Ch 16 - Conjugated Pi Systems and Pericyclic Reactions
1/48
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
49 Terms
diene
a compound possessing two C=C bonds
cumulated diene (allene)
diene in which the pi bonds are adjacent

conjugated diene
diene in which the pi bonds are separated by exactly 1 sigma bond
note: C=C pi bonds can also be conjugated with other types of pi bonds, e.g. C=O
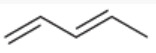
isolated diene
diene in which the pi bonds are separated by 2 or more sigma bonds
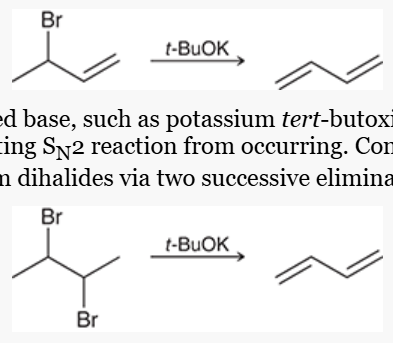
how to prepare a conjugated diene
from allylic halide via elimination
from alkyl dihalide via 2 successive elimination reactions with a bulky base
single bond length in a conjugated diene
shorter than a typical C-C bond (e.g. in butadiene, C-C length = 148pm compared to 153 pm)
because C-C in conjugated diene is formed from overlap of 2 sp2-hybridized orbitals while regular C-C is formed from overlap of sp3-hybridized orbitals (more s character)
stability of conjugated dienes
more stable than isolated dienes
stabilization energy associated with conjugated dienes = 15 kJ/mol
s-cis conformer
conformer in which the disposition of the 2 pi bonds (in reference to the connecting single bond) is cis-like (dihedral angle = 0*)
p orbitals effectively overlap to give a continuous, conjugated pi system
(picture shows this conformer of 1,3-butadiene)
significantly higher in energy than the s-trans conformer → only 2% of the molecules assume this conformation
p orbitals of the s-cis conformer of 1,3-butadiene
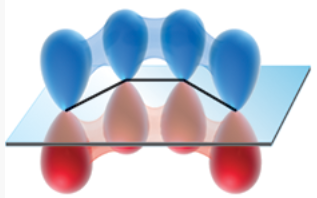
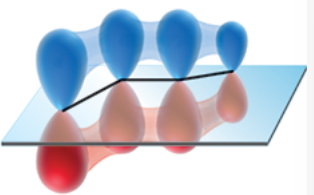
s-trans conformer
conformer in which the disposition of the two pi bonds (with regard to the connecting single bond) is trans-like (dihedral angle of 180*)
p orbitals effectively overlap to give a continuous, conjugated pi system
(picture shows this conformer of 1,3-butadiene)
significantly lower in energy than the s-cis conformer → ~98% of the molecules assume this conformation
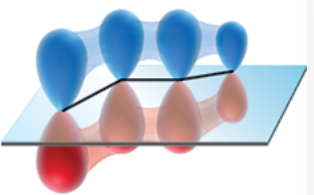
p orbitals of s-trans conformer of 1,3-butadiene
molecular orbitals
result when overlapping p orbitals combine
electrons occupying one of thee are associated with the entire molecule (unlike atomic orbitals, in which the electrons are associated with just 1 atom)
can be bonding (lower in energy), antibonding (higher in energy), or nonbonding
HOMO
highest occupied molecular orbital
highest energy bonding MO
a frontier orbital
contains highest energy pi electrons, which are most readily available to participate in a reaction
LUMO
lowest unoccupied molecular orbital
lowest energy antibonding MO
