Exam II Review
1/99
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
100 Terms
What are types of mutation?
physical
chemical
spontaneous
What are somatic mutations?
occur in any cell outside of sperm and egg
not passed on to children
accumulates over a lifetime
shorter lifespan means more mutations
What are germline mutations?
mutations that occur in the gametes
passed to offspring
older parents are more likely to pass new germline mutations
What controls mutation?
age
generation time - species that reproduce quickly, go through more DNA replication in shorter time, more opportunity for mutation)
SNPs
single nucleotide polymorphisms; one letter difference in the DNA
What is an example of a SNP?
sickle cell trait - in the hemoglobin beta gene, an A is changed to a T, causing glutamic acid to turn to valine
Structural Variants (SVs)
a large scale change involving 50 bp or more
What are types of SVs?
deletion
insertion
inversion
duplication
translocation
What is an example of an SV?
Hemophilia A - an inversion in the F8 gene on the X chromosome; body cannot produce functional blood-clotting proteins
What percent of the human genome are SNPs?
0.078%
What percent of the human genome are insertion-deletions?
0.069%
What percent of the human genome are SVs?
0.19%
What percent of the human genome are inversions?
0.397%
What percent of the human genome are multi-CNV?
0.232%
What is the goal of the International HapMap Project?
Identify genetic variants of common diseases.
When was the International HapMap Project launched?
2002
Was the International HapMap Project publicly or privately funded?
Public and Private (Japan, Canada, China, U.S., U.K.)
Haplotype
a specific set of alleles (DNA variations) that are located physically close to each other on a single chromosome and are inherited together as a single unit from one parent.
an overview of the International HapMap Project
-blood samples were collected from the Yorubas in Nigeria, Japanese, Han Chinese and US residents with ancestry from Northern and Western Europe.
-map haplotypes, not individual mutations
-find haplotypes that are different between healthy and diseased
-various SNP microarrays were used for genotyping
What was the goal of the 1000 genome project?
sequence >1000 genomes or to find variants with frequency >1%
When was the first and last sequence of the 1000 genome project sequence?
first sequence in 2008, last sequence in 2013
an overview of the 1000 genome project
-first project to sequence the genomes of a large amount of people
-3 phases
-sequence >1000 genomes or to find variants with frequency >1%
What data was found from the 1000 genome project?
-2504 individuals from 26 populations (low sequence coverage + exom data) 24 individuals sequence to high coverage
-88 million variants (84.7 million SNPs, 3.6 million short insertions/deletions, and 60,000 SVs)
-first genome map of variants larger than SNPs (revealed roles of SVs in gene expressions + diseases)
-more than 99% of SNP variants with a frequency of >1%
-cell lines and DNA available for further use
What was the goal of the “All of Us” project?
sequencing 1 million people by 2026
an overview of the “all of us” project
-funded by NIH
-first dataset (100,000 people) released in 2022 (enrollment began in 2018)
-413,000 individuals enrolled last year (2023), 250,000 sequence 46% are minority racial or ethnic group
-the database includes some participants’ survey responses, electronic health records and data from wearable devices
-145 new candidate factors discovered for type 2 diabetes.
What did Lewontin and Hubby do?
They introduced the summary statistic heterozygosity (H) and proportion of polymorphic sites (P)
alloenzymes
enzymes used to “track” genetic variation via electrophoresis
heterozygosity
probability of having different alleles
proportion of polymorphic sites
percent of the genes that actually have the variants
what are the formulas for measuring genetic variation? (h, H, P)
h = 1 - Σxi2 ( i = alleles at the loci)
H = 1/n(Σhi) ( i = no. of loci)
P = p/N (p = number of polymorphic loci, N = total loci)
average values found in early studies
-mean H = 0.12 (about 12% of an individual’s genes are heterozygous)
-mean P = 0.3 (about 30% percent of all genes locations in a population have more than one version of an allele)
What is Hardy Weinberg Equilibrium?
If the following conditions are met allele and genotype frequency will not change:
p² + q² = 1
heterozygotes = 2
homozygotes = p², q²
where p is the frequency of allele A and q is the frequency of allele a.
What are the assumptions of Hardy Weinberg Equilibrium?
-no natural selection
-random mating
-infinite population size
-no mutation
-no gene flow
What are Ne and Nc?
Nc → census population → all
Ne → effective population → breeding
What does Ne say about genetic drift?
-small Ne means higher genetic drift (random chance can easily wipe out certain alleles, genetic drift is stronger than selection meaning a bad mutation might spread due to bad luck)
-large Ne means there is more room for different mutations to coexist (good at weeding out deleterious mutations)
What are DNA markers?
a specific segment of DNA that is between genes; shows variation between individuals in a population
-non gene markers are DNA markers
-should have >= 2 alleles
What are the three major types of DNA markers?
