How do Cancers Begin/ How does air pollution promote lung cancer development?
1/44
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
45 Terms
Questions to ask for understanding cancer development
what are the pre-cancer events?
what drives the transition from normal to cancer?
what genomic events occur early vs late in tumor evolution?
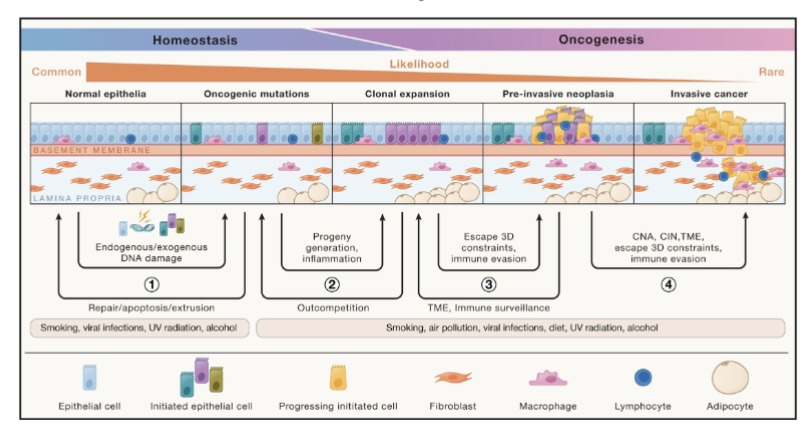
Early Studies on Clonal Expansion in (apparently) Normal Tissues
clonal expansion can occur in tissues that look normal under the microscope; somatic evolution begins way before cancer is clinically visible
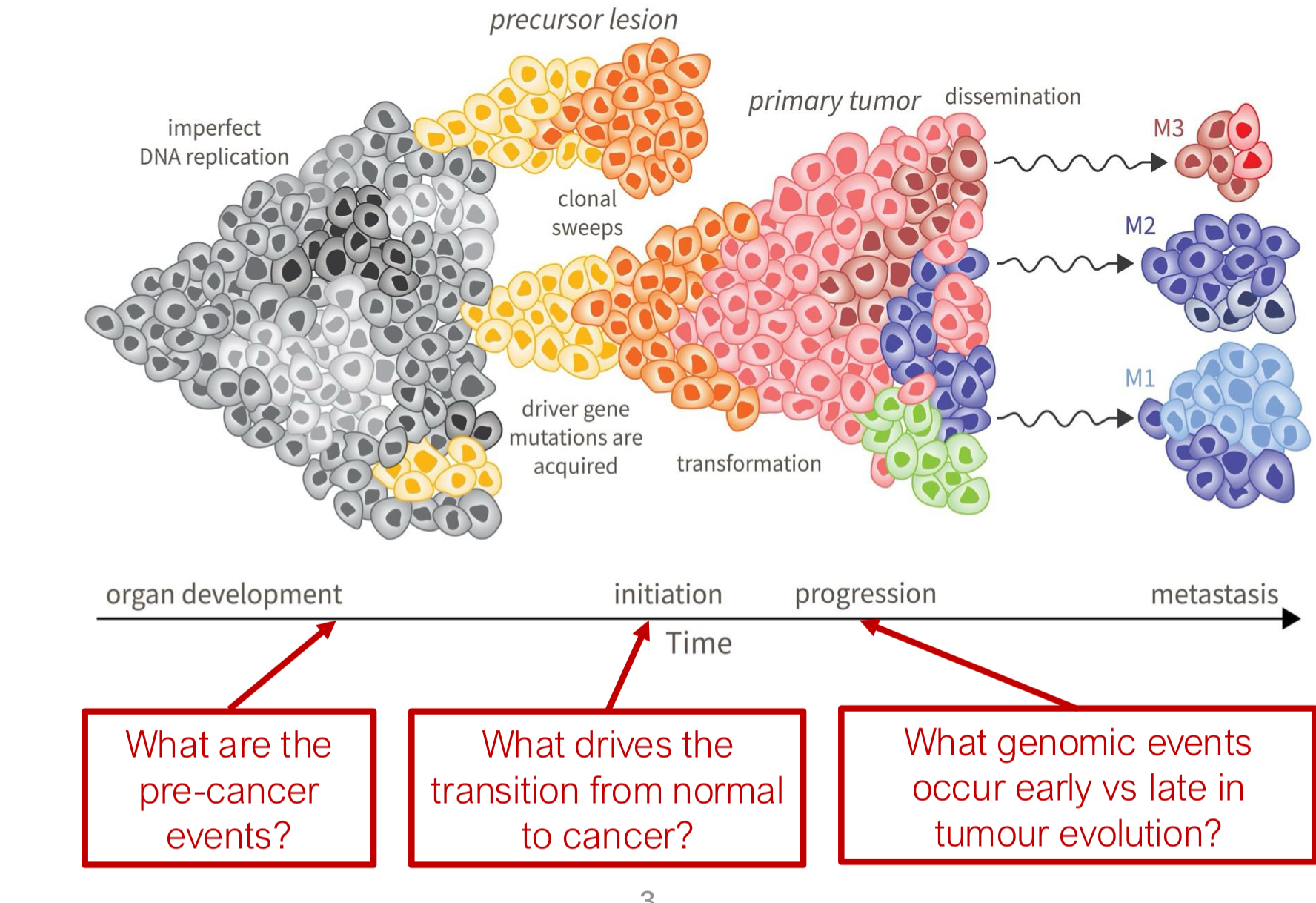
1986/1987: presence of skewed chromosome X inactivation (not 50:50), documented in healthy individuals with a normal blood count (particularly older people)
one X chromosome is randomly inactivated in each each cell early in development (supposed to happen); normally results in 50:50 mixture
50% of cells inactive maternal X, 50% inactive paternal X
considered if age was a factor
1994-2001: TP53 (tumor suppressor) in apparently normal epithelium
finding TP53 mutations in normal tissues suggested cancer-associated mutations before a tumor even forms
Early Studies on Clonal Expansion in (apparently) Normal Tissues FIGURE
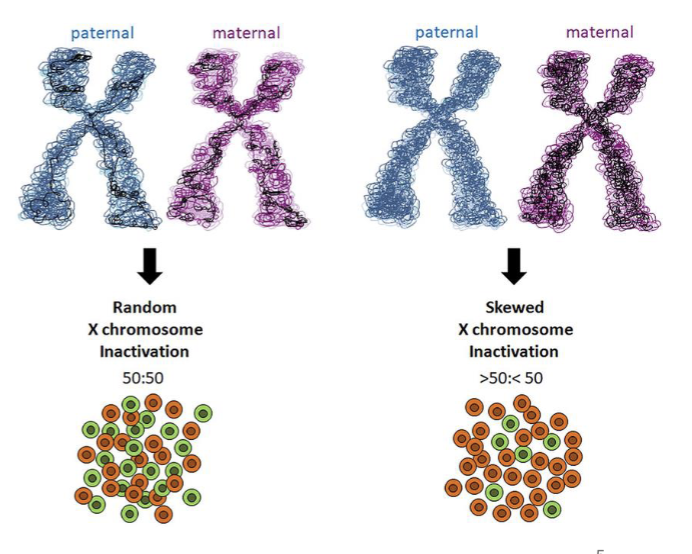
Clonal Expansion in normal tissues in various organs: Skin + Oesophagus
looking at pre-malignant state
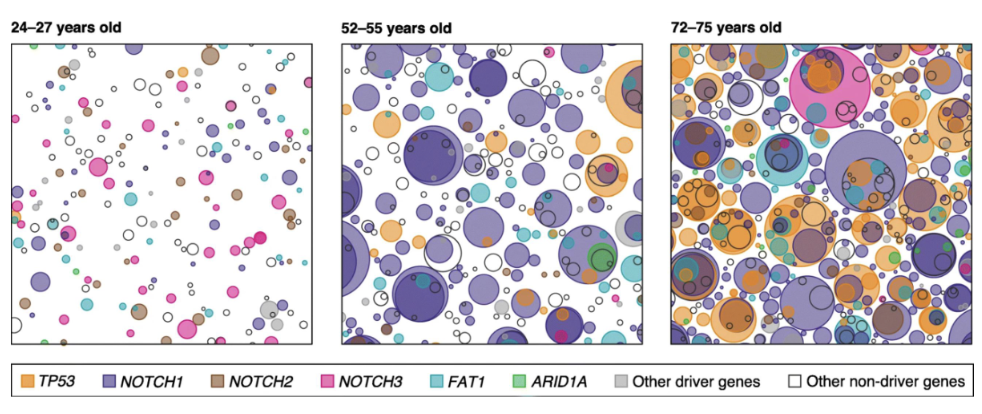
in the skin and oesophagus, clones carrying driver mutations (associated w/ oncogenicity) emerge as early as infancy and increase in their number and size with ageing
accelerated by exposure to UV light, alcohol and/or tobacco (lifestyle impact)
