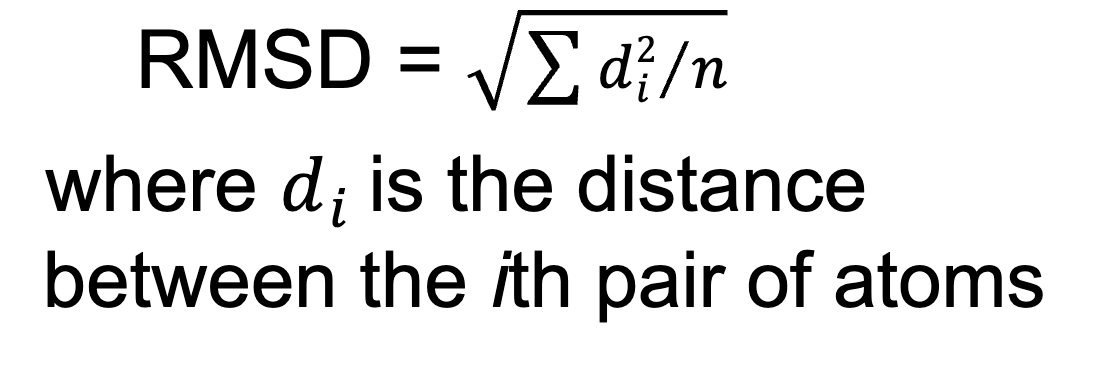Bioinfomatics Study Guide Exam 2
1/121
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
122 Terms
5 Stages of Phylogenetic Analysis:
selection of sequences: BLAST search results, protein families from Pfam, NCBI HomoloGene etc
multiple sequence alignment of homologous protein or nucleic acid sequences
specifying models of nucleotide or amino acid substitution
tree building: distance-based methods, max parsimony, max likelihood & Bayesian inference
tree evaluation
Distance-based methods:
analyze pairwise sequence alignments & use the distances to infer the relationships between all the taxa
UPGMA (unweighted-pair group method with arithmetic mean)
Neighbor-joining
Maximum parsimony:
a character-based method in which columns of residues are analyzed to identify the tree with the shortest overall branch length that can account for the observed character differences
Maximum likelihood & Bayesian inference:
model-based statistical methods to infer the best tree that can account for the observed data
Popular Software Tools for Phylogeny:
MEGA (molecular evolutionary genetics analysis)
PHYLIP (PHYLogeny inference package)
PAUP (phylogenetic analysis using parsimony)
TREE-PUZZLE (max likelihood method)
MrBayes (bayesian estimation of phylogeny)
__ comprises all the RNA transcripts synthesized by an organism
transcriptome
Proteome:
the entire set of proteins translated
Metabolome:
refers to the sum total of all the low-molecular-weight metabolites
Experimental approaches of gene expression
DNA microarrays
RNA-seq
“Large p, small n” problem:
gene expression studies typically measure the expression levels of 10s of 1000s of genes in only a few samples
DNA microarrays & RNA-seq have been widely used to __
identify which genes are significantly up/down-regulated (differently expressed)
Hypothesis testing:
inferential statistics; assign confidence to the discovery of regulated genes
Exploratory statistics:
define distances between genes; perform unsupervised analyses (clustering, PCA)
Classification:
perform supervised analyses (linear discriminants, support vector machines)
Affymetrix platforms for human microarrays
HG-U133 Plus 2.0:
54, 120 probe sets
multiple probe sets for some genes
HG-U133A:
27, 722 probe sets
well-characterized genes (RefSeq)
HG-U133B:
22, 577 probe sets
representing EST clusters
In RNA sequencing, lower # of reads = __
lower expression
In RNA sequencing, higher # of reads = _
higher expression
Sample-level/global normalization:
to remove the systemic bias in the data so that meaningful biological comparisons can be made
unequal quantities of starting RNA
experimental/technical variations
Normalization is based on the assumption that __
the total intensity distribution is comparable between 2 samples & the expression of a subset of genes is assumed to be constant
Why do we use sample-level/global normalization?