a frontier orbital
contains no electrons, but is the lowest energy MO capable of accepting electron density
excitation
when a compound absorbs energy
causes a change in the identities of the frontier orbitals
electrons can be promoted from HOMO to LUMO
electrophilic addition of HX across 1,3-butadiene
diene reacts with 1 equivalent of HBr to produce 2 alkyl halides
mechanism: protonation (forms a carbocation), followed by nucleophilic attack
note: protonation creates the more stable, resonance-stabilized, allylic carbocation instead of primary carbocation
products are products of 1,2-addition and 1,4-addition
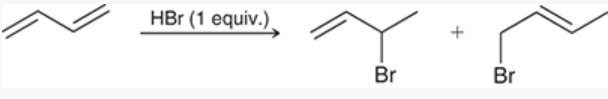
1,2-addition
reaction involving the addition of 2 groups to a conjuted pi system in which 1 group is installed at C1 and the other at C2
e.g. 2-bromo-3-butene (starting from 1,3-butadiene), H and Br are installed to C1 and C2
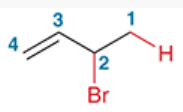
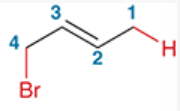
1,4-addition
reaction involving the addition of 2 groups to a conjugated pi system in which one group is installed at C1 and the other at C4
e.g. 4-bromo-2-butene (starting from 1,3-butadiene), H and Br are installed at C1 and C4
addition of Br2 across 1,3-butadiene
1,3-butadiene reacts with 1 equivalent of Br
forms a 1,2-adduct and a 1,4-adduct
ratio of products is temperature-dependent (71% 1,2-adduct and 29% 1,4-adduct at 0*C, 15% 1,2-adduct and 85% 1,4-adduct at 40*C)
1,2-adduct forms more rapidly because of a proximity effect → at lower temperatures, there is insufficient energy for it to be converted back into allylic carbocation; reaction happens under kinetic control
1,4-adduct happens under thermodynamic control, because it is lower in energy → predominates at equilibrium
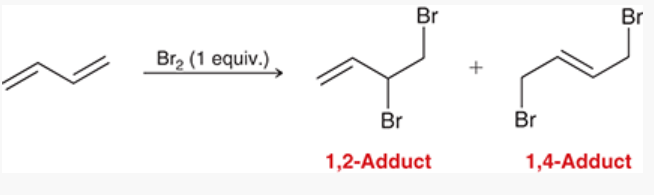
pericyclic reactions
concerted reactions that do not involve either ionic or radical intermediates
3 groups: cycloaddition rxns, electrocyclic rxns, and sigmatropic rearrangements
characteristics:
reaction proceeds via a concerted process (no intermediates)
reaction involves a ring of electrons moving around in a closed loop
reaction occurs through a cyclic transition state
polarity of solvent generally does not have a large impact on the rate/yield of the reaction (suggests that transition states bears very little/no partial charge)
cycloaddition reactions
a group of pericyclic reactions
2 pi systems join together to form a ring
2 pi bonds are converted into 2 sigma bonds
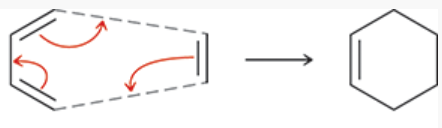
electrocyclic reactions
a group of pericyclic reactions
a conjugated polyene undergoes cyclization
1 pi bond is converted into a sigma bond