clones increase in number w/ age
mutations accumulate over time → some mutations give cells a growth advantage → cell divides more → its descendants form a clone → clone expands
Clonal Expansion in normal tissues in various organs: Skin + Oesophagus FIGURE
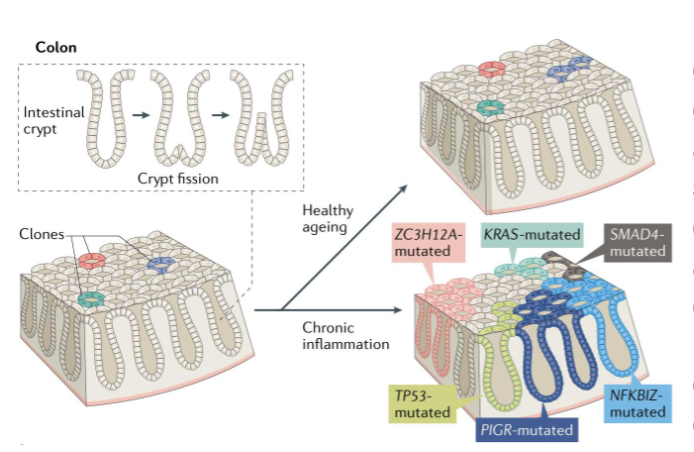
Clonal Expansion in normal tissues in various organs: Colorectal Epithelium
in the colorectal epithelium, clonal expansion occurs only occasionally throughout adult life
during the long-standing inflammation caused by ulcerative colitis, almost the entire colorectal epithelium is remodelled by numerous clones harboring driver mutations
i.e chronic inflammation alters microenvironment → more permissive for alterations to drive oncogenesis
a schema of crypt fission is also depicted (the colon is organized into structures called crypts)
crypts that develop mutations will split (fission) and now both daughter crypts carry the mutation
Clonal Expansion in normal tissues in various organs: Colorectal Epithelium FIGURE
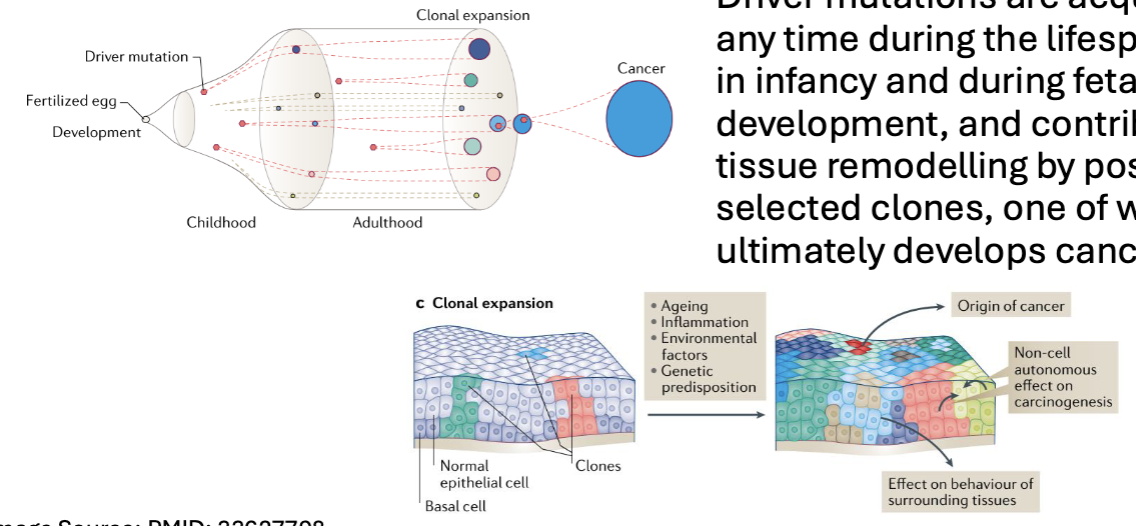
Positively Selected Clones in Normal TIssues
Cancer often arises from clones that were already expanding in normal tissue long before a tumor formed (already had growth advantage)
driver mutations are acquired at any time during the lifespan, even in infancy and during fetal development
driver mutations contribute to tissue remodelling by positively selected clones, one of which ultimately develops cancer
affected cells gain fitness advantage → divides more than neighbors → its descendants replace surrounding cells → normal tissue becomes a mosaic of competing clones
