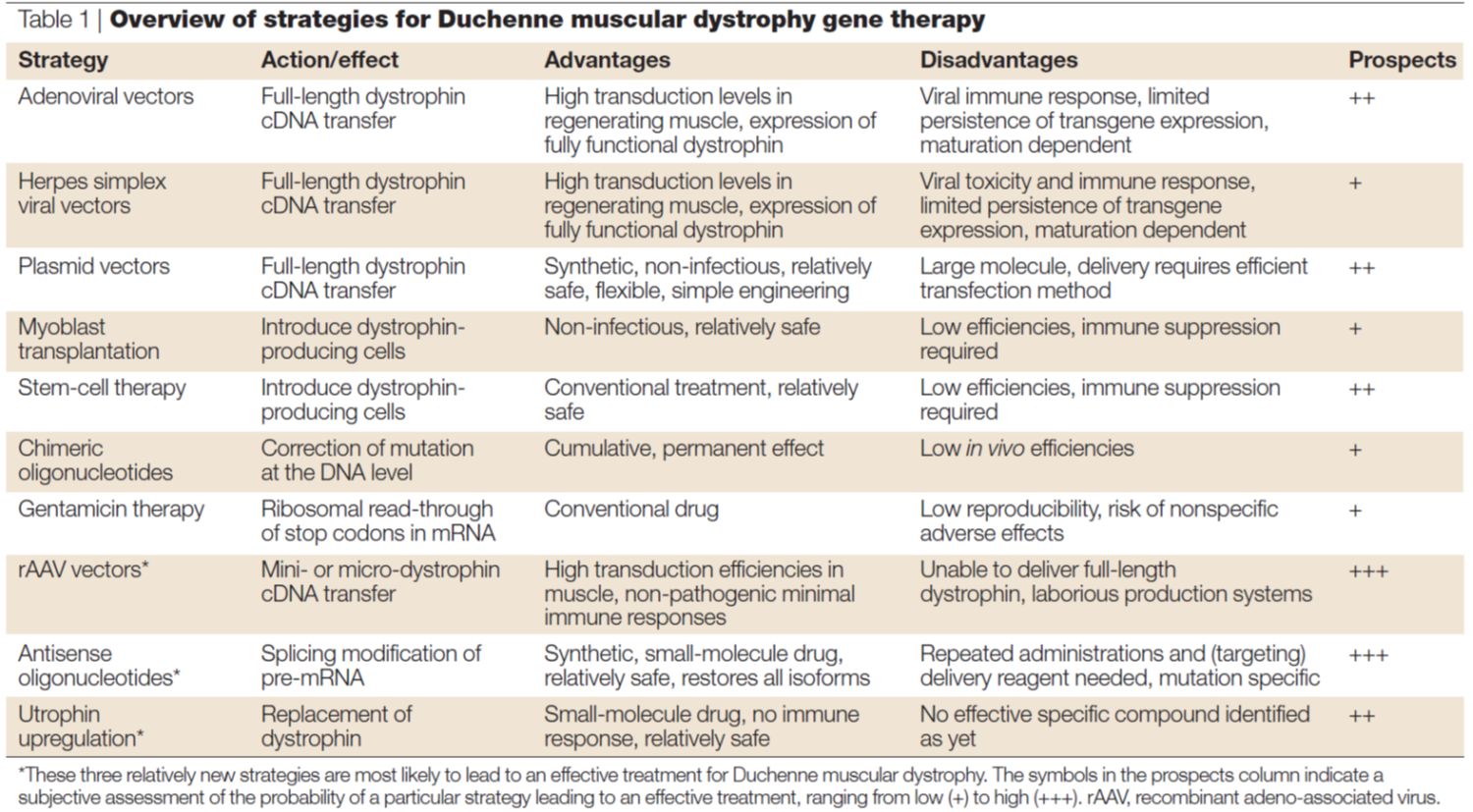
Gene therapy successes
1/40
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
41 Terms
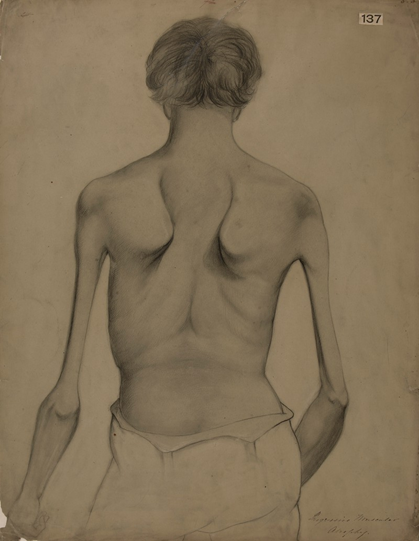
Duchenne muscular dystrophy
•X-linked recessive disorder associated with severe progressive muscle weakness.
•Degeneration and regeneration of skeletal and cardiac muscles
•Associated with severe progressive muscle weakness
•Affected boys cannot walk by 10 years of age
•Life expectancy is around 20 to 35 years, death due to heart or respiratory failures. As muscle degenerates it is replaced by stem cells and this keeps on happening until the stem cells run out.
•Due to mutations in dystrophin gene
Dystrophin
•The dystrophin gene is one of the longest human genes identified – 2.4 million base pairs with 79 exons
•Intronic sequences accounts for 99.4% of the gene
•8 tissue-specific promoters that lead to different spliced forms.
•Codes for linear cytoplasmic protein that supports muscle fibres - allows muscle fibres to line up together during movement.
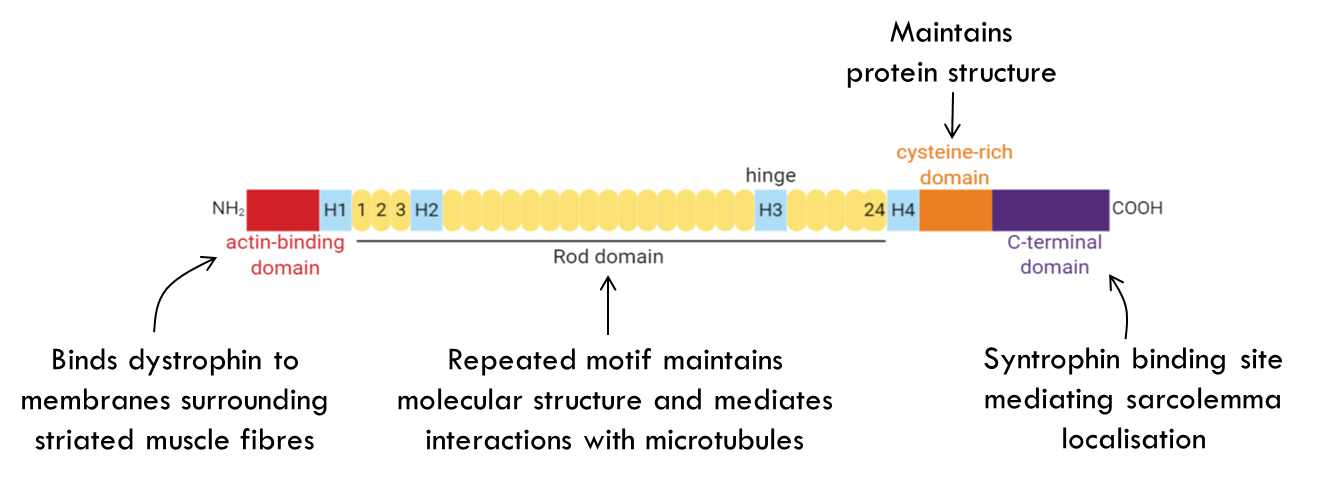
Starting from the left you have the actin binding domain which binds to actin filaments that surround the striated muscles. On the C terminal domain we have a functional domain that binds to syntrophin which localises it to the sarcolemma.
In the middle you have repeated rod domains which maintains molecular structure and also mediates cell interactions with microtubules in the cell.
Dystrophin function
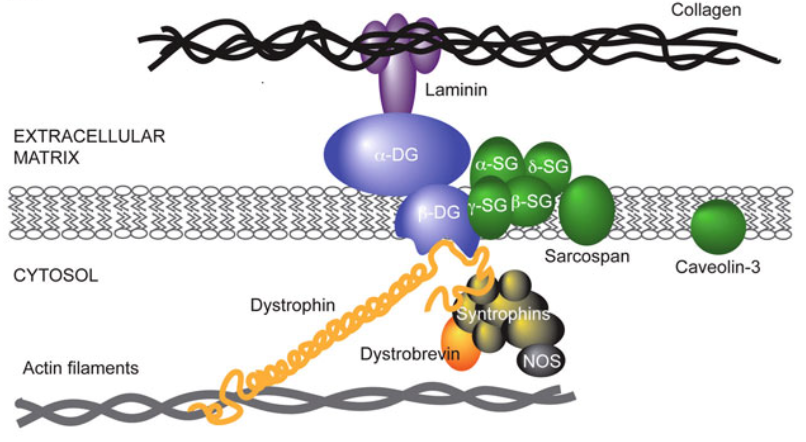
•
•Major protein in the dystrophin-associated protein complex (DAPC)
Attached to actin in the muscle and DAPC in cell membrane.
•The DAPC connects cytoskeleton of muscle fibres to extracellular matrix through the cell membrane
•Important for muscle fibre strength and flexibility
Without functional dystrophin you have muscle cells degenerating as they are stretched and damaged with use.
Mutations in dystrophin
DMD is recessive so you need both copies of the gene to be mutated.
Types of mutations leading to DMD:
•Deletion hotspots around exons 45-53 (cluster I) or exons 2-20 (cluster II)
•Missense mutations distributed throughout, introducing premature stop codons
•Duplications
1/3 of DMD cases are due to new mutations rather than family hisotry making it difficult to screen.
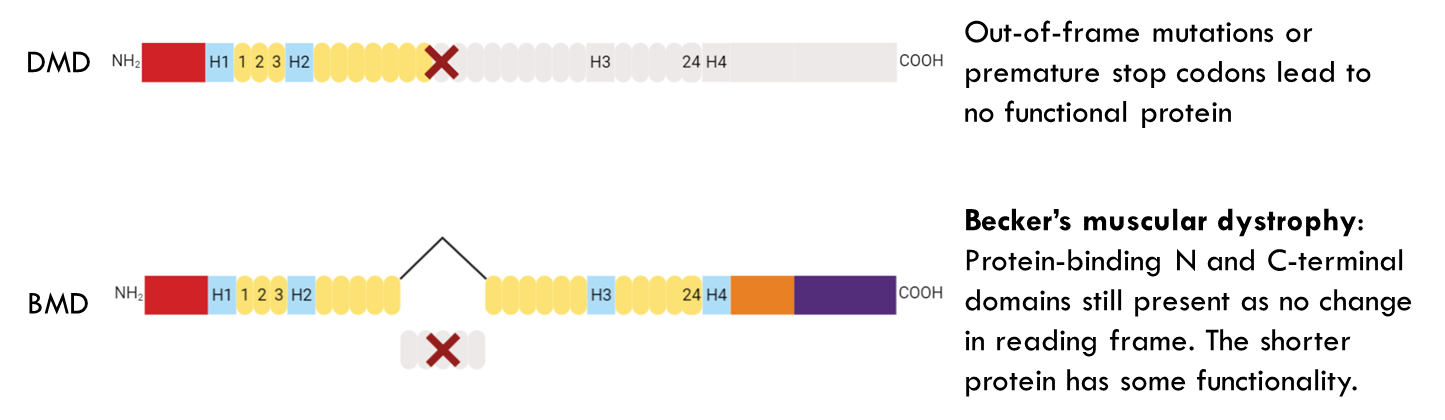
People with Becker’s tend to live longer and can maintain active lifestyles.