to allow valid cross-sample comparison & to minimize non-biological variation
Sample-level/global normalization method:
normalize all values for a sample so that median = 1
normalize to positive control genes
normalize to a constant value
Gene-level normalization:
rescales all genes to the same normalized value range & thus enables comparison of relative expression levels
Z transformation:
for each gene, calculate the z scores of the expression values
zxi = (xi - -x-) / σx
__ normalizes distribution
log transformation
In RNA-Seq Data Preprocessing and Normalization, experimental design needs to include __
sufficient replicates to measure biological variablity
RNA-Seq Data Preprocessing and Normalization steps:
experimental design
RNA acquisition
data acquisition
mapping
summarization
normalization
RPKM/FPKM (Reads/Fragments per Kilobase of transcript per Million mapped reads):
counts are first normalized for sequencing depth
counts are then normalized for gene length
FPKM for paired-end RNA-seq data
TPM (transcript per million):
proposed as an alternative to RPKM/FPKM
technology-independent measure of expression
Mapping software used to align unmapped reads to a reference genome is __ in RNA-Seq Data Preprocessing and Normalization
Tophat
Mapping software used to align millions of short reads to a reference genome is __ in RNA-Seq Data Preprocessing and Normalization
Bowtie
Advantages of RNA-seq
not limited to detection of known gene transcripts
little to no background signal
can detect large dynamic range of expression levels
RNA-seq can reveal information about __ resulting from alternative splicing methods
different transcript isoforms
RNA-seq can be used to discover __ such as lncRNAs
novel transcripts
Bowtie:
extremely fast, general purpose short read aligner
Tophat:
fast splice junction mapper for RNA-seq reads; aligns reads to the genome using Bowtie & discovers splice sites
Cufflinks:
assembles transcripts
Cuffcompare:
compares transcript assemblies to annotation
Cuffmerge:
merges 2 or more transcript assemblies
Cuffdiff:
finds differentially expressed genes and transcripts & detects differential splicing and promoter use
Bioconductor
an open source software project, based on R, to provide tools for the analysis of high-throughput genomic data
GEO2R
a web-based tool for comparing two or more groups of Samples in a GEO Series to identify differentially expressed genes. performs comparisons on original submitter-supplied processed data using R packages from Bioconductor
MeV (MultiExperiement Viewer)
a versatile tool (web-based or standalone) for expression data analysis, with sophisticated algorithms for statistical analysis, clustering, visualization, and classification
Experimental design
Compare normal vs diseased tissue, cells ± drug, early vs late development
RNA preparation
Isolate total RNA or mRNA
Microarrays
Fluorescently label cRNA samples and preprocessing (normalization, scatter plots)
RNA-seq
Make cDNA library for each sample and and align reads to genome or gene models; assemble transcripts
Inferential statistics
Identify significant regulated transcripts e.g. using ANOVA
Exploratory analyses
Scatter plots, principal components analysis