one of these clones may eventually acquire a mutation that leads to malignancy
driver mutations can continue to persist in tissues w/o being detected by the immune system
mutations that affect an essential gene in combination with factors* and microenvironment harbors oncogenesis
Positively Selected Clones in Normal Tissues
Histologically Normal Tissues Harbor Cancer-Associated Mutations
we accumulate mutations as we age
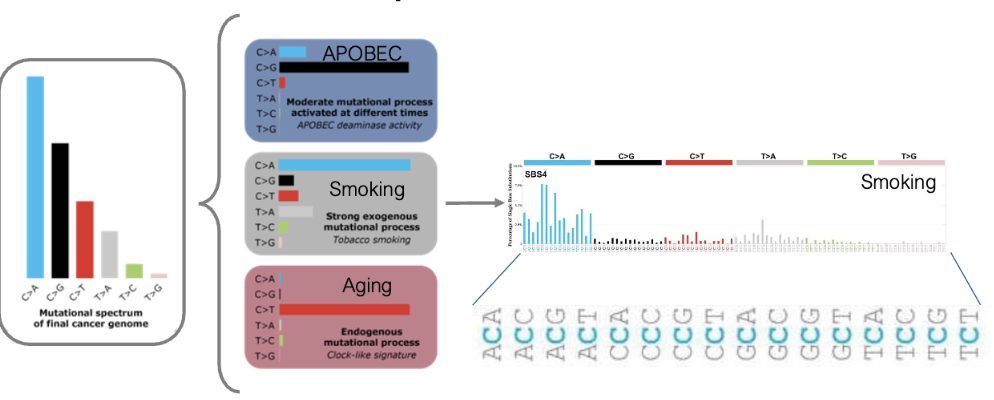
Mutational Signatures
mutational signatures provide clues as to how mutations were acquired (what damaged the DNA?)
mutational signatures: a count of types of mutations you see in a tumor
can reveal environmental exposures, inherited DNA repair defects, predict therapy response and show ongoing mutational processes
tri-nucleotide context is considered (i.e neighboring bases considered (upstream/downstream))
Mutational signatures decode the evolutionary history of a tumor by analyzing the pattern and context of mutations, revealing the biological processes that generated them
if a particular signature is highly represented in a tumor, that mutational process was very active in the tumor’s history
Mutational Signatures FIGURE
Mutational Signatures provide clues as to how mutation were acquired FIGURE
we see diff base changes that we can attribute to diff cancers
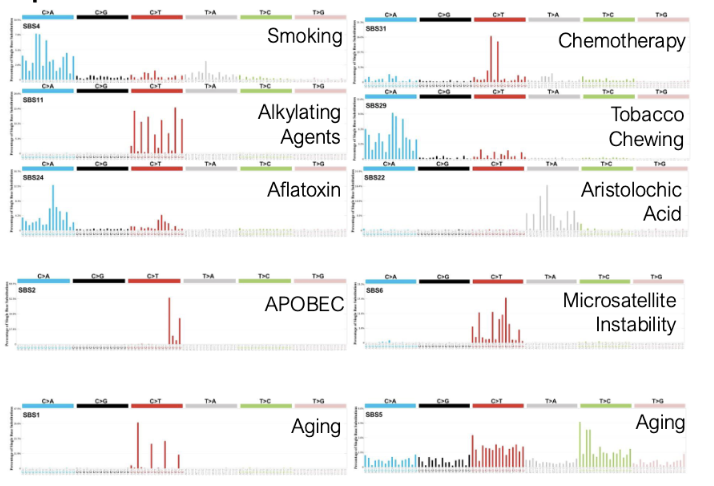
Mutational Processes → Mutational Signatures
mutational processes result in distinct mutational signatures
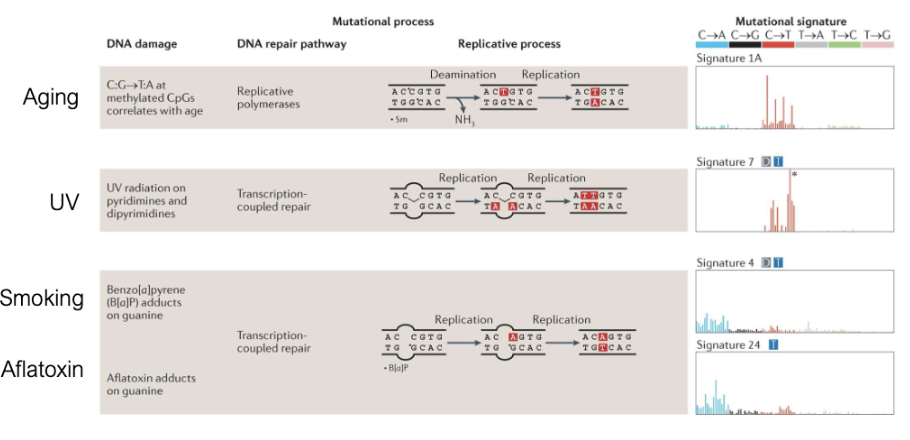
We Accumulate Mutations Throughout Life
mutations can come from diff sources
we assign mutation to diff mutation signatures
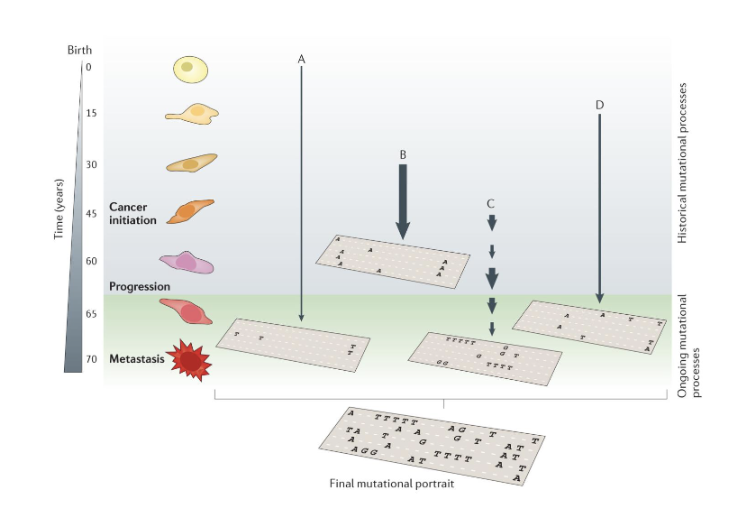
How do Cancers Begin?