remaining pi bonds change location
the new sigma bond joins the ends of the original pi system → creates ring
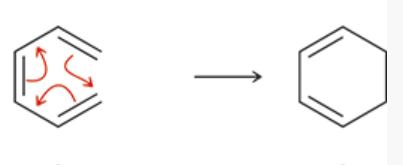
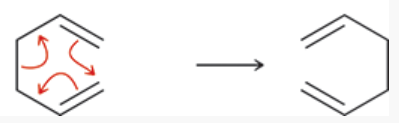
sigmatropic rearrangement
a group of pericyclic reactions
1 sigma bond is formed at the expense of another
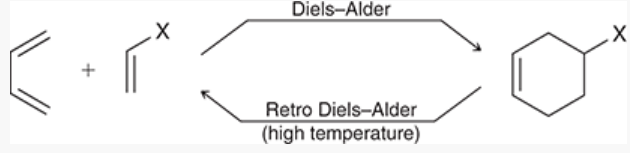
Diels-Alder reaction
a [4+2] cycloaddition
1 pi system involves 4 atoms, the other involves 2 atoms
product: substituted cyclohexene
arrows can be drawn clockwise or counterclockwise
only occurs when the diene is in s-cis conformation (when diene is in s-trans, the ends of the diene are too far apart to react with the dienophile)
energy diagram has just 1 peak, representing transition state
![<p>a [4+2] cycloaddition</p><p>1 pi system involves 4 atoms, the other involves 2 atoms</p><p>product: substituted cyclohexene</p><p>arrows can be drawn clockwise or counterclockwise</p><p>only occurs when the diene is in s-cis conformation (when diene is in s-trans, the ends of the diene are too far apart to react with the dienophile)</p><p>energy diagram has just 1 peak, representing transition state</p>](https://knowt-user-attachments.s3.amazonaws.com/6bb8f145-9c72-4632-b564-d1aeba0dbee9.png)
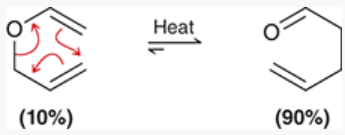
retro Diels-Alder reaction
reaction performed at high temperature to reverse a Diels-Alder
substituted cyclohexene breaks into 2 pi systems
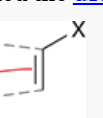
dienophile
compound that reacts with a diene in a Diels-Alder reaction
Diels-Alder reaction proceeds much more rapidly and with higher yield if this group has an electron-withdrawing substituent (e.g. carbonyl, nitrile, ester, carboxylic acid)
if this compound is a 1,2-disubstituted alkene, the reaction proceeds stereospecifically (cis alkene produces cis disubstituted ring, trans alkene produces trans disubstituted ring)
can also be triple bonds
endo positions
positions that are syn to the larger bridge in a Diels-Alder reaction that produces a bicyclic structure
generally highly favored (often is the exclusive product) because favorable interaction exists between electron-withdrawing substituents and the developing pi bond
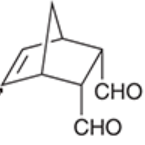
exo positions
positions that are anti to the larger bridge in a Diels-Alder reaction that produces a bicyclic structure
generally not favored (many times not formed at all) because the electron-withdrawing substituents and developing pi bond are too far apart
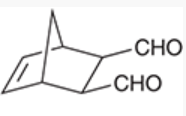
regioselectivity of Diels-Alder reactions
if either diene or dienophile is symmetrical, only 1 possible regiochemical outcome