Other analyses
Classification, co-regulated genes
Biological confirmation
Independently confirm that genes are regulated e.g. by RT-PCR
Deposit data in a database
GEO, ArrayExpress, ENA, SRA
Fold change
Uses a fold change threshold (e.g., 2-fold) to select genes; does not take into account the biological and experimental variability
Statistical tests
Such as t test and ANOVA; require a number of replicates for each condition
Bonferroni correction
Bonferroni correction:
Set the significance cutoff, p' = α / N, where α is the false positive rate, and N is the number of genes
If you have 10,000 genes in your dataset, with 5% of false positives, p' = 0.05 / 10000 = 0.000005 (5 E - 6)
Calculate the adjusted p value: Padjusted = p * N
False Discovery Rate (FDR)
Rank all the genes by significance (p value) so that the top gene has the most significant p value
Start from the top of the list, and accept the genes if: p less than or equal to (i/N)q
i = the rank of the gene in the list, N = the number of genes in the dataset, q = the desired FDR
ANOVA (ANalysis Of VAriance)
Used to find significant genes in more than two conditions
Clustering Analysis
Divide a dataset into a few groups (clusters)
Homogeneity
Objects in the same cluster are similar to each other
Seperation
Dissimilar objects are placed in different clusters
Expression Vector
Each gene can be represented as a vector in the N-dimensional hyperspace, where N is the number of samples
Euclidean distance
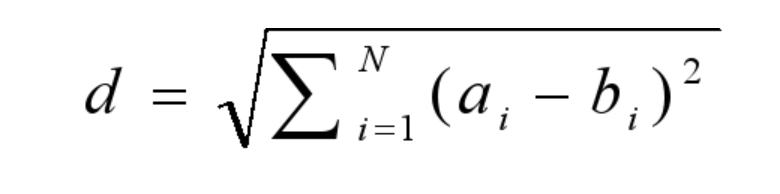
Vector angle
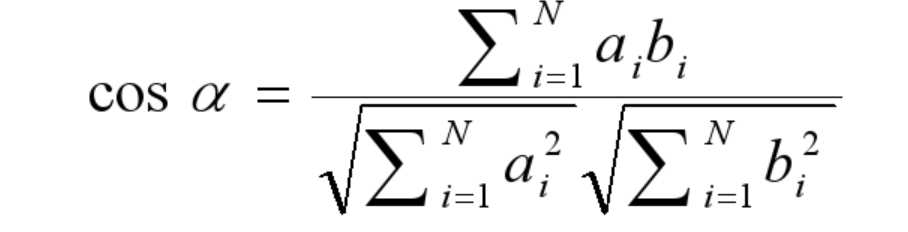
Pearson’s correlation coefficient
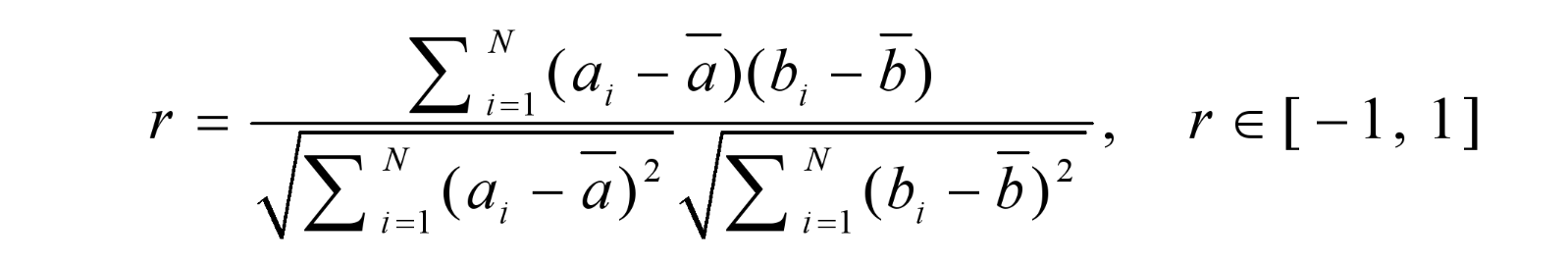
Initialization (Hierarchical Clustering Algorithm)
Each object is a cluster
Iteration (Hierarchical Clustering Algorithm)
Merge two clusters which are most similar to each other until all objects are merged into a single cluster
Hierarchical Clustering
Results are often visualized using a tree (called dendrogram) with color-coded gene expression levels. Can be applied to genes, samples, or both
Initialization (k-Means Clustering)
User-defined k (# clusters) randomly place k vectors (called centroids) in the data space
Iteration (k-Means Clustering)
Each object is assigned to its closest centroid re-compute each centroid by taking the mean of data vectors currently assigned to the cluster until the cluster centroids no longer change
Self-Organizing Map (SOM)
The user defines an initial geometry of nodes (reference vectors) for the partitions such as a 3 x 2 rectangular grid
During the iterative “training” process, the nodes migrate to fit the gene expression data
The genes are mapped to the most similar reference vector
Gene Co-expression Network Analysis
A gene co-expression network is an undirected graph, in which each node is a gene, and each edge represents a significant co-expression relationship between two nodes
It can be constructed by looking for pairs of genes which show a similar expression pattern across various samples
Does not attempt to infer the causal relationships between genes, and thus is different from a gene regulatory network
How to Assess Whether a Gene Ontology (GO) Term Is Enriched in a Gene List?