The TRACERx 421 Cohort
large prospective study of 421 patients with early-stage non-small cell lung cancer
purpose:
To track tumour evolution over time using multi-region sequencing.
To study intratumour heterogeneity.
To distinguish clonal vs subclonal mutations.
To understand how heterogeneity impacts relapse, metastasis, and survival.
The TRACERx 421 Cohort: Tumor Phylogenies Figure

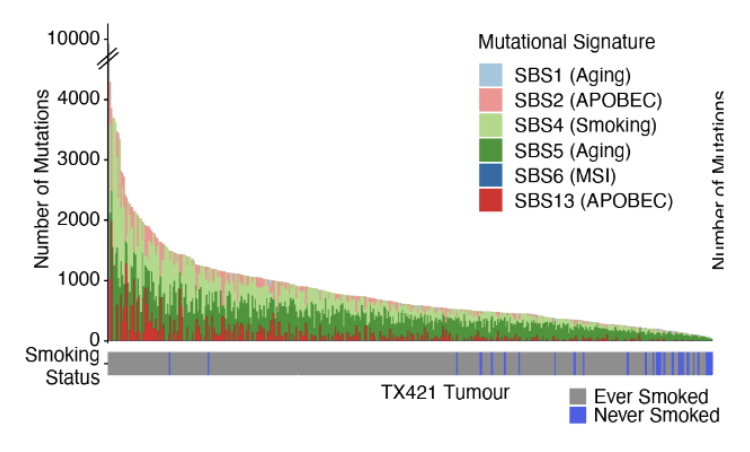
Smokers: SBS4 Mutational Signature
majority of smokers lung cancers harbor the SBS4 mutational signature
in the TRACERx421 cohort, majority of smokers’ lung cancers harbored SBS4
tumors from never smokers shows little to no SBS4 contribution
Smokers: SBS4 Mutational Signature FIGURE
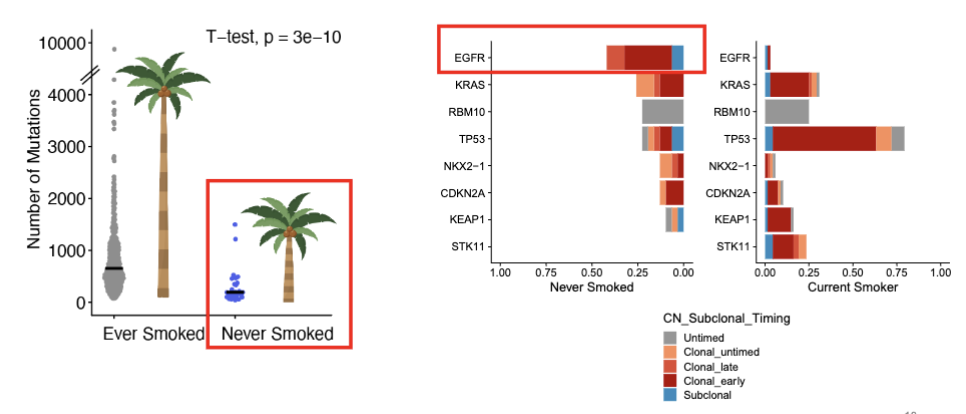
Lung Cancers in Never Smokers
lung cancers in never smokers have 3x fewer mutations and are frequently mutated in EGFR (epidermic growth factor receptor; always on to receive signal to divide and grow)
in the figure, we see a small trunk = less clonal mutations which led to initiation of cancer
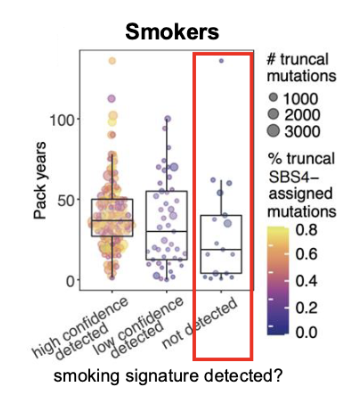
Problem: Not all smokers have the SBS4 mutation
7-8% of smokers with a long smoking history have no smoking mutations (SBS4) detected
smoker lung cancers (w/o smoking signature) are similar to never smoker lung cancers (the next figure)
question: how are these cancers (few mutations + EGFRm) initiated
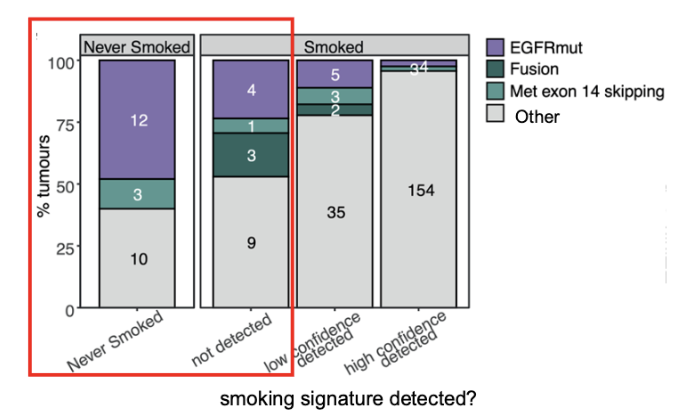
Problem: Not all smokers have the SBS4 mutation (2nd Figure)
Air Pollution
99% of people who live in places where air pollution levels exceed WHO Guidelines