Why is DMD suitable for gene therapy?
•DMD is a monogenic recessive disorder
•Mapped to mutations in a single gene
•Currently no cure for DMD
•Physiotherapy, corticosteroids, creatine medication to improve muscle strength and slow progress of muscle weakening
•Translarna (Ataluren) available to some patients with nonsense mutation – allows ribosome to read-through premature stop codon in mRNA. In this case the disease must be caused by a nonsense mutation and its only really affective at the beginning stages of the disease → given to patients below 5 years.
Approach 1: Exon skipping
•Exon skipping occasionally occurs during splicing of primary transcript, observed in revertant fibres in DMD when splicing occurs and it repairs this mechanism. However it is relatively rare.
•Can be induced with antisense oligonucleotides that mask splice sites. Skipping the exon in the middle can still have some functional benefits. Look at diagram notes.
•Protein with partial deletions can still have functional benefits
•ON1 binding to exon 44 improved phenotype in mouse model
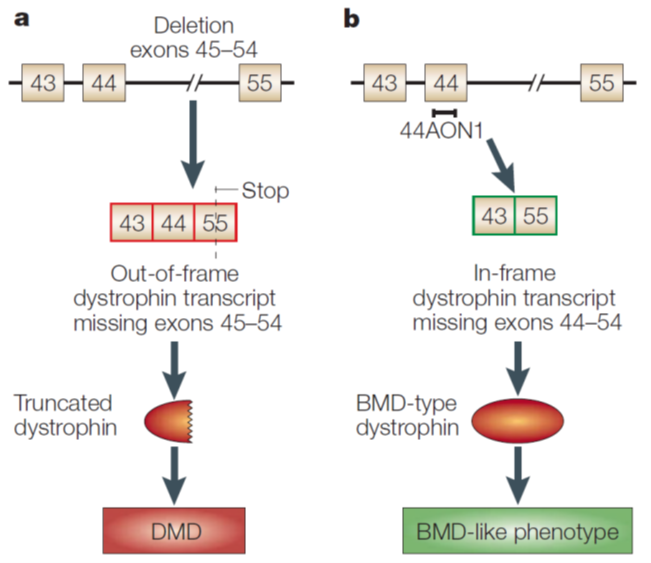
Here from 45 to 54 there is a deletion causing an out of frame mutation leading to DMD in mice. However when you add antisense oligonucleotide (44AON1) to exon 44 it is skipped and therefore exon 43 splices to the next available exon which is 55. This leads to a protein that is shorter. → Beckers muscular dystrophy like phenotype
Study
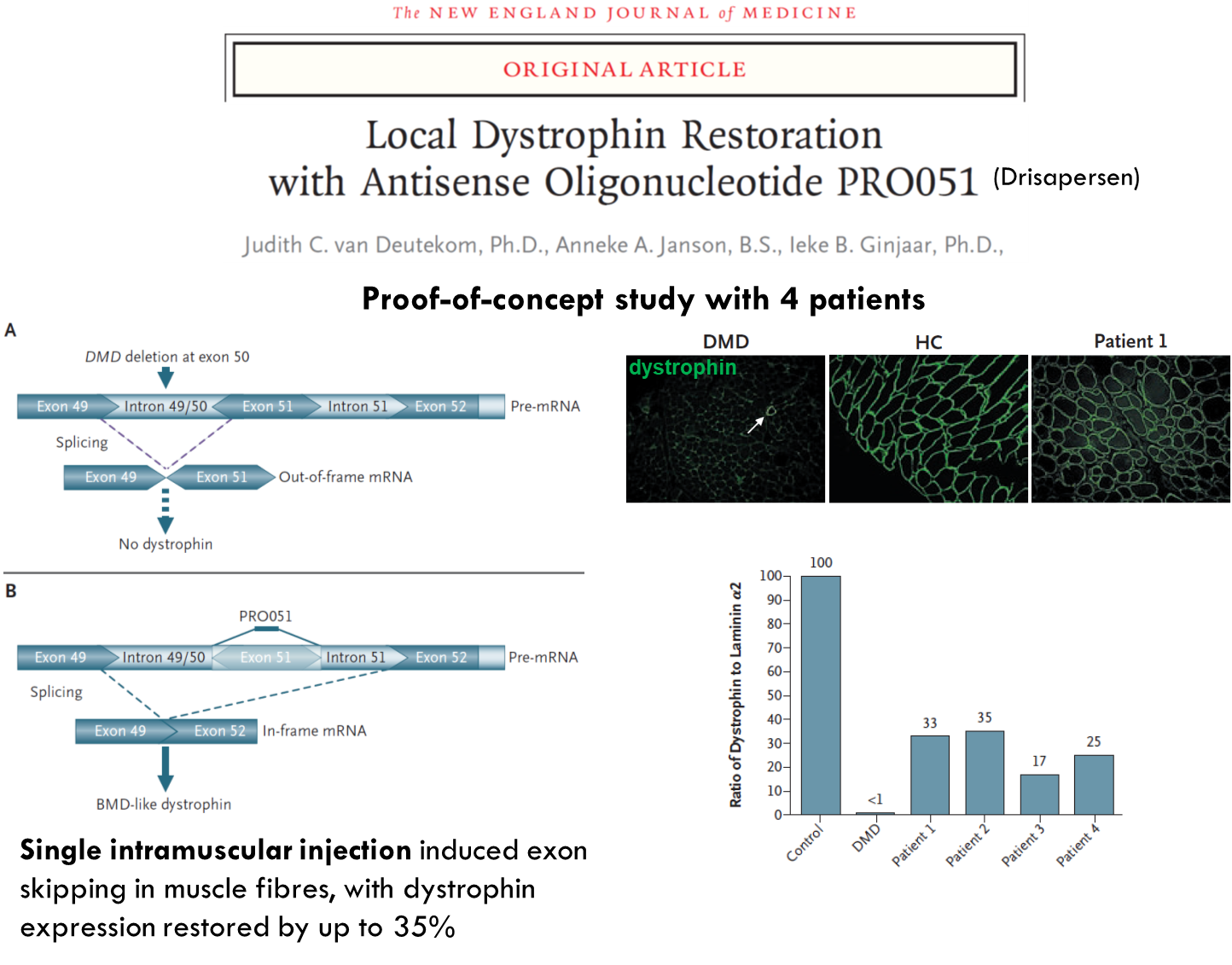
What about in humans?
These patients had deletion in exon 50 → non functional dystrophin. Used antisense oligonucleotide which targeted splice site in exon 51. This leads to it being skipped. You get splicing of exon 49 to 52 which produces a shorter dystrophin protein. Can only be used for the percentage of patients who have deletion in exon 50.
Continues
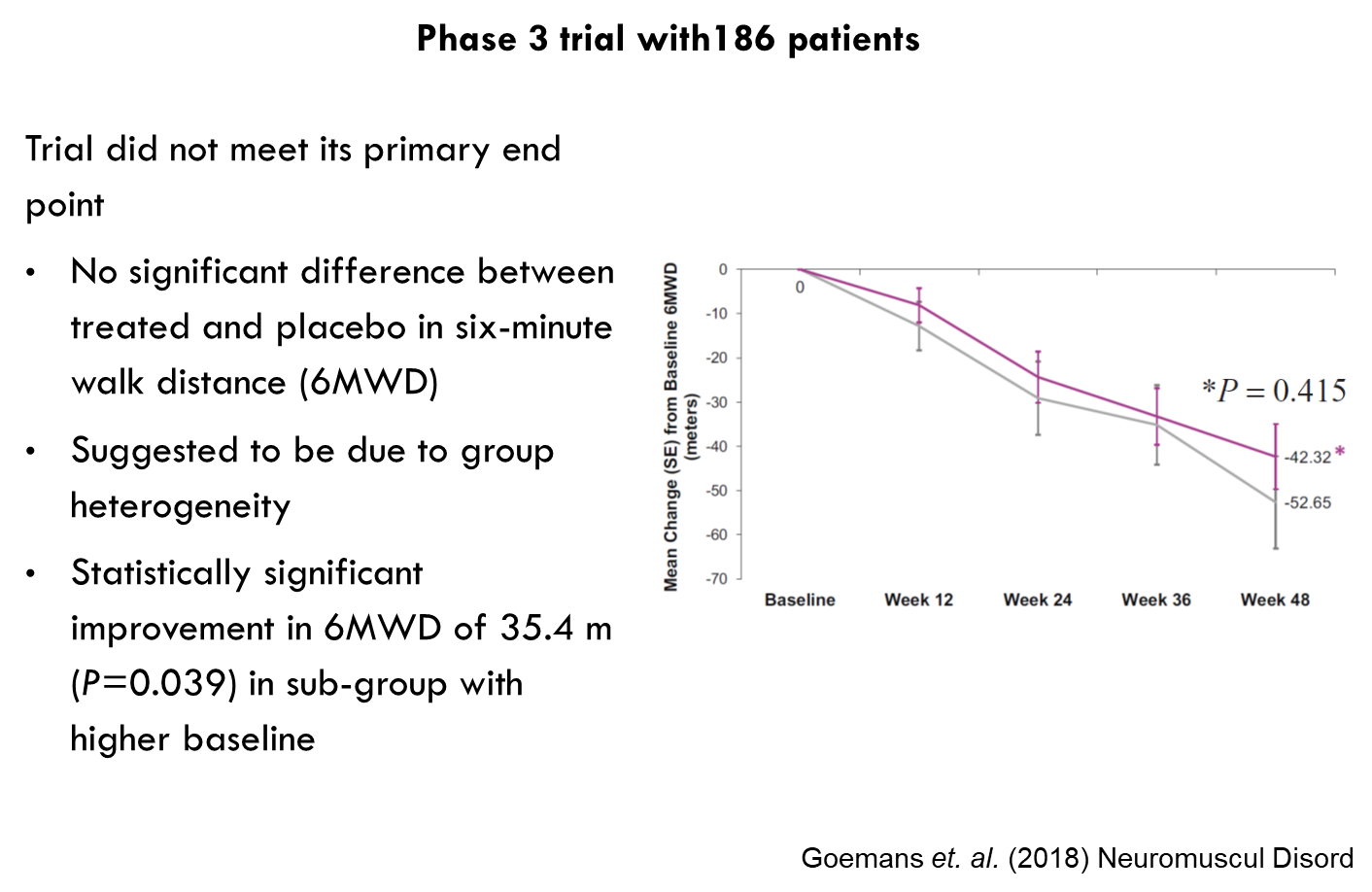
Continued
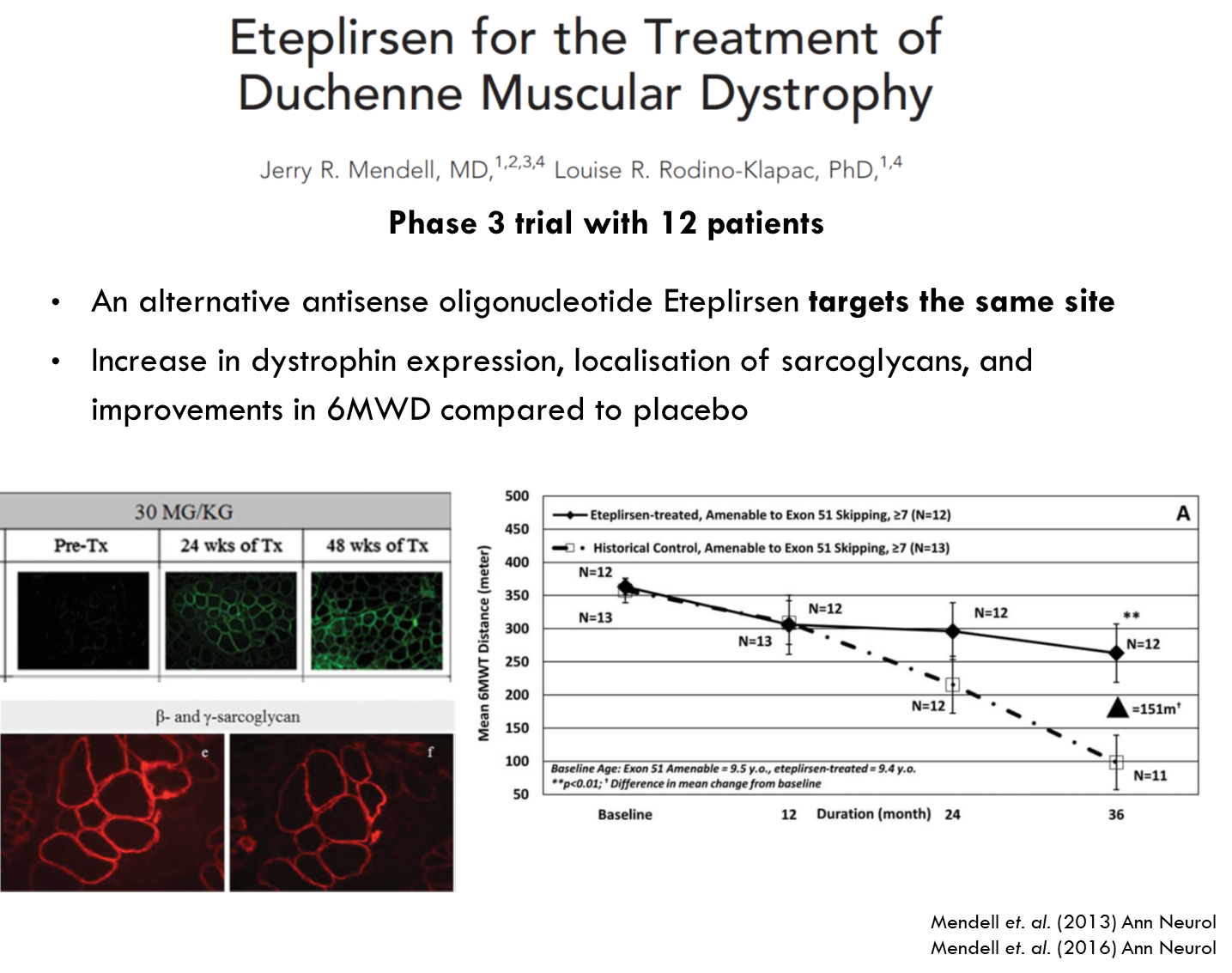
Only difference is it uses a different backbone for the oligonucleotide.
Obstacles with oligonucleotide drugs