-Restriction Fragment Length Polymorphism (RFLP)
-Simple Sequence Length Polymorphism (SSLP)
-Single Nucleotide Polymorphism (SNP)
Restriction Fragment Length Polymorphism (RFLP)
concept: uses restriction enzymes that cut DNA only at specific sequences
variation: if one person has a mutation at that cut site, the enzyme won’t cut it.
result: you run the DNA on a gel, the fragments will be different lengths
Simple Sequence Length Polymorphism (SSLP)
concept: focuses on repetitive sequences
variation: each person would have different amounts of repeats
result: highly variable among people
KNOW THE ADVANTAGES AND LIMITATIONS OF VARIOUS METHODS INCLUDING LEWONTIN AND HUBBYS
In-situ, synthesized array
oligo synthesized using photolithography
How do synthesized arrays work?
light is used to activate specific spots of a glass slide. when the light hits a spot, a specific DNA base is “glued” (forms a covalent bond with the linker molecule on the glass slide) by repeating this they form a short DNA strands (oligos) directly on the slide
What is the capacity of synthesized arrays?
500,000 SNPs can be tested on one chip
Who was synthesized arrays developed by?
Affymetrix
self assembled arrays
-instead of growing on the DNA slide, the DNA is synthesized on tiny polystyrene beads, and deposited in wells etched on a glass surface.
what is the capacity of self assembled arrays?
2.5 million SNPs
who are self assembled arrays licensed and sold by?
Illumina
What are the advantages of microarrays?
-high throughput (faster)
-standardized
-cost effective
what are the disadvantages of microarrays?
-no SVs
-expensive equipment
-discovery bias (only find what you are looking for)
what are some criteria for SNP filtering?
-high missing frequency
-not in hardy weinburg equilibrium
-low minor allele frequency (MAF) (<1% or 5%)
-strand consistency
-exclusion of HapMap SNPs
what are some criteria for sample filtering?
-low call rates (ie people with several missing genotypes)
-high heterozygosity levels
-sex and race mismatch
what is RAD-seq?
-uses restriction enzymes to cut DNA at specific “anchor” points. you only sequence the DNA right next to those cuts.
-reduced representation sequencing strategy
Where is RAD-seq used?
widely used in non-model organism in relation to ecological evolutionary and conservation genomics
what are the advantages of RAD-seq?
-cost effective
-higher sequencing coverage per locus (high quality genotype calls)
-does not require a reference a reference genome
what are the two types of RAD-seq?
-original RAD-seq
-ddRAD
What are the steps of original RAD-seq?
digest (one enzyme)
ligate adapters
multiplex - samples are pooled together
shear - physically broken into smaller chunks
size select
end repair
A-tailing - add an A to the end
ligate y-adaptors
PCR
What are the steps of ddRAD-seq?
digest (two enzymes)
ligate adapters
multiplex
size select
PCR
what are the limitations of RAD Seq
allele dropout - when a mutation (SNP) at a restriction site prevents the enzyme from cutting the DNA, causing that specific version of a gene to be missed during sequencing. This leads to a "null allele," which can trick researchers into thinking an individual is homozygous when they actually have two different versions of that gene.
what does a Phred quality score (Q) represent?
It is a property assigned to each nucleotide base call that represents the probability that the base was called incorrectly
Why do quality scores typically decrease toward the end of a sequencing read?
Because sequencing becomes asynchronous within a cluster (dephasing/signal decay), making the "images" noisier and more error-prone over time
What is the formula for a Phred Score (Q)?
Q = -10log10Pe where Pe is the probability of error
What is a common QV score used?
20
What is the role of a Basecaller in NGS?
It converts platform-specific raw data (like fluorescent light signals or images) into actual nucleotide sequences (A, C, T, G) and their associated Phred scores.
Which tool is the industry standard for checking the overall quality of a raw sequencing run?
FastQC
What is the primary "alignment problem" when mapping NGS reads to a reference?
read aligners need to accommodate variation which looks similar to sequencing errors.
Why are long reads preferred for hypervariable regions of the genome?
to provide enough context to ensure the read is mapped to the correct unique location
What are the two main types of algorithms used for read alignments
-Data compression algorithms (BWT-based)
-Hash-based algorithms
What is the Burrows-Wheeler Transformation (BWT) used for in bioinformatics?