the incidence of lung cancer in never smokers appears to associated with average PM2.5 levels
pm2.5: small particles that can deeply penetrate the lungs, enter bloodstream, cause inflammation, lead to respiratory issues
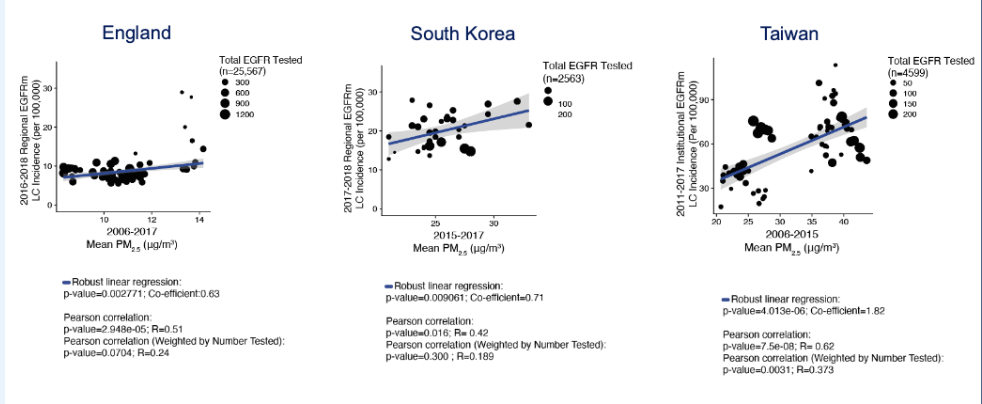
Is there a relationship b/w EGFRm lung cancer and PM2.5 exposure?
PM2.5 air pollution has become increasingly implicated in lung cancer
air pollution is associated w/ LCINS (lung cancer in never-smokers)
many never smoker lung cancers are EGFR mutant
epidemiological studies suggest that PM2.5 exposure may contribute to the development of EGFR-mutant lung tumors
therefore EGFRm lung cancer isn’t caused solely by random mutations or genetic predisposition; environmental exposures may act as mutagenics or inflammatory trigger
Is there a relationship b/w EGFRm lung cancer and PM2.5 exposure? FIGURE
Does Pollution Trigger Tumor Iniitation of Mutant Clones? Inconvenient Truths
TRACERx: 7-8% of smokers with a long smoking history have no smoking mutations detected
BRAF V600E in melanoma is not a UV light induced mutation
Normal healthy tissue harbors mutant clones with a cancer driver mutations w/ no evidence of cancer
17/20 Environmental Carcinogens tested in mice, without directly causing DNA mutations
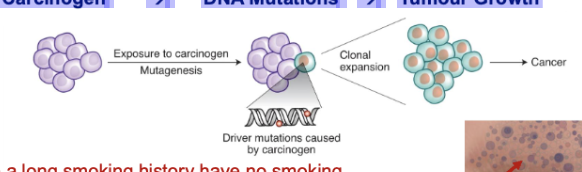
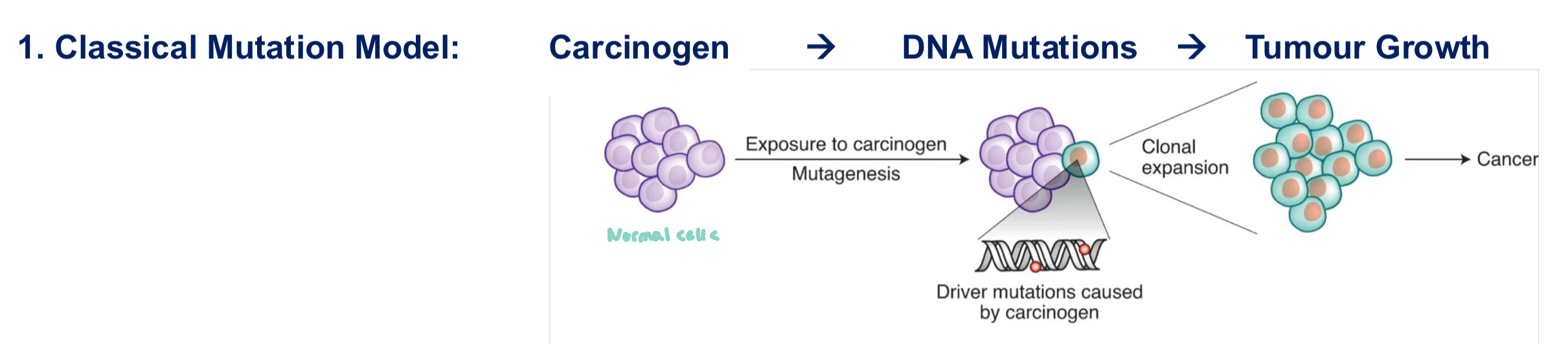
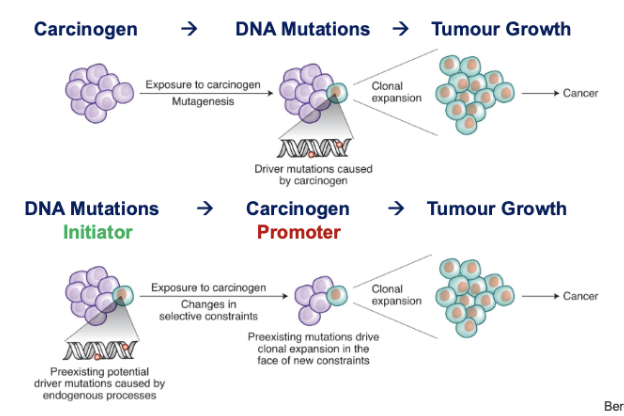
Classical Mutation Model vs Tumor Promotion Model
Tumor Promotion Model:
cells that harbor pre-existing mutations (acquired somatically, random mutations)