Compare gene with one already saved in the database
Database for Annotation, Visualization, and Integrated Discovery (DAVID)
Can be used to extract biological meaning from large lists of genes
DAVID’s functional enrichment analysis of a gene list is based on a modified version of the Fisher’s exact test
How to Represent a Promoter Motif?
Multiple sequence alignment
Consensus: e.g., TATAAAA (the TATA box)
Position Weight Matrix (PWM): relative frequencies of nucleotides at different positions
Sequence logo: information content of each site (a measure of intolerance for substitution)
PWM Representation of a Motif
A motif is assumed to have a fixed width, W
In the PWM, pnk is the probability (relative frequency) of nucleotide n in column k
Background probability: pn0 is the probability of n in the background (i.e., outside the motif)
Equal distribution: pA0 = pC0 = pG0 = pT0 = ¼
Pattern matching
Scanning a nucleotide or protein sequence for matches to a known pattern
How to get better sensitivity and specificity is the major consideration
Pattern discovery
Given a set of sequences, discovering a pattern that is shared by the sequences.
It is unknown in advance about what is the pattern
Using search or learning approaches
A much harder problem than pattern matching
Multiple EM for Motif Elicitation (MEME)
Widely used for discovery of DNA and protein sequence motifs
It is based on the Expectation Maximization (EM) algorithm with several extensions
MEME is now complemented by the GLAM2 algorithm which allows discovery of motifs containing gaps
Protein Structure
Proteins are very complex molecules with diverse functions
Levels of protein structure:
Primary structure
Secondary structure
Tertiary structure
Quaternary structure
Simple images highlighting specific features are useful:
Space-filling models
Ribbon cartoon models
Protein Primary Structure
Amino acid sequence of a polypeptide chain
20 amino acids, each with a different side chain (R)
Peptide units are building blocks of protein structures
The angle of rotation around the N−Cα bond is called phi, and the angle around the Cα−C′ bond from the same Cα atom is called psi
Protein Secondary Structures
Local structures as a result of hydrogen bond formation between the carbonyl and N-H groups in the polypeptide backbone (backbone interactions)
Types of secondary structures:
Alpha helix
Beta sheet
Loop or random coil
Secondary structure formation is influenced by several properties (e.g., size and charge) of amino acid side chains
Alpha Helix
Most abundant secondary structure
3.6 amino acid residues per turn, and hydrogen bond formed between every fourth residue
Proline (with no N-H group) and glycine (too small) do not foster alpha helix formation
Beta Sheet
Two or more polypeptide chains line up side by side
Hydrogen bonds formed between adjacent strands
The chain directions can be same (parallel sheet), opposite (antiparallel), or mixed
Antiparallel beta sheets are more stable than parallel beta sheets
Loop or Coli
Regions between alpha helices and beta sheets
Various lengths and 3D configurations
Often functionally significant (e.g., part of an active site)
Supersecondary Structures (Motifs)
Many proteins contain supersecondary structures (motifs) with combinations of alpha-helices and beta-sheets
In the beta-alpha-beta motif, two beta-strands and one alpha-helix are connected by loops
Protein Tertiary Structure
The unique three-dimensional structure formed by a globular protein
Stabilized by hydrophobic interactions, hydrogen bonds, and other interactions
Important Features of Tertiary Structures
Many polypeptides fold in a way to bring distant amino acid residues in the primary structure into close proximity
Globular proteins are compact because of efficient packing as the polypeptide folds
Enough hydrophobic surface must be buried, and the interior must be sufficiently packed
Buried polar atoms must be hydrogen-bonded to other buried polar atoms
Large globular proteins often contain several compact units called domains
Protein Domains
Structurally independent segments that have specific functions
The core 3D structure of a domain is called a fold
A certain type of 3D arrangement of secondary structures
Domains are classified on the basis of their core structure:
Alpha: composed exclusively of alpha - helices