Method of administration
•Lack of effective distribution away from the injection site. Can have good dystrophin recovery at site of injection but not necessarily in other muscles in the body.
•>300 muscles make up the total musculature in the human body
•Systemic delivery system required - How this can be achieved is using viral vector.
Approach 2:
Adeno-associated viral (AAV) vectors
Systemic delivery of an AAV vector with sequence coding for
•Antisense RNA
In mouse models this RNA molecule has been successfully delivered to restore normal levels of dystrophin expression.
•Dystrophin-like proteins (insert size limit of 4.5 kb) → This is also being considered. Since dystrophin is so long there is a limit to how much the viral vector can fit and deliver.
Alternatives to dystrophin?
Preservation of N- and C-terminal domains are crucial for function
•Becker's muscular dystrophy caused when shorter protein is expressed leads to milder symptoms
•Micro- and mini-dystrophin can be genetically engineered where the rod domain in the middle is deleted. Key protein binding domains are still retained.
•Utrophin is a protein homologous to dystrophin, found in the sarcolemma only in foetuses, so generally stops being expressed once foetus develops. Can lead to a partial restoration of function.
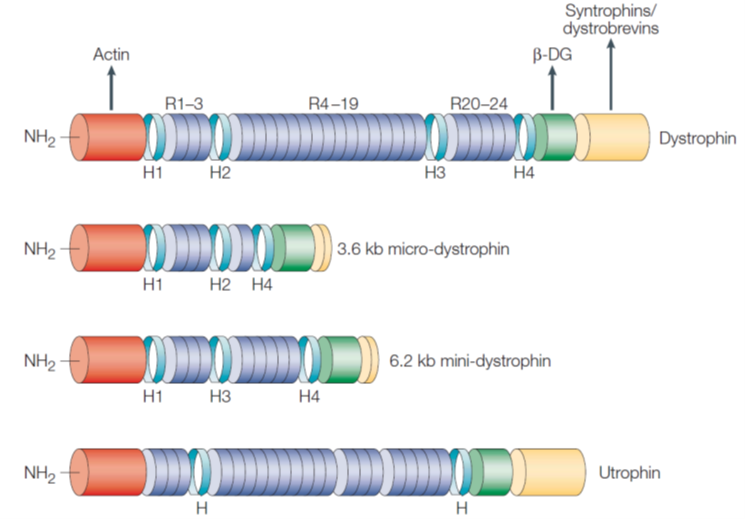
AAV expressing micro-dystrophin
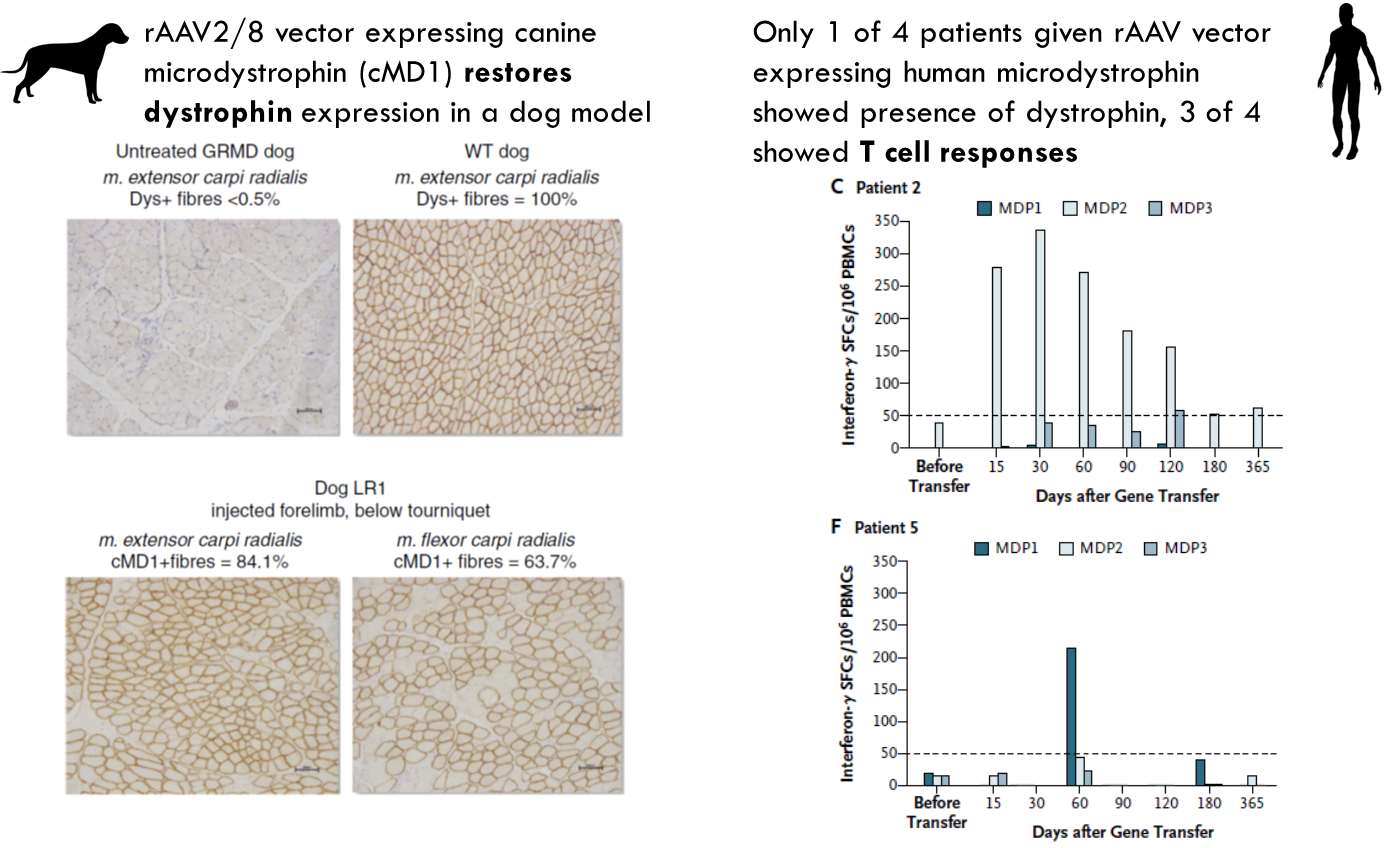
In the dog it seemed to restore function of dystrophin. However 1 in 4 patients had this same response while 3 in 5 had a t cell response where interferon gamma was being produced. This suggests that this is potentially killing the cells expressing the protein → immune system is recognising vector or the protein as foreign molecule.
Obstacles to AAV vectors
Potential problems:
•High dose of virus needed to express required amounts of protein
•Immune responses against AAV may limit effectiveness of subsequent deliveries
•
How can this be overcome?
•Transient immunosuppression by giving cyclosporin or corticosteroids.