carcinogen provides a condusive microenvironment to promote pre-exsting mutations to initiate (instead of inducing more mutations)
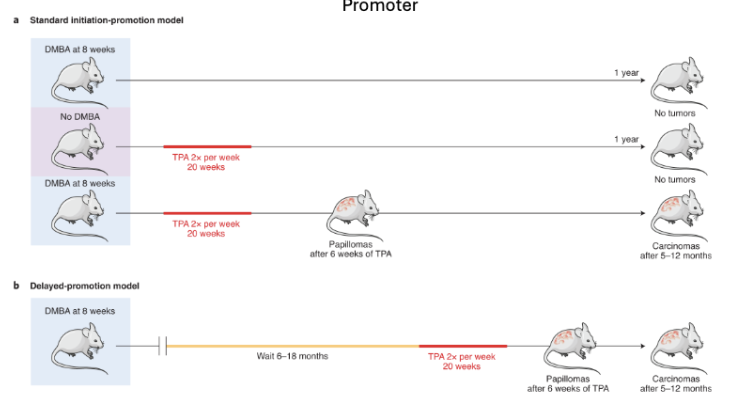
Tumor Promotion Mice Experiment
DMBA: carcinogen known to induce mutations
TPA: tumor promoter (induces conducive environment)
Standard Initiation-Promotion Model: you need both mutation (initiation) and promotion close together to get tumors (mutation or promotion alone is not enough)
Delayed Promotion Model: Initiated (mutated) cells can remain dormant for months to years; only need a later promoting event to expand into tumors
Tumor Promotion Mice Experiment FIGURE
Air Pollution (PM) in Mouse Models
air pollution (PM) promotes cancer in mouse models w/ pre-existing mutations
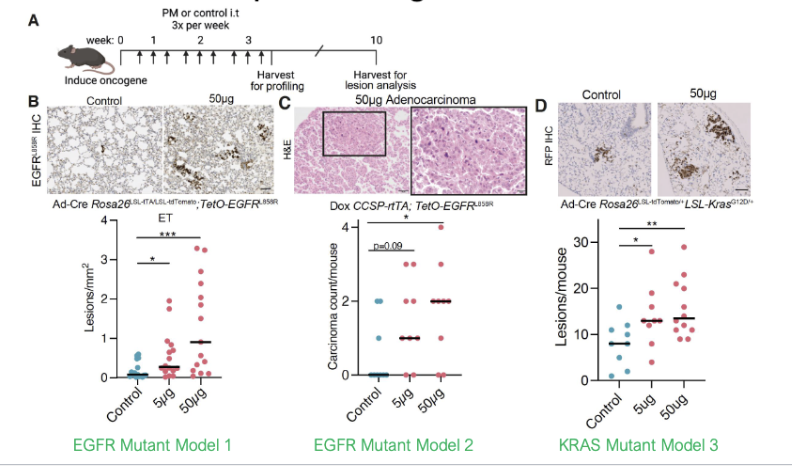
Does PM2.5 drive an environmental carcinogen signature and DNA mutagenesis?
mutation profiles obtained from WGS of mouse tumours look the same (no significant diffs)
tumour mutation counts do not significantly differ b/w control and pollution exposed mice
air pollutants accelerate age mutations (could be considered a promotional mutation)
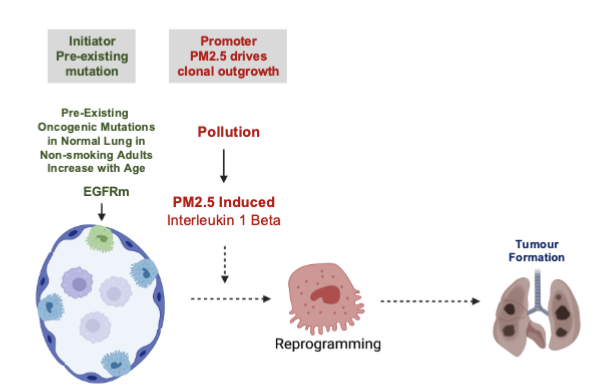
How does Pollution Initiate Cancer without Directly Causing DNA Mutations
RNA seq of mouse tumors: tdTOM (control) and EGFR-L858R (oncogenic driver mice)
exposed to PBS control (TC) or air pollution (EA)
testing if air pollution changes gene expression or interact w/ the EGFR mutation
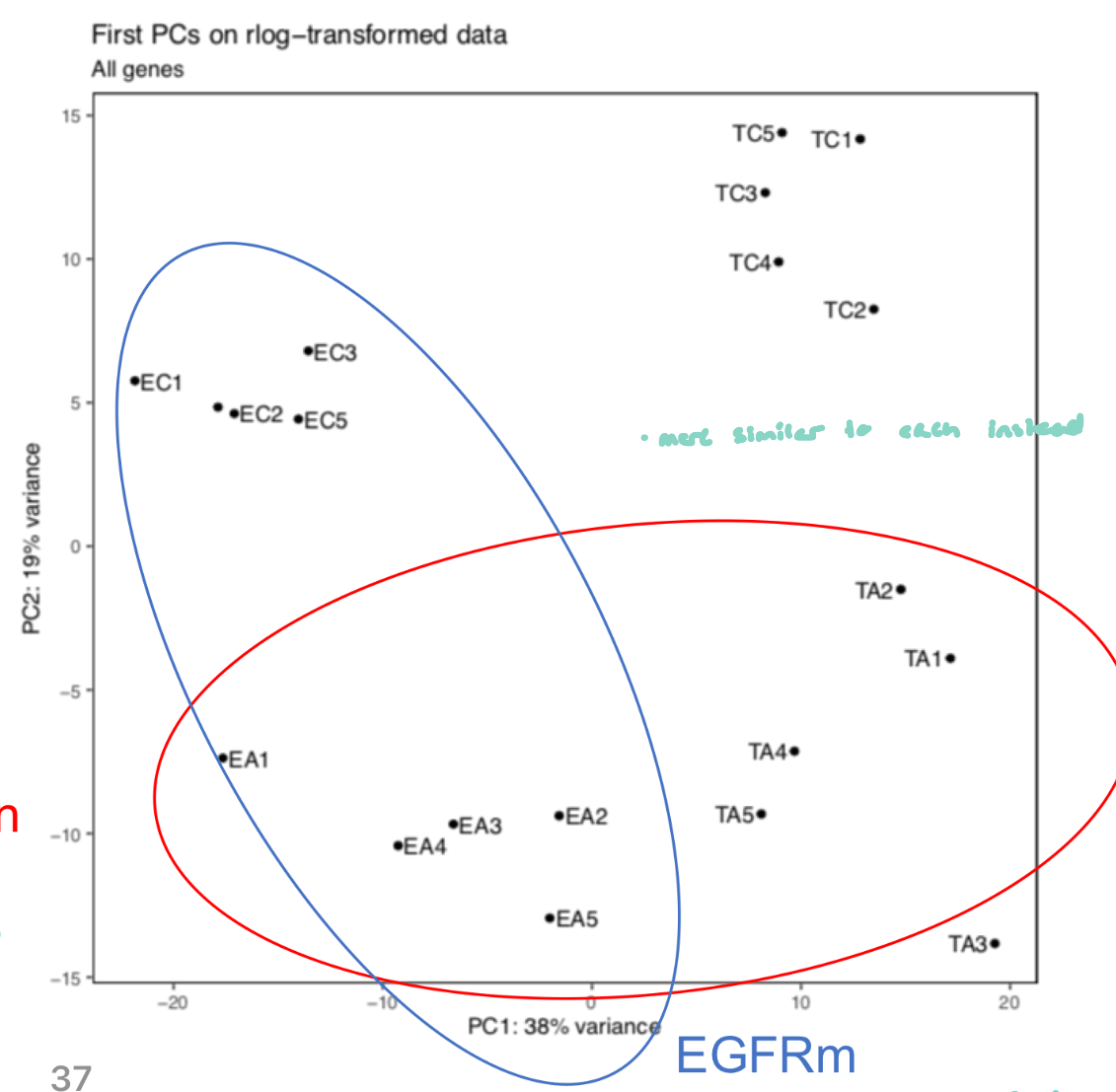
Principal Components Analysis (PCA)