Beta: consists of antiparallel beta -strands
Alpha/beta : contains various combinations of alpha -helices and beta -strands
Protein Quaternary Structure
Two or more polypeptide chains (subunits) form a larger protein complex
Protein subunits are often held together by non-covalent interactions, including hydrophobic interactions (most important), electrostatic interactions, and hydrogen bonds
Important for understanding protein-protein interactions
Unstructured Proteins (Regions)
Some proteins are partially or completely unstructured
Unstructured proteins (regions) are referred to as intrinsically disordered proteins (regions)
Over 30% of eukaryotic proteins are partially or completely disordered and have a variety of functions
The disordered segments (e.g., KID domain of CREB) may be involved in searching out binding partners
X-Ray Crystallography
Basic steps: Expression/purification, Crystallization, X-ray diffraction, Structure solution
Advantages: High-resolution structures, large protein complexes or membrane proteins
Disadvantages: Requirement for crystals, molecules in a solid-state (crystal) environment
Nuclear Magnetic Resonance (NMR)
Reveals information on the distances between atoms in a molecule, and these distances can be used to derive a 3D model of the molecule
Advantages: No requirement for crystals, proteins in a liquid state (near physiological state)
Disadvantages: Limited by molecule size (up to 30 kD), inherently less precise than X-ray crystallography, membrane proteins may not be studied
Cryogenic Electron Microscopy (Cryo-EM)
A beam of electrons is fired at a frozen protein solution. The emerging scattered electrons pass through a lens to create a magnified image on the detector, from which their structure can be worked out
Advantages: No crystal requirement for large complexes, structure remains in native state (no dehydration)
Disadvantages: Relatively low resolution (but improving), 3D structure reconstruction from 2D images
Protein Data Bank (PDB)
Established at Brookhaven National Laboratory in 1971, initially with seven structures
Was moved to the Research Collaboratory for Structural Bioinformatics (RCSB) in 1998
The RCSB PDB (https://www.rcsb.org/) has been the primary repository for 3D structural data of proteins, nucleic acids, and complexes
The Worldwide PDB (wwPDB, http://www.wwpdb.org/) was formed in 2003 to maintain a single PDB archive of macromolecular structural data
RCSB PDB
PDB supports services for structure submission, search, retrieval, and visualization
By 3/6/2026, PDB contains 250,441 experimental structures and 1,068,577 computed structure models
Search RCSB PDB
Basic search using a PDB identifier or keywords: A PDB ID consists of one number and three letters (or numbers) (e.g., 4HHB for a human hemoglobin structure; pdb_00004hhb)
Advanced search with specific attributes or data types, sequence search (BLAST/PSI-BLAST, or FASTA)
Protein Structure File Formats
PDB supports the download of protein structural data in the following text file formats:
PDB file format: outdated but human-readable
PDBx / mmCIF: simple and consistent data representation for exchanging and archiving structural data, used by PDB to store its files
PDBML / XML: a modern and robust file format
Access to Structures through NCBI
MMDB (Molecular Modeling Database)
Structures obtained from PDB
Data in NCBI’s ASN.1 format
Integrated into NCBI’s Entrez system
Cn3D (“see in 3D”): NCBI’s protein structure viewer
VAST (Vector Alignment Search Tool): for direct comparison of 3D protein structures to identify structural neighbors
RasMol and RasTop
RasMol: An open-source software package, which was a breakthrough in 3D
structure visualization. It is widely used to view 3D protein structures.
Structure file formats supported by RasMol:
PDB file format
mmCIF file format
RasTop: Provides a graphical user interface to RasMol
Other 3D Visualization Tools
Jmol: An interactive web-browser Java applet to view chemical structures in 3D, and JSmol is a JavaScript-based extension
Cn3D: Can be used for interactive exploration of 3D structures, sequences, and alignments
Swiss-Pdb Viewer (DeepView): Probably the most powerful freely available molecular
modeling and visualization package. Supports homology modeling, site-directed mutagenesis, structure superposition, etc.
Root-mean-square deviation (RMSD)