•Recombinant AAV that are less immunogenic
Study
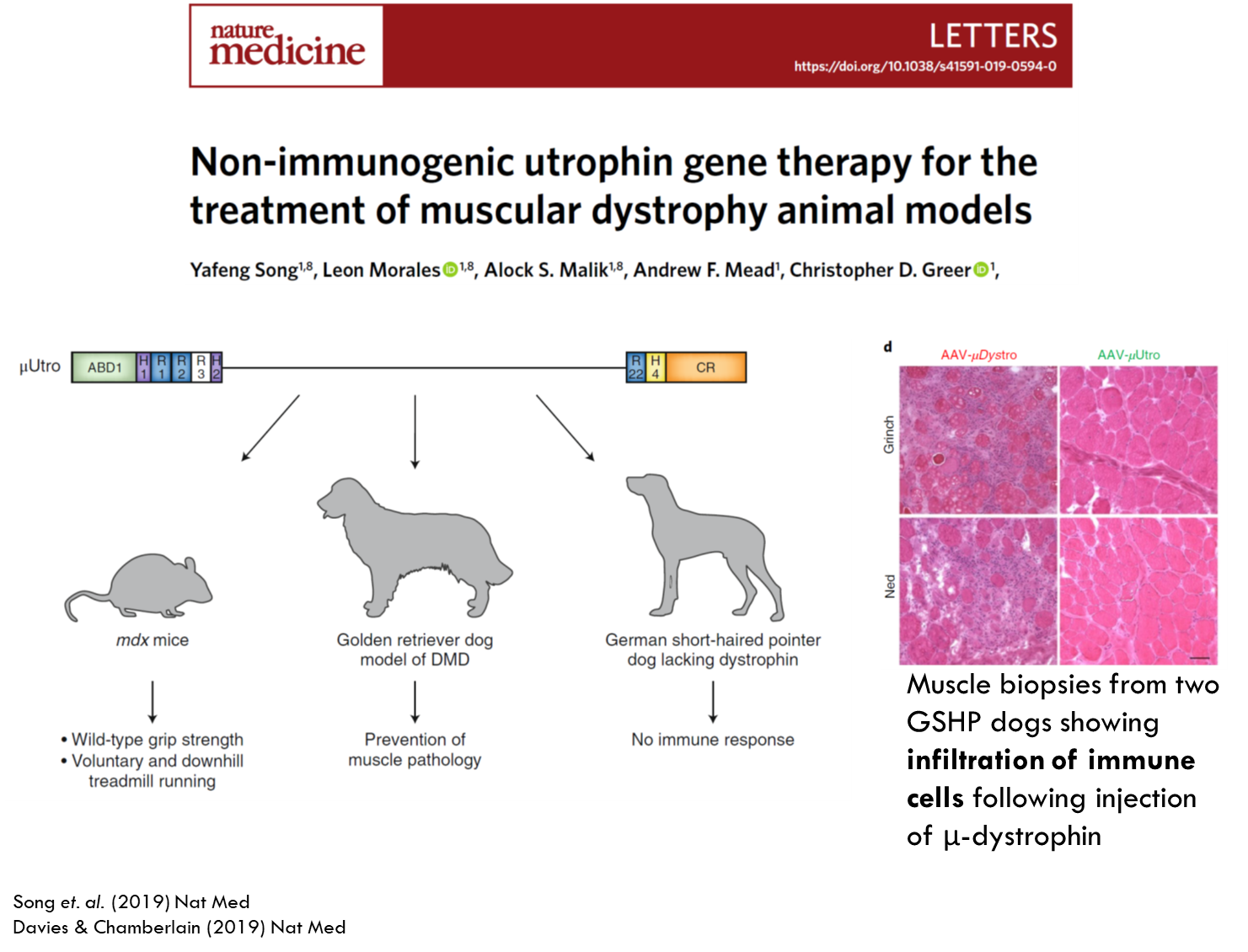
Genetically engineered utrophin to make it even smaller and inserted it into the HIV vector and tested it in 3 models. In the mouse model there was an increase in muscle strength. The same happened with the golden retriever dog model. German short haired dog model was used to looks at immune responses. This model had complete deletion of dystrophin. Therefore any small amount of expression of dystrophin could activate the immune response. Purple dots indicate immune cells and when using micro utrophin they saw very little immune response.
Other approaches
Stem cell therapy is also being used to transfer cells into the patient that produce dystrophin.
CRISPR/CAS9 can also be used.
Cystic fibrosis
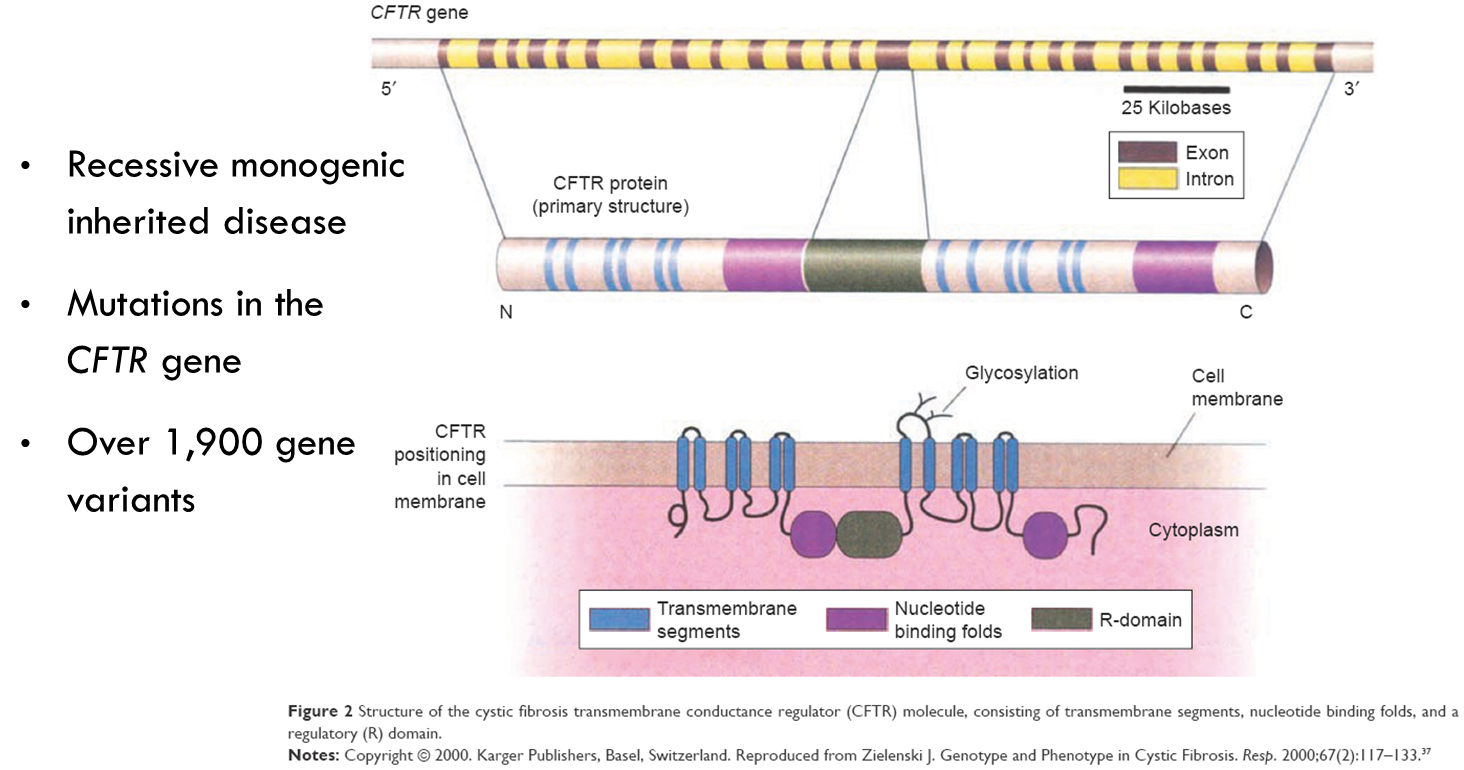
CTFR lies in cell membrane and allows passage of chloride ions.
Why is CF suitable for gene therapy?
•CF is a monogenic disorder
•Mapped to mutations in a single gene → therefore could potentially target that gene.
•
•CFTR modulators target the underlying cause of the disease
•But only work on some mutations
•Gene replacement therapies are most promising hope for a cure
Approach 1:
Adenoviral vector to deliver CFTR gene
•Ex vivo transfer of CFTR gene using adenovirus and retrovirus vectors caused cells to produce the CFTR protein
•Adenoviruses cause common colds
•Genetically engineered to non-harmful versions that carried the target gene
•Adenovirus Sequence does not integrate into host genome
Study
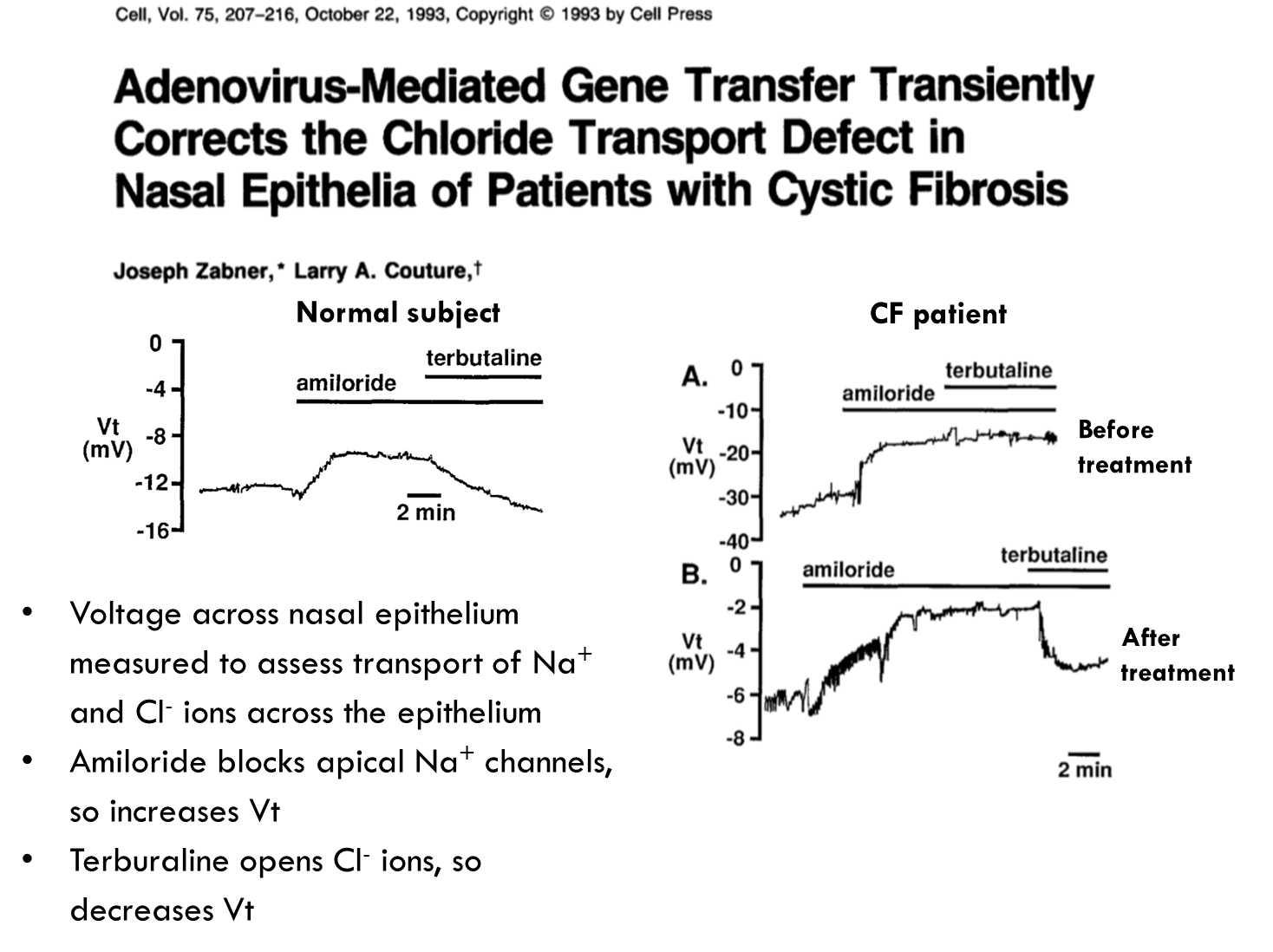
Here they looked at if they could transfer the CFTR gene into the nasal epithelium of patients. They found that they could do it for short periods of time.