reduces thousands of gene expression variables into a few major axes that capture the biggest differences between samples
X-axis = PC1 and Y-axis =PC2; each dot is one sample (one mouse tumor)
PC=principal component
PC1: Captures the largest source of variation in the dataset
PC2: Captures the second largest source of variation
samples that cluster together are similar in gene expression, share many characteristics
samples that are far apart are transcriptionally diff
value of PCA is not important, it's how its grouped
PCA: RNA Seq of Mouse Tumors
PC1: 38% variance EGFRm drives variance
PC2: 19% variance Air pollution drives variance
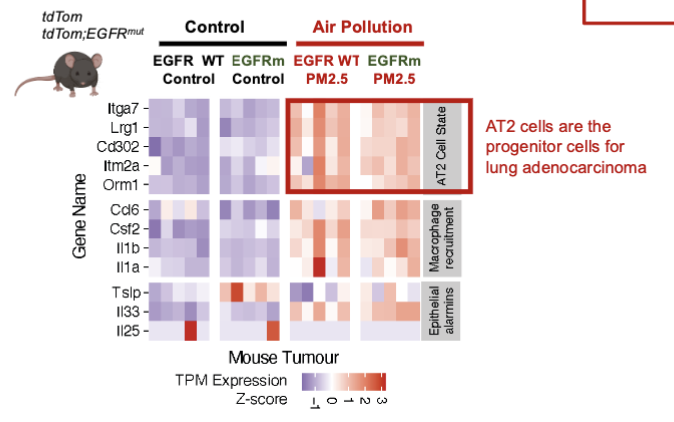
Pollution induces an AT2 transcriptional Signature
air pollution induces an AT2 transcriptional signature in lung tumors
causes changes in the microenvironment that promotes progenitor-cells for lung tumors
air pollution also affects immune cells (macrophages), not just AT2 cells
can lead to suppressing anti-tumor responses and promoting AT2 cell proliferation
RNA-seq of tumors revealed dysregulated pathways
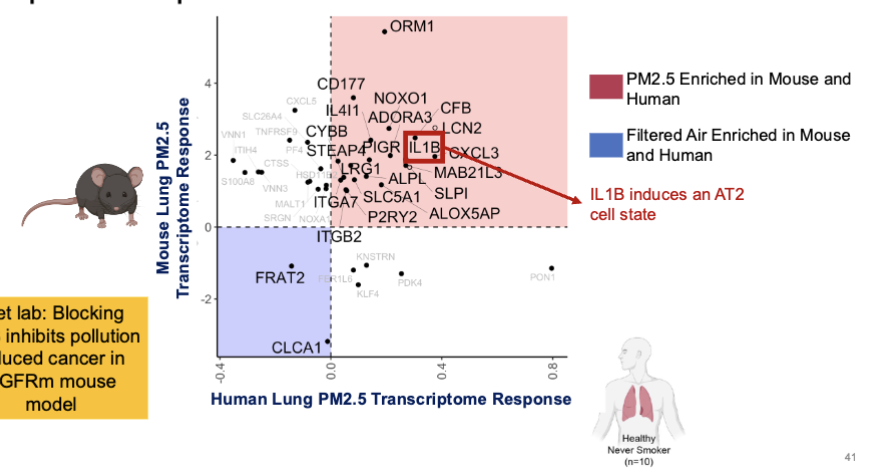
Exploring the effects of pollution exposure on human lungs
PM2.5 exposure levels equivalent to those frequently encountered in major Asian cities
Bronchial brushing taken from healthy individuals 24 hrs after exposure to 300µg/m3 for 2 hours
compared to brushings from same individual after filtered air
IL1B
IL1B is upregulated in both human and mouse pollution exposure experiments
blocking IL1B inhibits pollution induces cnacer in EGFRm mouse model