if both are unsymmetrical, there are 2 possible regiochemical outcomes
to predict major product, consider charge distribution in both diene and dienophile. Draw resonance structures and find electron-rich or poor areas
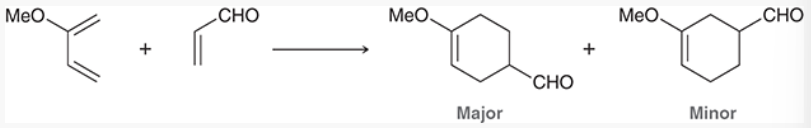
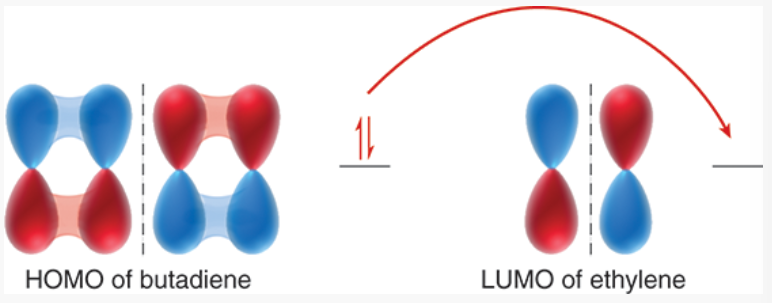
MO description of cycloadditions
Diels-Alder happens when electron density is transferred from the HOMO of the diene to the LUMO of the dienophile
because the dienophile usually has an electron-withdrawing substituent, it is treated as electron-poor → empty orbital that accepts electron density
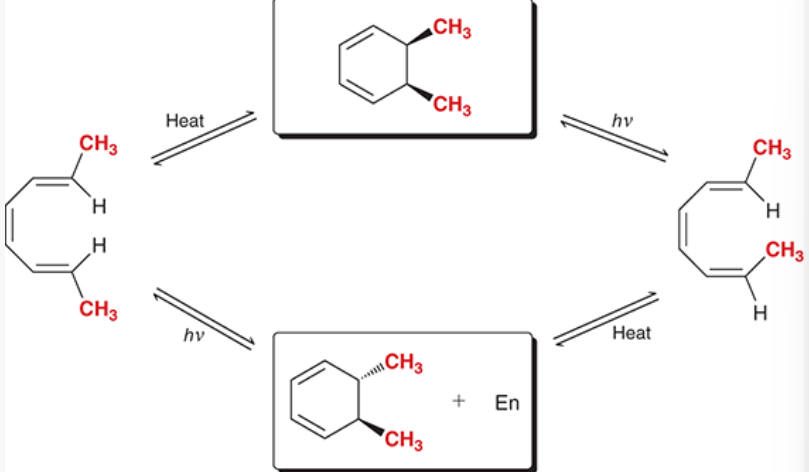
converting between stereochemical outcomes of an electrocyclic reaction for a 6-membered ring
start with non-ring pi system with up-down substituents (see picture) → add heat → cis disubstituted cyclohexene → add light → open pi system with up-up substituents
start with open pi system with up-up substituents → add heat → trans disubstituted cyclohexene → add light → open pi system with up-down substituents
both pathways are reversible
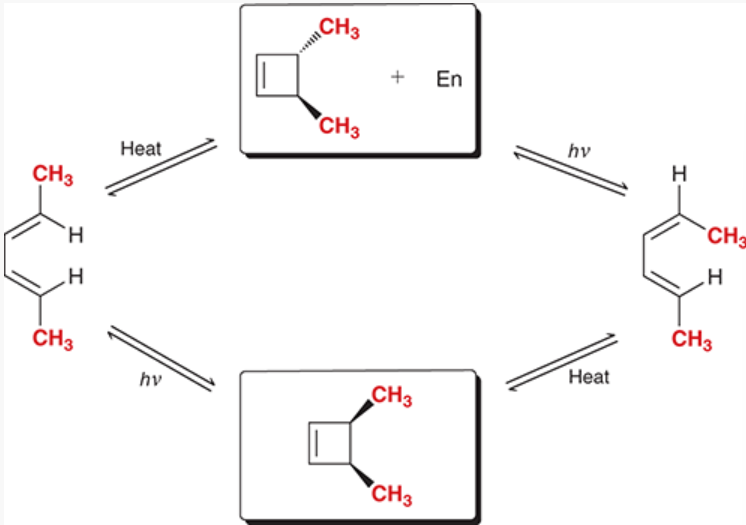
converting between different stereochemical outcomes of an electrocyclic reaction for a 4-membered ring
start with open pi system with up-down substituents → add heat → trans disubstituted cyclobutene → add light → open pi system with down-down substituents
start with open pi system with down-down substituents → add heat → cis disubstituted cyclobutene → add light → open pi system with up-down substituents
disrotary
rotation in which one set of lobes rotates clockwise, the other rotates counterclockwise
lobes that interact with each other must exhibit the same sign in order to form a new sigma bond
groups whose lobes rotate this way will have cis relation in the product
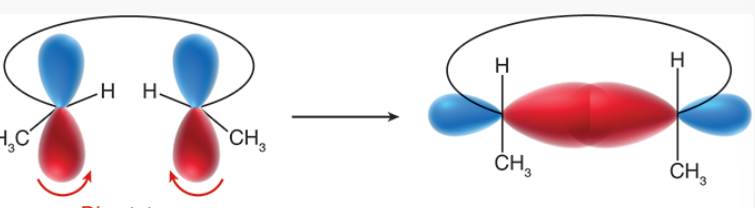
conrotary
type of rotation in which both sets of lobes rotate the same way (clockwise or counterclockwise)
lobes that interact with each other have opposite signs
groups whose lobes rotate this way will have trans relation in the product
rotation of pi systems under different conditions
under photochemical conditions:
system with 6 pi electrons will undergo conrotary ring closure
system with 4 pi electrons will undergo disrotary ring closure
under thermal conditions:
system with 6 pi electrons will undergo disrotary ring closure
system with 4 pi electrons will undergo conrotary ring closure
notation for a sigmatropic rearrangement
numbers in brackets indicate the number of atoms separating the bond forming and bond breaking (two numbers for two pathways)
e.g. [3,3] or [1,5] sigmatropic rearrangement
(picture is of [3,3] rearrangement)
