MIB- lecture 5 - Virulence factors immune system
1/17
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
18 Terms
Receptors of innate and adaptive immune response;
T cell receptors recognize mainly proteins
Formylated-peptide receptor (FPR1 and FPR2, both found on neutrophils) → N-formlylmethione is recognized in by this receptor → it has a different group compared to methionine → this is a start codon
In bacteria the methionine is formylated to N-formylmethionine
All bacteria start with this ‘start’ codon
So the receptor can easily recognize the bacteria
Toll-like receptor: detect and bind lipoproteints and also free DNA and RNA
Lectins and NOD-like receptors: recognize polysaccharides or glycans, saccharides
name different ways that immune cells combat virulence factors
Granulocytes contain lactoferrin/transferrin, involved in iron uptake
Antimicrobial peptides → kills bacteria by forming holes in the membrane
Lysozymes → can specifically cleave (peptidoglycan of bacteria) or hydrolyze bacteria and initiate cell dead
Effectors innate immune system
phagocytosis
antimicrobial peptides / lysozyme
reactive oxygen / nitrogen species
complement activation
iron / nutrient withholding
neutrophil granules
neutrophil extracellular traps; takes chromosome and throws it on bacteria and traps it (NETosis)
s. aureus, how has this pathogen developed specific mechanism to cope with immune defense system against it, explain CHIPS
They body protects against the S. aureus by
Coagulation: trapping pathogen and limiting spread
secretion antimicrobial peptides → lead to pores in bacterial membrane
Complements system activated
Phagocytosis by neutrophils and macrophages → neutrophils actively migrates to pathogen, where the concentration is the highest
all these immune cells know where to go because of chemotaxis and the FPR1/2 receptors
S. aureus block the migration of the neutrophils to protect itself from the immune system → produces CHIPS (chemotaxic inhibitory protein of S. aureus).
CHIPS (protein): binds to the formylpeptide receptor on the neutrophils → the neutrophils do not recognize it anymore and the migration is blocked
CHIPS bind to C5A receptor (complement system)
Explain the toxins S. aureus produces to kill immune cells
A-hemolysis (Cytotoxin) recognizes ADAM10 → Monomer binds to this in the membrane, this attracts all the other a-hemolysis monomer → create hole that lyse red bloods cells
LukED recognizes the receptor CCR5 → kills T cells, DC and macrophages
PVL (cytotoxin) → receptor is unknown → but does kill cells
is PVL a virulence factor for S. aureus? and how does PVL work
PVL forms pores in the membranes of white blood cells resulting in cell lysis, releasing inflammatory mediators
Patients infected with PVL+ strains have a worse survival rate than people without PVL → does impact virulence
mouse study → no different between PVL +/- → does not have affect on virulence in mouse model
PVL binds C5a receptor + leukocytes of humans → makes a hole in the membrane
thus virulence factors are also host specific not only cell specific
S. aureus and the complement system
different methods to evade complement system
Protein A SAK: binds to C1 so the antibody cannot bind, therefore the cascade of the classical pathway can not start. can also bind to C3B, stops opsonization pathway
SCIN: Directly binds to C2 and B (complement system) and inactivates these so the alternative and the classical pathway stops
CHIPS binds to C5a on the neutrophils
Understand: high amount of redundancy → multiple mechanisms that perform similar function
Facts about the protein being secreted by s. aureus interacting with complement;
small
have similar folding
homologous
Viruses also block complement
Influenza and astroviruses inhibit the binding of C1, the cascade of the complement can not start
Other viruses interfere with the formation of the membrane attack complex
herpesvirus adaptive immune response
Herpesviruses inhibit proteasome, so it cannot be degraded
With Us11, EBNA-1 and LANA-1
Herpesviruses inhibit TAP complex → prevent the transport from the degraded proteins to the chaperones
By Us6, BNFL2a, ICP47 and UL49.5
all results in the protein not being loaded on MHC class 1
anitgen variation / african sleeping sickness
Caused by protozoa of the species Trypanosoma brucei and transmitted by tsetse fly
Endemic in some sub-Saharan contries
two different stages of disease
Haemolymphopatic phase → fever, headache, joints pains
Neurological phase → confusion, reduced coordination
Infection cycle → disease diagnosed by looking at the blood where you see the worm (microscopy) → does not hide because it has VSG
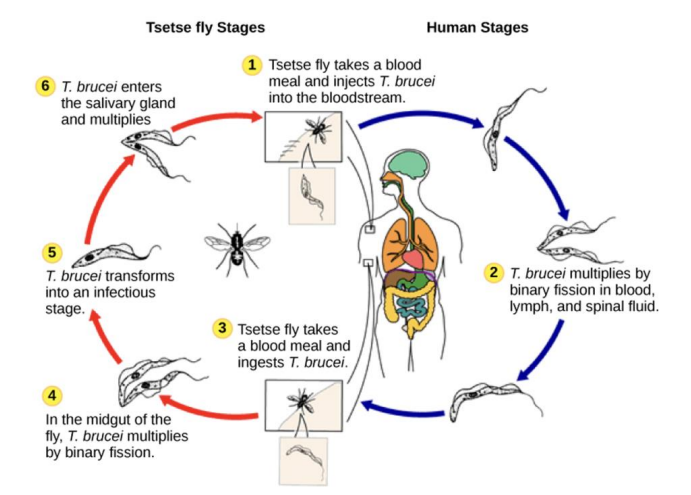
How the VSG coat helps Trypanosoma Brucei
VSG (variant surface glycoprotein) coat covers entire surface of parasite
highly variable surface glycoproteins
can be targeted by specific antibodies → pathogen can be cleared
The proteins switch, to avoid being seen → different waves of serotypes
VSG expression:
Pol I → transcribes active VSG gene
allelic exclusion: only one allel is expressed the other is silenced
telomere exchange → active VSG gene swapped with VSG gene on different chromosome (Gene not deleted!)