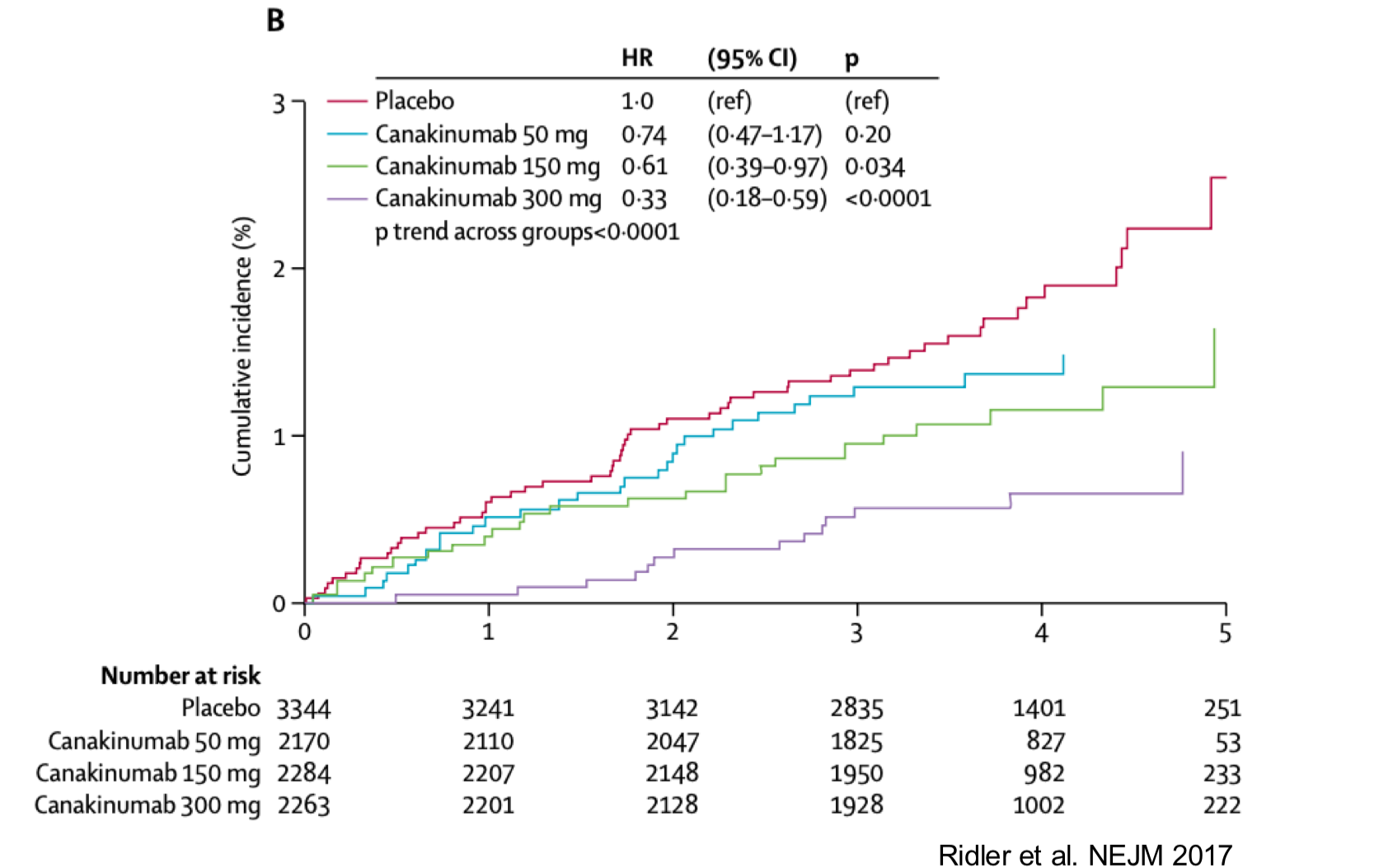
Clinical Trial of Canakinumab (anti-IL1B drug)
Clinical Trial of Canakinumab (anti-IL1B drug) reduced lung cancer incidence
suggests IL1b/air pollution-induced tumor growth may be prevented
Do EGFR and KRAS mutations exist in normal tissue?
Yes, these mutated cells can exist for years and be silent and don’t always form tumors
latent = present but inactive
cells w/ a driver mutation, not yet cancerous or expanding agressively, being kept in check by the environment
A tumor promoter must act on latent cells to become cancerous
tumor promoter: doesn’t necessarily cause new DNA mutations, but stimulates proliferation or survival of existing mutated cells
Latent Cells and Tumor Promoters Figure
Anthracosis
the asymptomatic, milder type of pneumoconiosis as caused by the accumulation of carbon in the lungs due the repeated exposure to air pollution or inhalation of smoke or coast dust particles
carbon deposits (anthracotic pigment)
EFRm clonal expansions are associated w/ anthracosis
anthracotic pigment (associated w/ pollution) is not associated w/ presence of EGFRm in normal tissue (acquiring the mutation), but the VAF of those mutations (expansions of those clones)
VAF = varient allele frequency, the proportion of DNA sequencing reads that carry a specific mutation at a given position
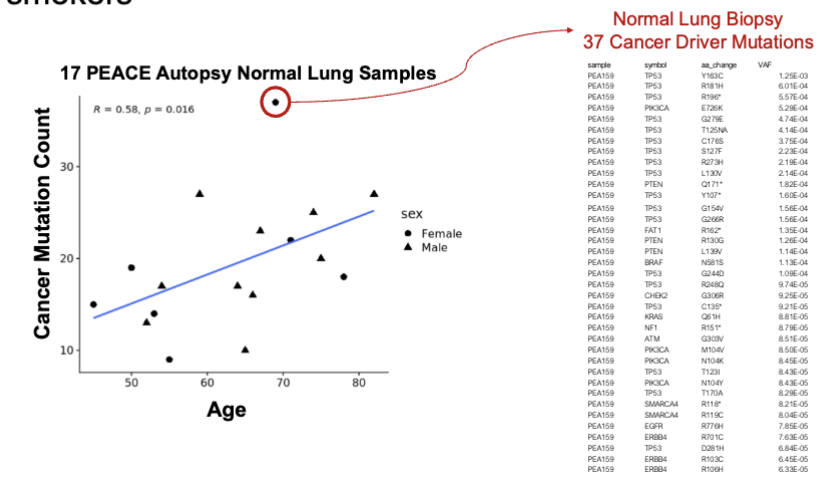
Mutations w/ Age in Non Smokers
mutations increase w/ age in normal lungs from never smokers
Lung Cancer Promotion by Air Pollutants Figure
A Vision towards cancer prevention