They saw how much of the CTFR was being expressed by looking at the voltage across the nasal epithelium, so they could see how much sodium ions etc were getting transported across the nasal epithelium. Amiloride was used to block sodium channels to increase voltage and terbutaline to open chloride ions and therefore decrease the voltage. When done with CF patients before treatment the amiloride causes voltage increase but when given terbutaline it doesn’t cause a decline. After treatment they found they could restore a decrease in voltage suggesting these patients could restore this decrease in voltage.
Disadvantages of adenovirus vectors
•Modest improvements observed, but...
•No dramatic changes in the long-term
•Sequence does not integrate, and so treatment must be repeated
•Host immune system was producing a response against the adenovirus and destroying the vector
Approach 2:
AAV vector to deliver CFTR gene
•Adeno-associated virus (AAV) does not cause human disease
•Many phase I trials indicated better safety profiles
•Two studies in 2004 and 2007 tested repeat-dosing of AAV vector containing the CFTR gene, delivered to the respiratory tract by inhaler
Study
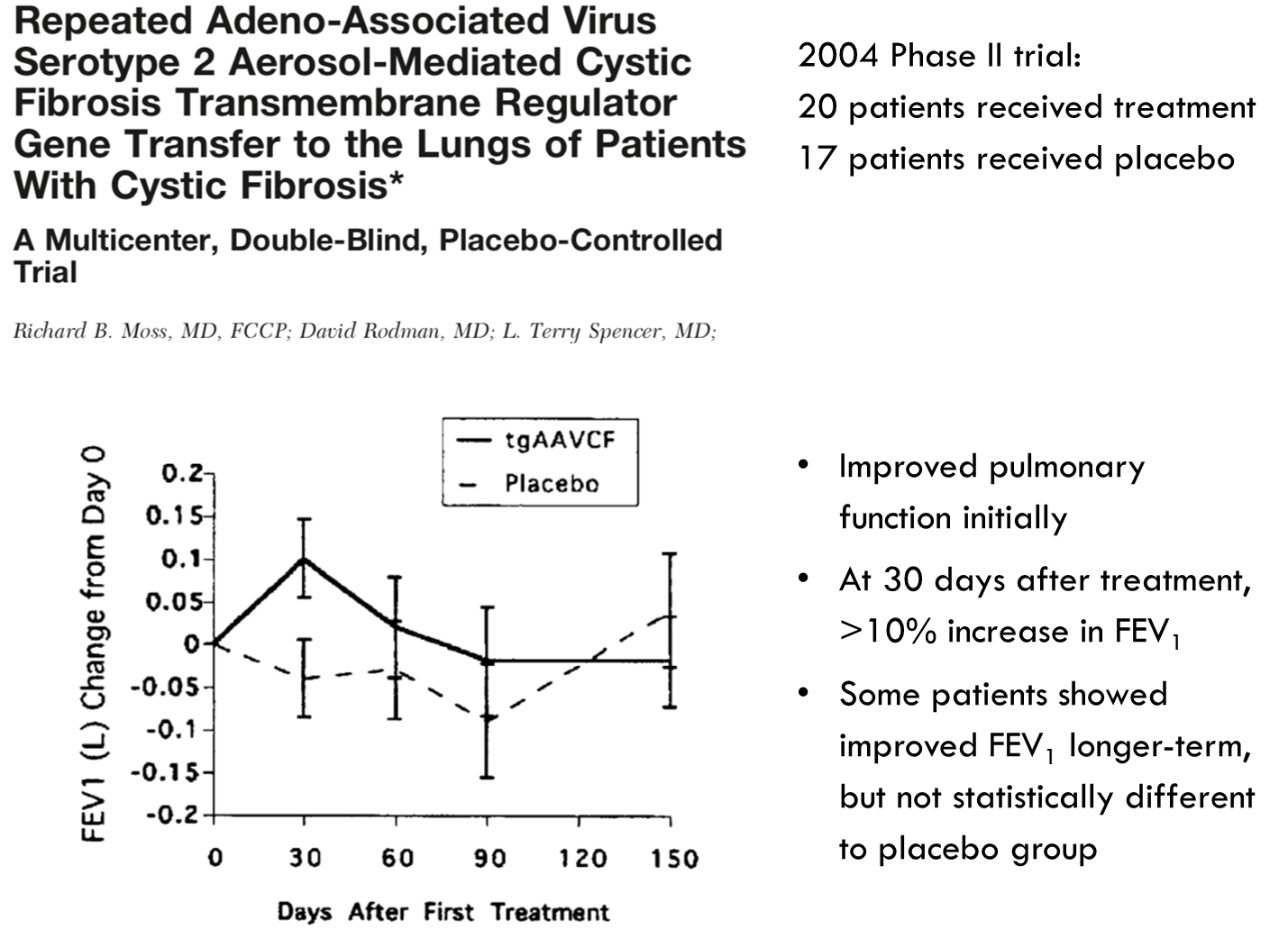
Study
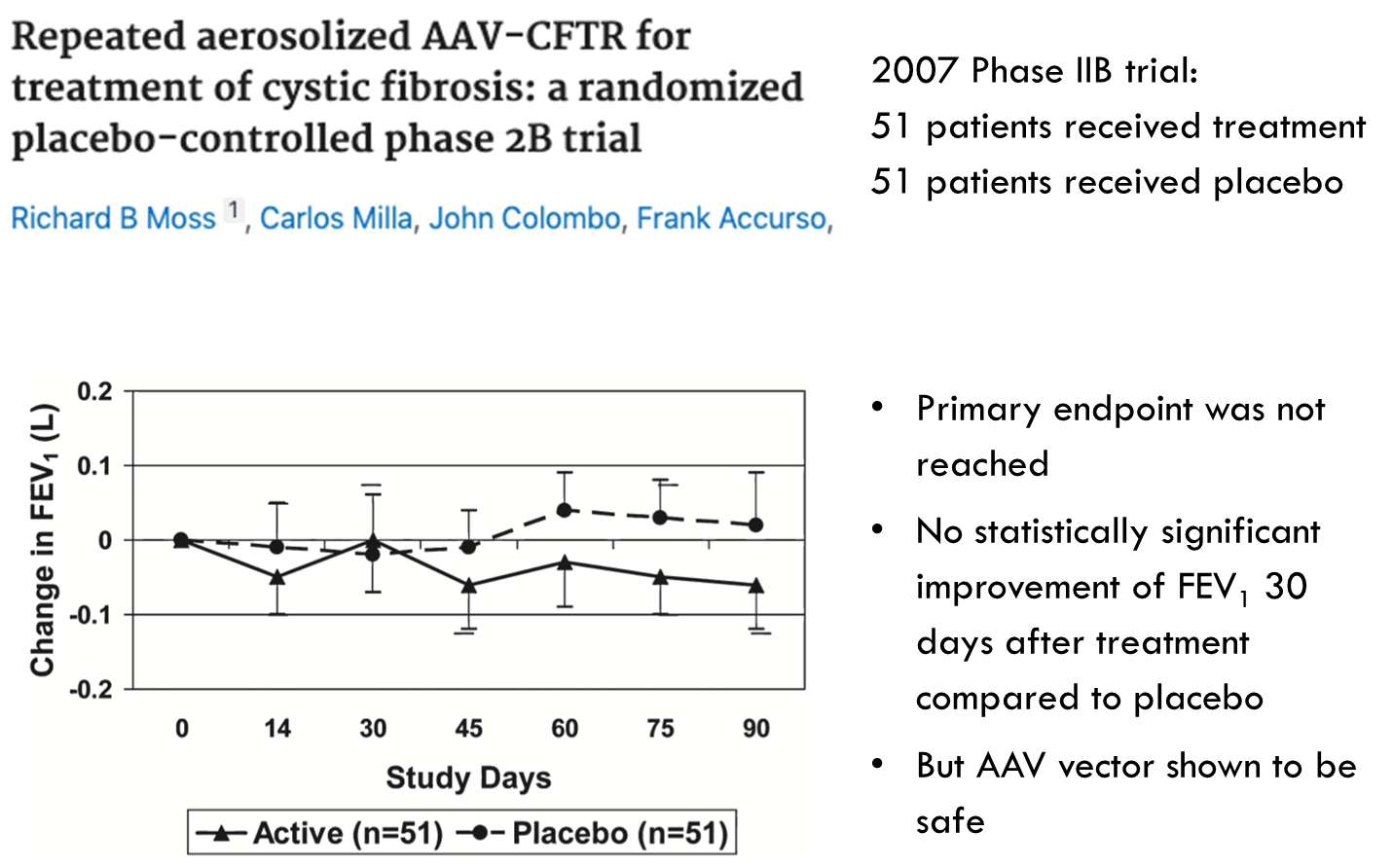
Improving delivery
•Next-generation AAV vectors that have increased infectivity to reach more cells in the lung
•Shorter CFTR gene to allow additional stronger promoter sequences in the vector
•Lentiviruses that integrate the target gene into host genome and therefore provide long term expression.
