired a distaste for alcohol - antioxidant prevented oxidation of alcohol - build up of acetaldehyle - very unpleasant - used as a treatment of alcohol addiction