Gene conversion → silent VSG gene is copied and replaces the active VSG gene (Gene deleted!)
Red Queen Hypothesis, and question if the adaptive immune system is superior
evolutionary change gives a temporary advantage. but after adaptation of the pathogen the end result will be similar
Superiority of the adaptive immune system
Invertebrates can live very long without having an adaptive immune system → they get bigger than humans
Non-vertebrates have much more TLRs than humans → Their TLRs are much more specific than those of humans
drawback adaptive immune system
Molecular mimicry → antigens produced are very similar to humans → this causes autoimmunity
Innate immune response - Iron
pathogens need iron as nutrient
host ensures that the amount of available iron is extremely low → the pathogens can counteract this → red-queen hypothesis
interplay between host and pathogen - iron
Human body full of iron (in red blood cells) →
a-haemolysin kills red blood cells by binding to ADAM10 → this way the iron of the red blood cells is flushed out → the host produces proteins to stop this
The human body releases haemopexin (HPX) bind to free haem and haptoglobin (HP) binds to free haemoglobin → no longer available to pathogen
Free iron (Fe 3+) is bound by transferrin (more in the blood) or lactoferrin (more on the mucosal surface)
The macrophages keep the pathogens in the phagosome to stop them from getting iron → NRAMP1 (iron pump, pumps it out of phagosome)
Effect of iron overload
storage disease- hemochromatosis. These patients have more iron in the blood
more prone to have infections of opportunistic pathogens
bacterial response to iron withdrawal
Transferrin → binds free iron
Neisseria meningitidis, produces protein TbpB that captures transferrin, takes up iron from the host
Disadvantage for pathogen: Binding of Tf/Lf by TbpB is highly species specific (might bind human but not other vertebrates)
production of siderophore to capture iron
Siderophore is iron carrier (binds free iron) → the pathogen has a specific receptor so that the iron can be taken up again (against concentration gradient)
enterobactin and aerobactin
siderophores produced by E. coli
Enterobactin is strongest siderophore → removes iron bound to transferrin
aerobactin (pathogenic strain), binds iron less strong than enterobactin
Why would pathogenic strain have a less effective siderophore?
human cells produce lipocalin-2 (binds enterobactin, preventing E. coli from using it)
The purification of lipocalin-2 always resulted in a brown product → it was contaminated by enterobactin bound to iron
KO of lipocalin 2 → severe infection of E. coli cells in mice
Aerobactin is not bound by lipocalin 2 allowing e. coli to bypass this immune defense
→ host is expressing lipocalin, so enterobactin is then useless. So the bacteria has developed a new strategy to capture iron (aerobactin), that is not countered by the host yet
→ But tear lipocalin (in tear liquid) is binding a wide range of siderophores → the new counteract of the host
counteract of the body against lentiviruses
APOBEC3G (in non permissive cells) packaged in new virus cells, deanimates cytosine → converting it to uracil. viral DNA unreadable, no viable virus produced
Vif (HIV gene) binds to APOBEC3G flags ubiquitin, is degraded
Host pathogen interaction in mammalian evolution
genes related to host-pathogen interaction have changed
LCT → protein against digesting milk.
SLC45A2 → important in skin colour, less sunlight in the north
TLR genes
MHC genes
Autophagy regulation (ability to remove internal infection in a cell)