•Lipid nanoparticles instead of viral vectors
Approach 3: CRISPR/Cas 9 gene editing
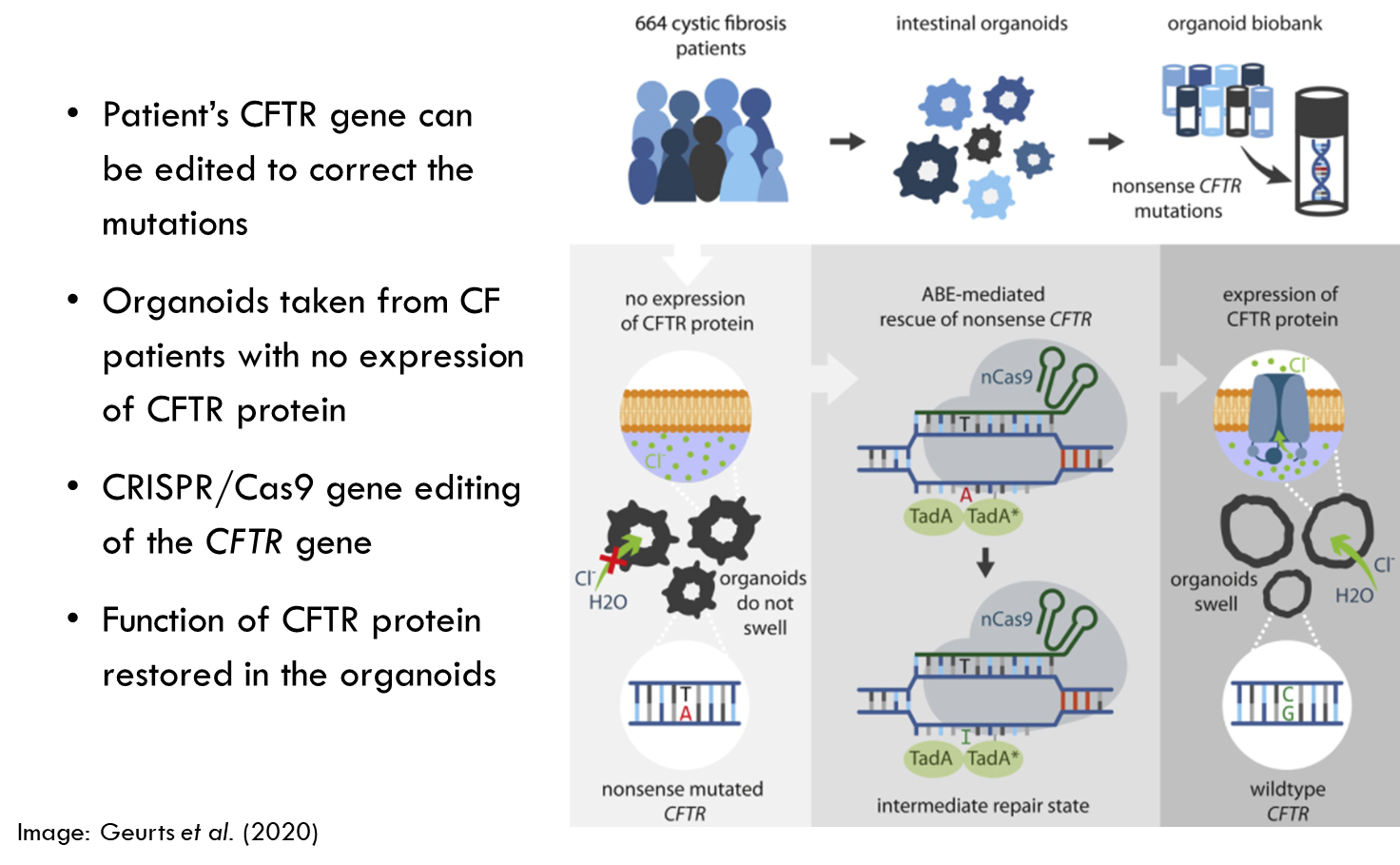
Future of CF gene therapy
•New AAV vectors and non-viral delivery methods, and CRISPR approaches are being researched
•Delivery of gene therapy to the deepest parts of the lungs is the main challenge → inhalers won’t reach right down to the bottom of the lungs.
•Long-term cure requires delivery to lung stem cells
•Safety profile of CRISPR gene editing requires testing – will there be off-target effects?
Human immunodeficiency virus (HIV)
•37 million people infected with HIV globally, with ~2 million new infections each year
•Single stranded RNA lentivirus
•Infects CD4 T cells, leading to their depletion and AIDS
•Integrates into host genome, and persists in memory T cell reservoirs. Therefore why its so hard to cure.
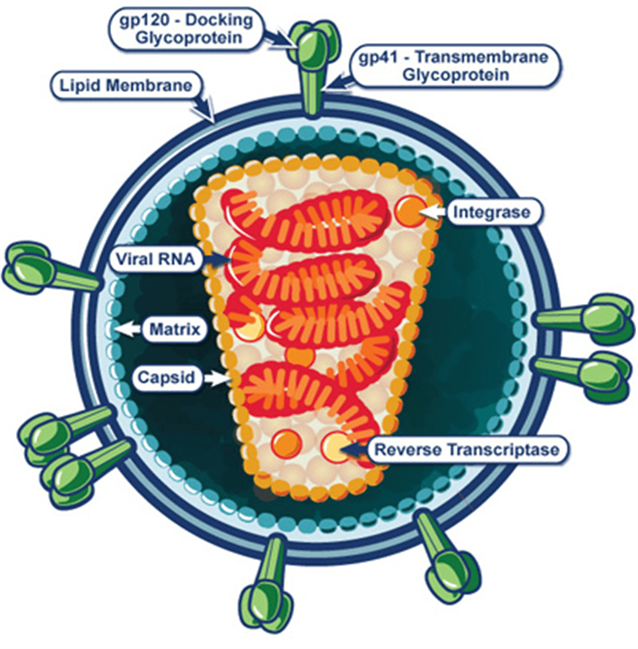
HIV genome
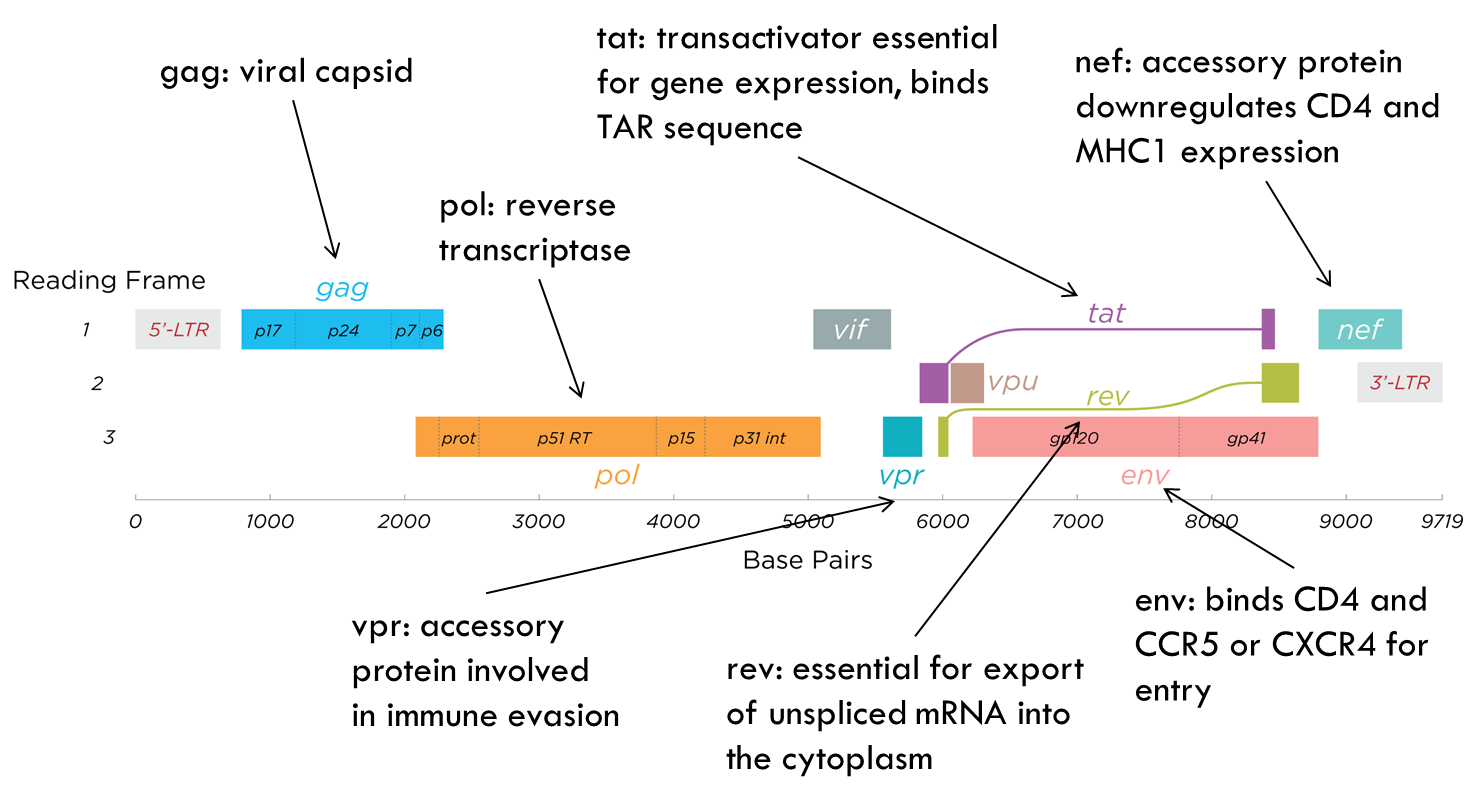
Genome is very simple → only 9 genes.