It is a data compression algorithm that makes aligners (like BWA or Bowtie) extremely fast and memory-efficient, especially when dealing with repetitive DNA
Compare BWT aligners (e.g., BWA) vs. Hash-based aligners (e.g., Stampy)
BWT
faster
memory efficient
great for large datasets and repeats
Hash-Based
more sensitive and accurate
slower
more memory intensive
Name two popular BWT-based aligner tools
BWA and Bowtie
Name three Hash-based alignment tools
MaQ, Novolalign and Stampy
Why do Quality Value (QV) scores often need to be recalibrated?
Raw scores may not represent the true base-calling error rate
How are raw quality scores recalibrated?
By mapping reads to invariant sites (areas known not to vary) in a reference genome to see how often the machine "calls" a mutation that isn't actually there.
What is SNP calling?
identifying polymorphic site
What is genotype calling?
assigning genotypes to individuals
What is the "Old Approach" to genotype calling and its main limitation?
It simply counts the number of alleles at a site (using a 20-80% threshold). Its limitation is that it requires very high coverage (>20X) to be accurate
What characterizes the "Modern Approach" to genotype calling?
It uses a probabilistic framework that incorporates uncertainty, allele frequencies, and Linkage Disequilibrium (LD) information
In the context of SNP calling, what does the Genotype Likelihood (P(X|G)) represent?
The probability of observing the sequencing data (X) given a specific true genotype (G), calculated using base quality scores of each read multiplied over all reads
What is a Genotype Prior (P(G)) represent?
The probability of a genotype existing at a site before looking at the sequencing data, often based on population allele frequencies or Linkage Disequilibrium
How do researchers decide which genotype to assign to an individual using the Posterior Probability?
They choose the genotype with the highest posterior probability or use the ratio between the highest and second-highest as a confidence score.
What is the formula used to combine priors and likelihoods in modern SNP calling?
Bayes' Formula
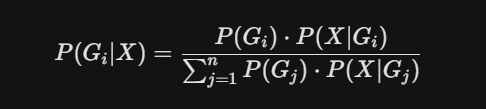
What is a Genotype Prior?
A fancy term for "what is the probability of observing a certain genotype" before even looking at the specific sequencing reads for that individual
How is the prior determined for a Single Sample if no database is available?
You assign equal probability to all possible genotypes (e.g., 1/3 for AA, 1/3 for Aa, 1/3 for aa) to avoid biasing the results
Allele frequency from multiple samples ___ genotype prior calculation.
improves
Which mathematical model is often used to estimate priors when analyzing multiple samples?
Hardy-Weinberg Equilibrium
Let’s assume genotype likelihood of AT & AA are equally large, but allele frequency of A is 1%. What would be your genotype call with & without the allele frequency data?
without - You would likely call the genotype uncertain or a tie (50/50) between AT and AA. Since the machine sees "A" and "T" reads as equally likely, it has no reason to doubt either one
with - You would call the genotype TT
How does Linkage Disequilibrium (LD) help in genotype calling?
It uses known "haplotype blocks" (neighboring SNPs that are usually inherited together) to predict a missing or low-quality genotype based on the clear genotypes surrounding it
What is Imputation in the context of NGS data?
The process of "filling in" missing genotype data by using LD status and reference haplotypes to make highly educated guesses.
When is data filtering technically unnecessary during SNP calling?
Unnecessary if posterior probability of all sites are accurate
What are some criteria used for filtering data?
-deviation from HWE
-low quality score
-systematic score difference between minor and major allele
-abberant LD pattern
-extreme read depth
-strand bias
-Batches of 1000 genomes data were discarded if they showed high discrepancy with HapMap data
Why is "Deviation from HWE" used as a filtering criterion?
Significant deviation from Hardy-Weinberg Equilibrium can indicate genotyping errors, such as an excess of heterozygotes due to mapping issues or paralogous sequences
Why is "Extreme read depth" flagged during data filtering?
Too low: Insufficient data to confidently call a genotype.
Too high: May indicate repetitive regions or duplicated sequences (paralogs) where multiple parts of the genome align to the same spot, causing false SNP calls
What is strand bias in the context of NGS filtering?
When a variant is only seen on the forward or reverse strand (it should ideally appear on both)
What is Systematic score differences in the context of NGS filtering?
When the quality scores for the major allele and minor allele differ significantly, suggesting the "variant" might just be a sequencing error
What do "Aberrant LD patterns" suggest during the filtering process?
Aberrant LD (unusually high or low correlation between neighboring SNPs) can signal assembly errors or incorrect mapping of reads to the reference genome
what is the typical pipeline for variant calling?

Why ancient DNA?
why can’t we sequence a T-rex genome?
What are the challenges of ancient DNA?
-small fragments (<200 bp)
-DNA degrades, nucleotides change
-bacterial DNA contamination
-human DNA
How is the contamination problem is solved with ancient DNA?