![<p>numbers in brackets indicate the number of atoms separating the bond forming and bond breaking (two numbers for two pathways)</p><p>e.g. [3,3] or [1,5] sigmatropic rearrangement</p><p>(picture is of [3,3] rearrangement)</p>](https://knowt-user-attachments.s3.amazonaws.com/3e2507fa-e17d-46bf-8fbf-c78bc6763fd8.png)
Cope rearrangement
a [3,3] sigmatropic rearrangement when all 6 atoms of the cyclic transition state are Cs
equilibrium favors formation of the more substituted alkene
![<p>a [3,3] sigmatropic rearrangement when all 6 atoms of the cyclic transition state are Cs</p><p>equilibrium favors formation of the more substituted alkene</p>](https://knowt-user-attachments.s3.amazonaws.com/784a959e-6148-4175-83fd-6b81db4adf95.png)
Claisen rearrangement
the oxygen analogue of a Cope rearrangement
commonly observed for allylic vinylic ethers and for allylic aryl ethers
equilibrium greatly favors product because of formation of a C=O, which is thermodynamically more stable than C=C
absorbance
the value log(I0/I) where I0 = intensity of the reference beam and I = intensity of the sample beam
plotted as a function of wavelength on a UV-vis absorption spectrum
lambda max
wavelength for maximum absorption
described by molar absorptivity, expressed by Beer-Lambert law
increases with more conjugation → indicates extend of conjugation present in a compound
effect of conjugated double bonds on lambda max
each additional conjugated double bond adds 30-40 nm
Beer-Lambert law
A=ϵCl
A = absorbance
ϵ = molar absorptivity
C = concentration of solution (mol/L)
l = sample path length (width of cuvette) (cm)

lambda max of a simple diene
217 nm
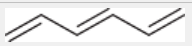
lambda max of a simple triene
258 nm
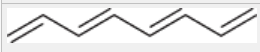
lambda max of a simple tetraene
290 nm
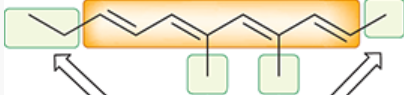
chromophore
region of a molecule responsible for the absorption of UV-vis light
(the conjugated pi system)
depicted in orange
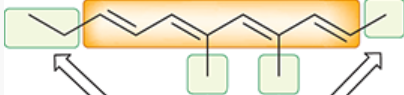
auxochromes
groups attached to the chromophore in a molecule
depicted in green
can have substantial effect on the value of lambda max
Woodward-Fieser rules
can be used to make simple predictions about lambda max
rules:
base value of conjugated diene = 217
each additional bond: +30
each auxochromic alkyl group+ +5
each exocyclic double bond: +5
homoannular diene: +39
how compounds appear colored
highly conjugated pi systems → possible for lambda max to be above 400 nm
compound will absorb visible light instead of UV → will be colored
e.g. lycopene, beta-carotene
a compound will be colored if it absorbs a specific color more strongly than its complementary color