Current therapies
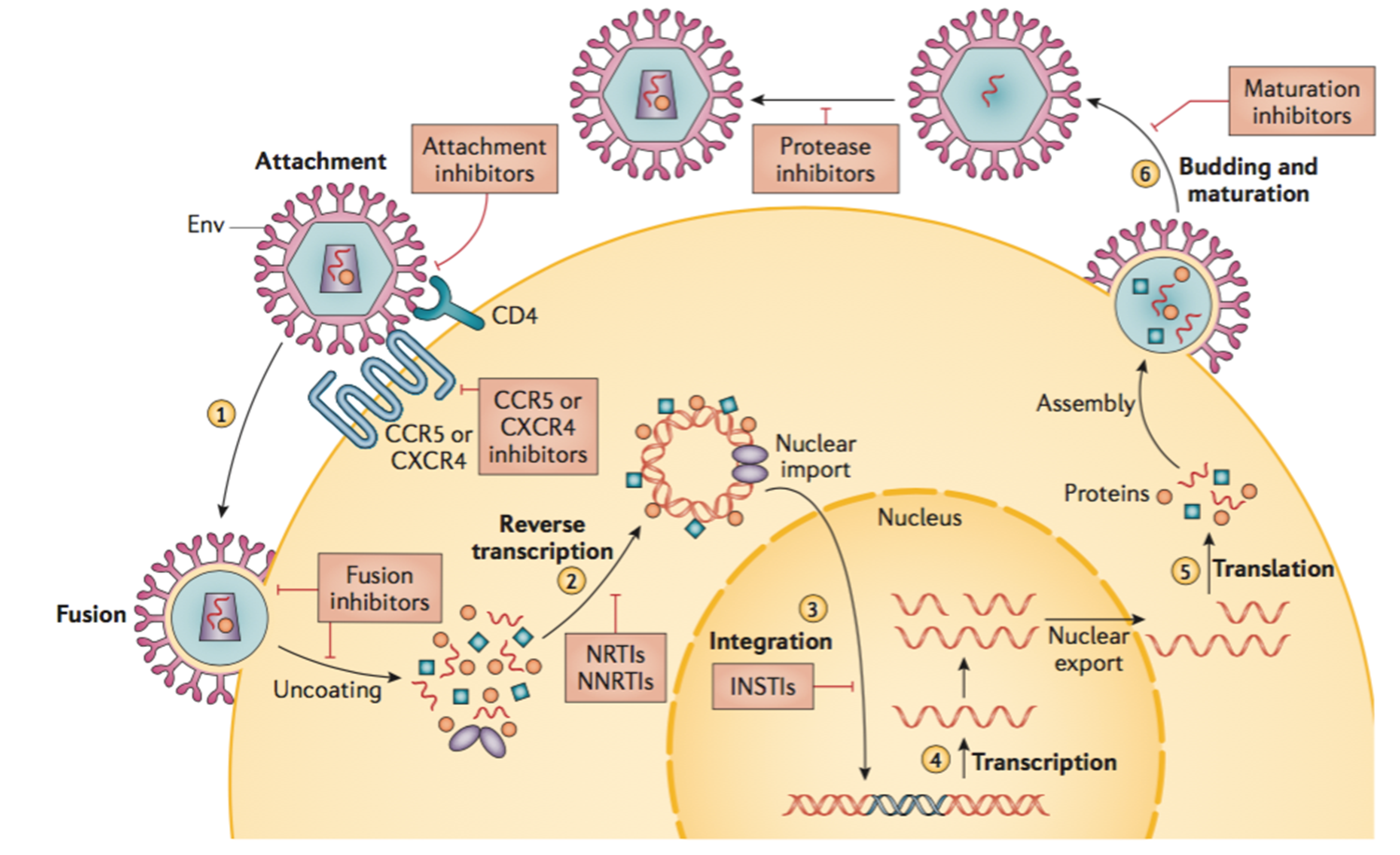
This diagram shows lifecycle of HIV.
Efficacy of current therapies
•Current therapies are highly effective
•
Obstacles:
•Requires adherence
•Development of resistance
•Viral genome is integrated into host genome and therefore current therapies are not a cure
How can HIV be treated with gene/cell therapy strategies?
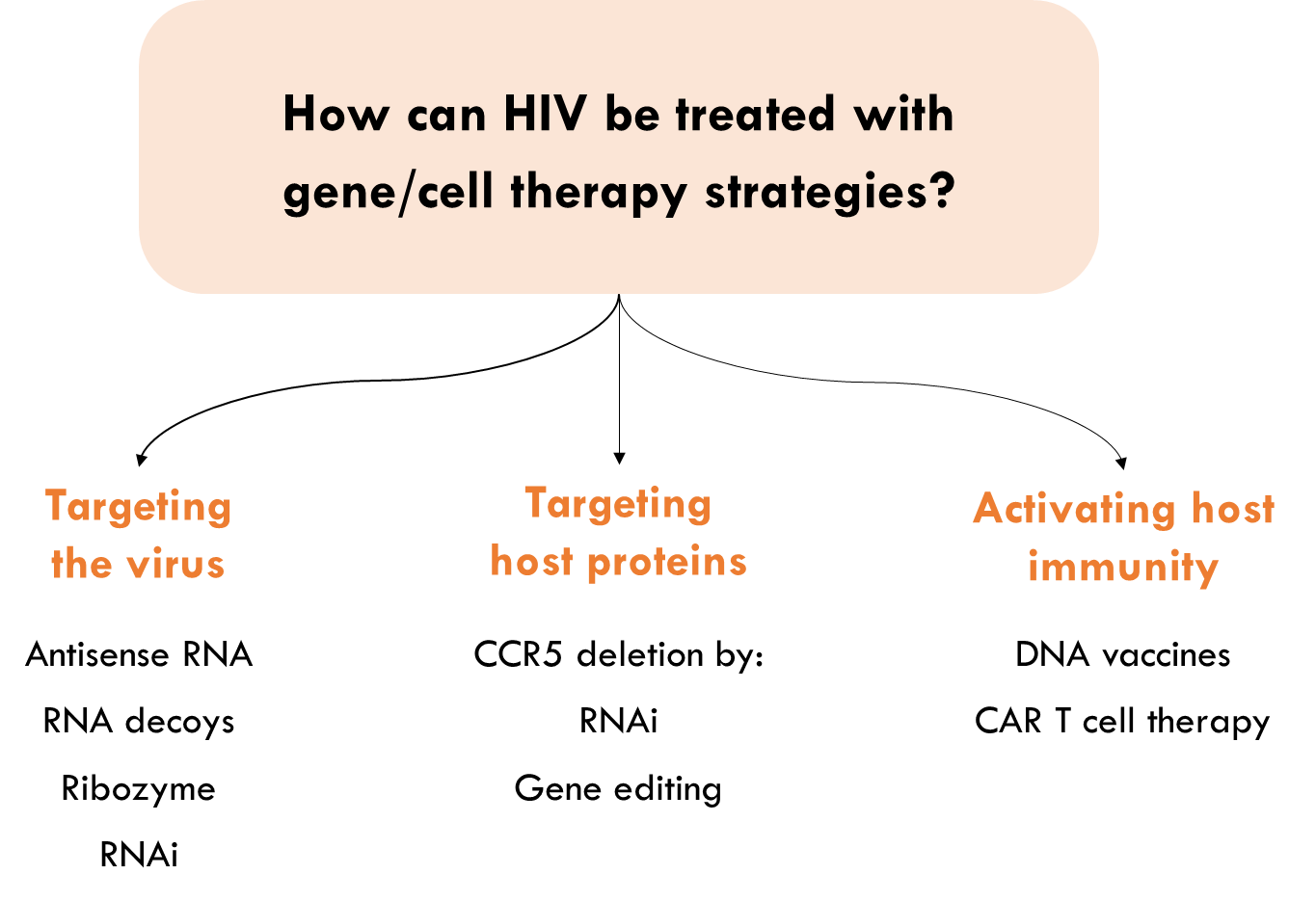
RNA-based therapies
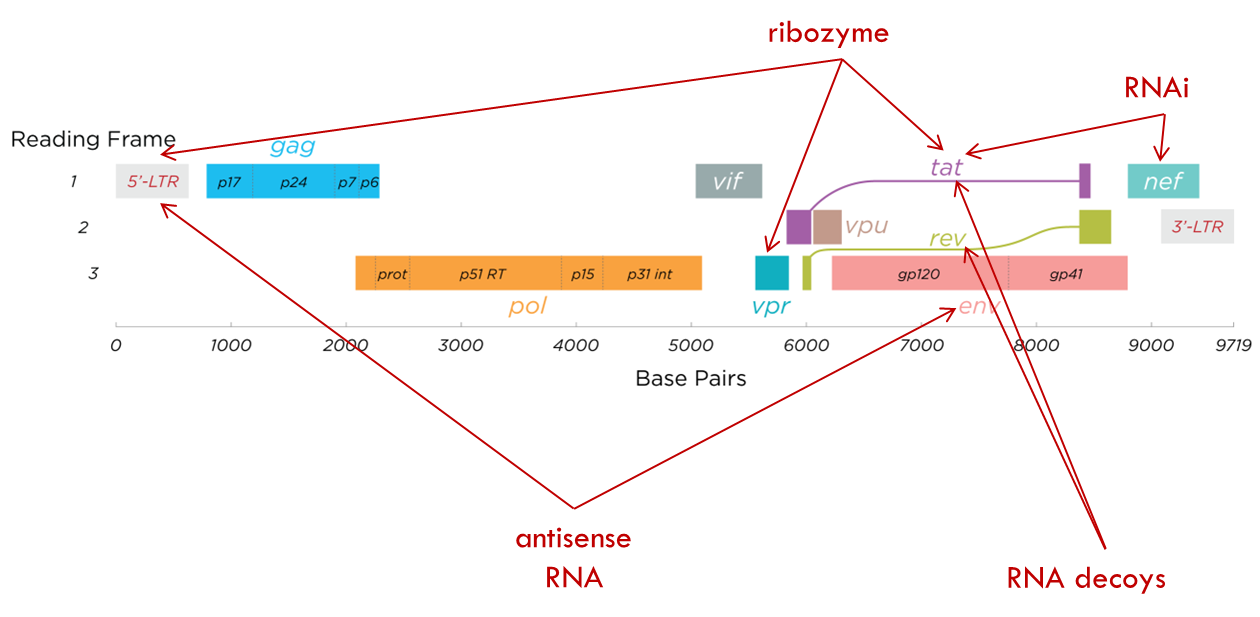
RNA based therapies target various parts of viral genome e.g. antisense RNA to target the envelope protein and 5 prime LTR which is very important for gene expression. We can also have ribozymes that target the 5 prime LTR as well or VPR or TAT. Just look at the diagram
Ribozyme targeting HIV tat/vpr
•First randomised Phase II trial using ribozymes
•Ribozyme targeting HIV-1 tat/vpr genes - OZ1. Remember that ribozymes are molecules of RNA that specifically cleave at a certain point.
•OZ1 delivered by MMLV retroviral vector to autologous CD34+ cells
•Why was a retroviral vector used?
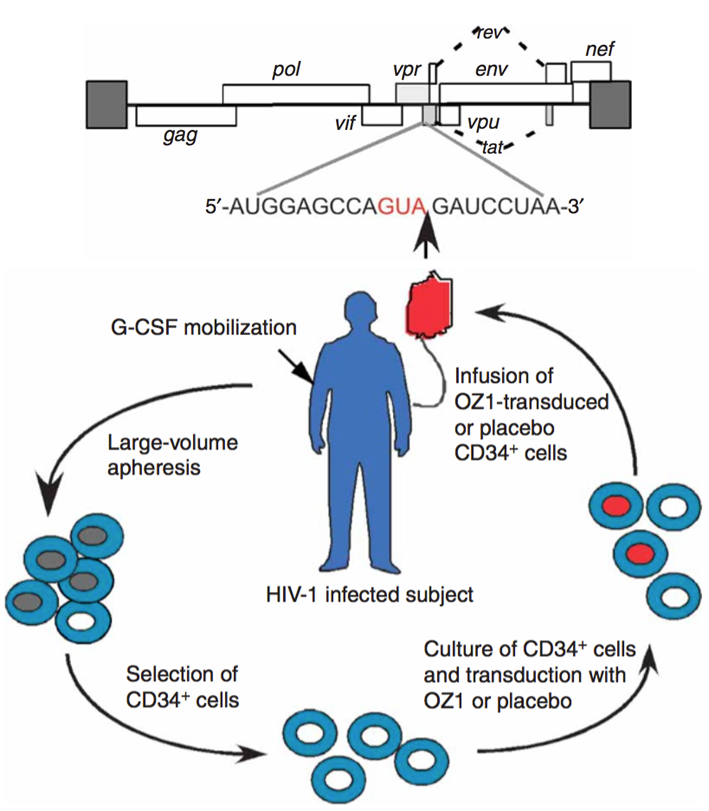
These patients were on heart therapy, but heart therapy was stopped to see effect of ribozymes.
Ribozyme targeting HIV tat/vpr
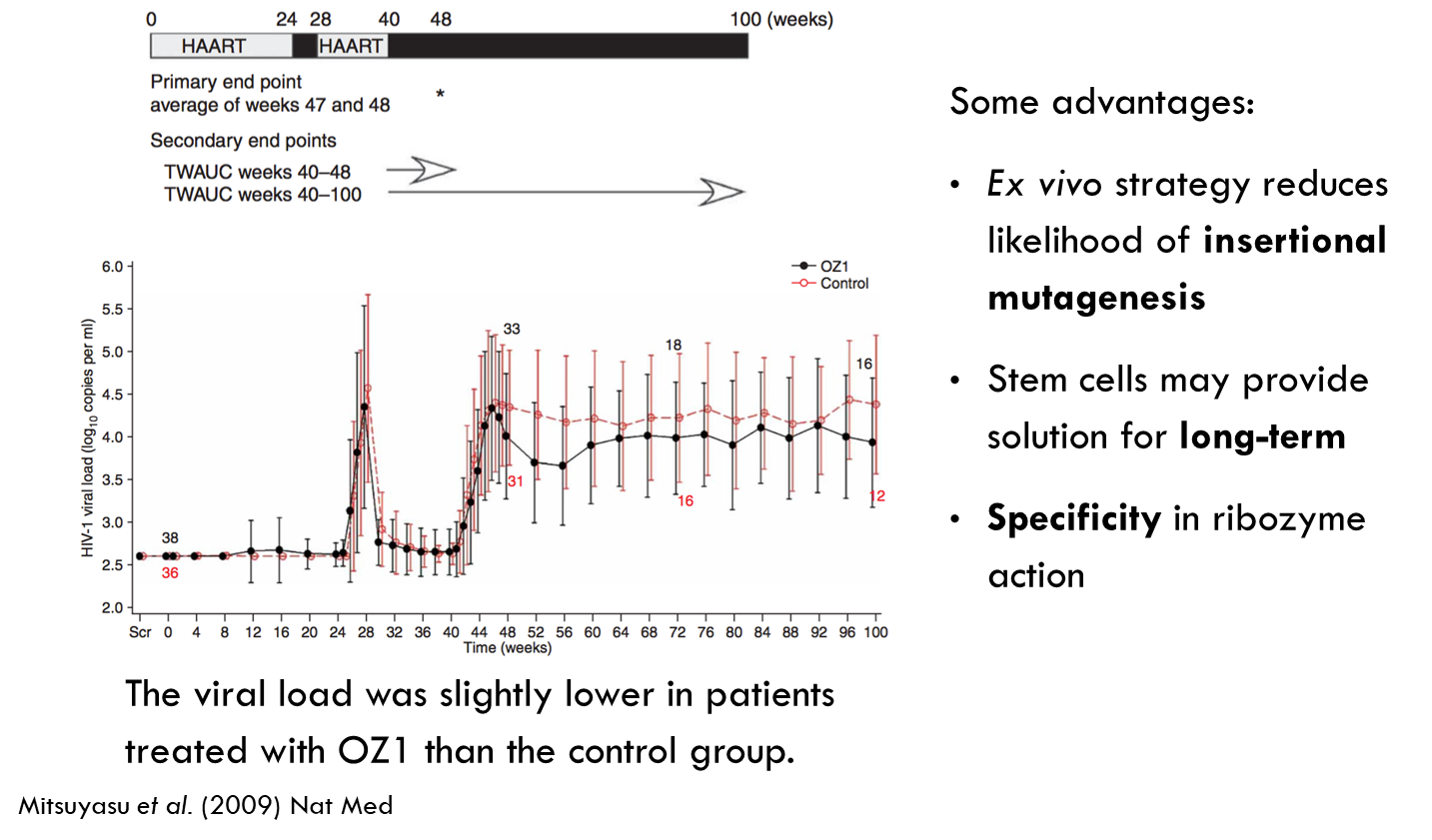
Red line is control and black line is treated group. Treated patients had slightly lower viral load.
Issues with targeting virus
What are potential obstacles with directly targeting the virus?
•HIV reverse transcriptase is error prone allowing the virus to mutate fast and escape the treatment.
•Allows virus to mutate fast and escape the treatment
How can this be overcome?
•Target host genes essential for HIV replication e.g. CCR5 and CXCR4 are cell surface receptors essential for viral entry
•Can target host genes in combination with viral genes to minimise escape
Targeting CCR5
Why is CCR5 a good target?
•CCR5 is a co-receptor required for entry of macrophage-tropic strains of HIV, usually responsible for transmission
•Up to 16% of Europeans have CCR5Δ32 mutation that introduces a premature stop codon and no expression on the cell surface
•Individuals with homozygous Δ32 mutations show resistance to M-tropic strains of HIV, and heterozygotes show delayed progression to AIDS
•N.B. There are some detrimental effects associated with West Nile Virus or tick borne encephalitis infections
Two patients “cured”!
Berlin and London patients
•Two HIV-positive individuals diagnosed with leukaemia or lymphoma
•After failed chemotherapy, allogenic haematopoietic stem cell transplant for treatment of cancer, and antiretroviral therapy was discontinued
•HSCT donors were CCR5 Δ32/Δ32
•Recipient HIV-positive cells eliminated before transplantation, donor cells cannot be infected with M-tropic HIV
•Graft vs host disease might also play a role
•Berlin patient has been HIV-free for 12 years, London patient for 2 years
RNA therapy targeting virus and host
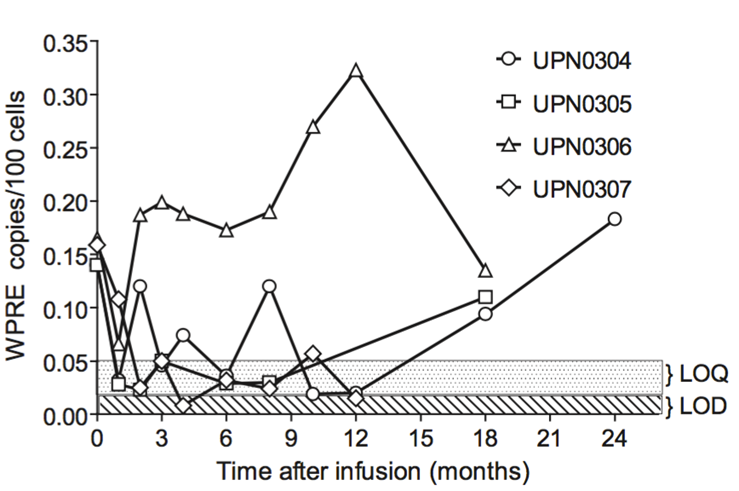
Phase I trial with AIDS patients undergoing autologous transplantation for lymphoma
Transduced with CD34+ cells modified with RNA-based gene therapy targeting viral and host proteins:
tat/rev short hairpin RNA
TAR decoy
CCR5 ribozyme
Integrated vector copies, siRNA and ribozyme levels detected in patients to varying degrees for up to 24 months
Targeting the latent reservoir
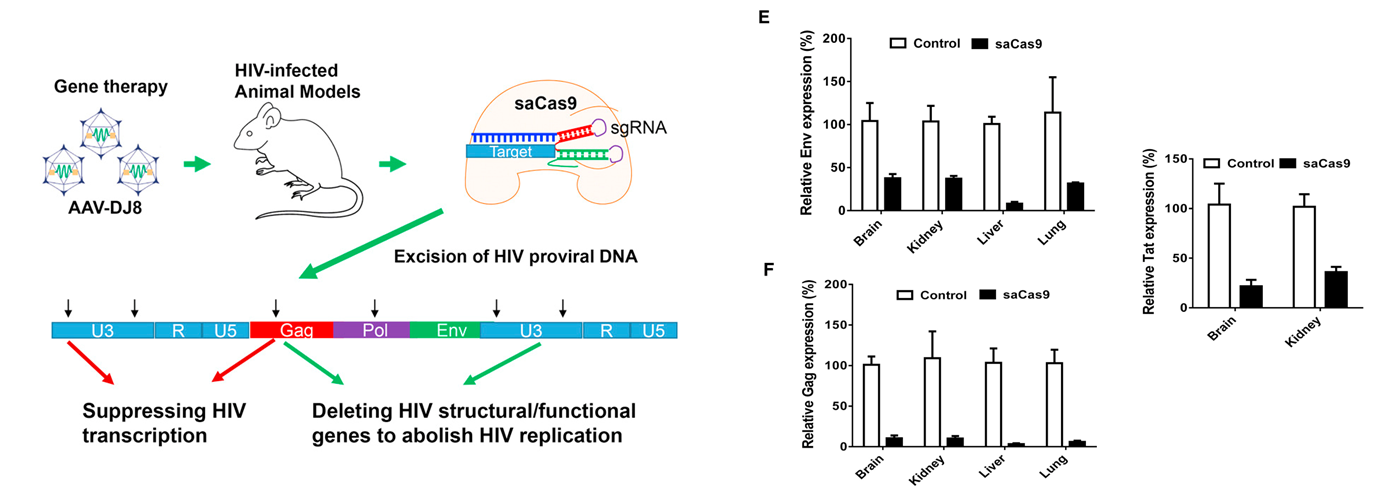
Multiplex single-guide RNAs and Cas9 directed against HIV-1 genome delivered to humanised mouse model by intravenous injection of AAV vector
HIV-1 RNA transcripts decreased in treated mice in various organs
DNA vaccines
•DNA vaccine directs the expression of HIV-derived epitopes
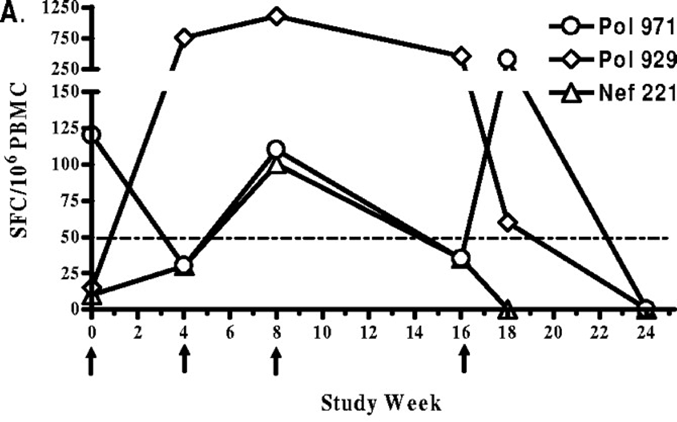
•Phase I clinical trial: injection of plasmid DNA containing conserved HIV sequences that are immunogenic
•~38% of patients showed specific responses to at least one epitope
Further improvements required:
•Delivery of DNA vaccines has to be optimised further
•Improved vaccines to trigger both antibody and CD8 T cell responses
•Sustained responses required