pharmacokinetics
1/47
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
48 Terms
Pharmacokinetics
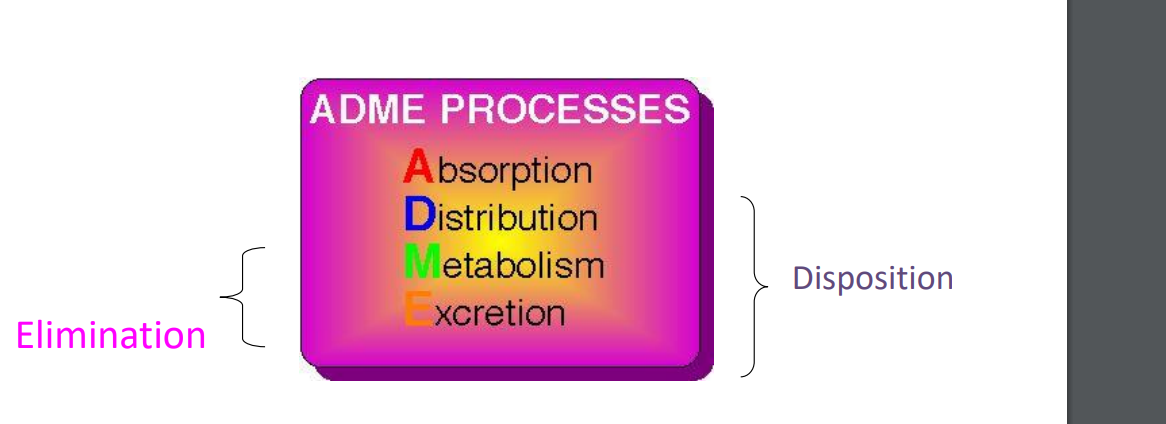
the study of handling of drugs by the body - what the body does to the drug
➢ time course of drugs and their effects in the body.
➢ allows the processes of drug absorption, distribution, metabolism and excretion (ADME ) to be quantified
• Clinical pharmacokinetics - application of PK principles for the safe and effective drug therapy in individuals
• Individualised drug treatment
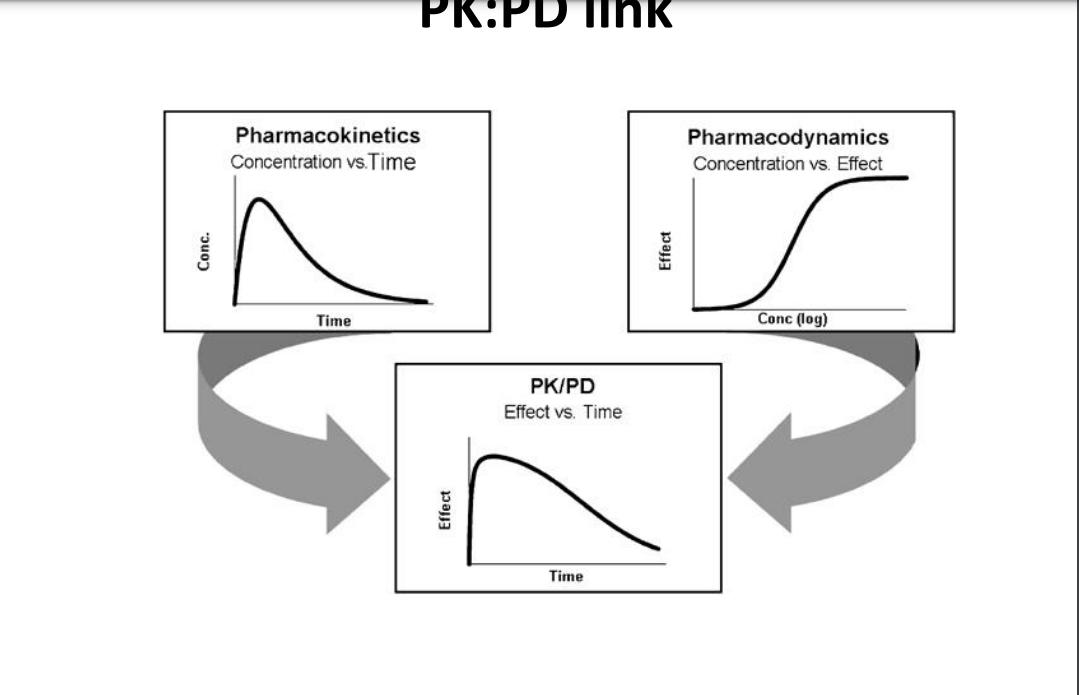
Pharmacodynamics
is a study of the time course of the drug effect in the body - what the drug does to the body ➢explains relationship between drug concentration and therapeutic effects
➢PD is important in determining the change in drug effect due to changes in PK
difference
what happens when drug is given
Absorption
Drug action - requires adequate concentration of the drug at the target tissue (site of action) Systemic effect – occurs via entry of the drug into the systemic circulation, requires absorption of the drug into the bloodstream (except if given intravenously) Topical or local effect – applied directly to site of action: e.g emollient for dry skin; antibiotic eye drops for infection, absorption should be minimal (avoid side-effects)
FACTORS AFFECTING ABSORPTION
Physical properties of the drug e.g. physical state, lipid or water solubility and pH of surroundings.
• Formulation e.g. Disintegration and dissolution rate, Particle size and surface area, Dosage form
• Physiological factors e.g. Changes in gastric motility/emptying First-pass effect Disease states, Presence of other substances: antacids, food, etc
Route of administration and drug absorption
➢ Enteral route – drug absorbed into systemic circulation via the GIT (oral/rectal)
➢ Parenteral route – all systemic routes other than oral or rectal. For example intramuscular, intravenous, subcutaneous, inhalation, sub-lingual etc
➢ Drugs given parenterally are generally absorbed more quickly than via oral administration
➢ Intravenous (i.v.) injection – only route not requiring drug to be absorbed into the circulation
Distribution
The movement of drugs around the body
• Once absorbed, drug molecule may spend some time in one organ, then be carried by the blood to another organ and so on.
Factors affecting drug distribution
Tissue permeability of the drug
• Organ or tissue perfusion
• Plasma protein binding of drug
• Other factors – age, pregnancy, disease, obesity etc
Elimination
Irreversible removal of the drug from the body The primary routes are
• Metabolism: destruction of the drug by chemical alteration (usually in the liver)
• Excretion: Removal of the chemically unaltered drug from the body in the kidneys
• Some drugs - combination of the above
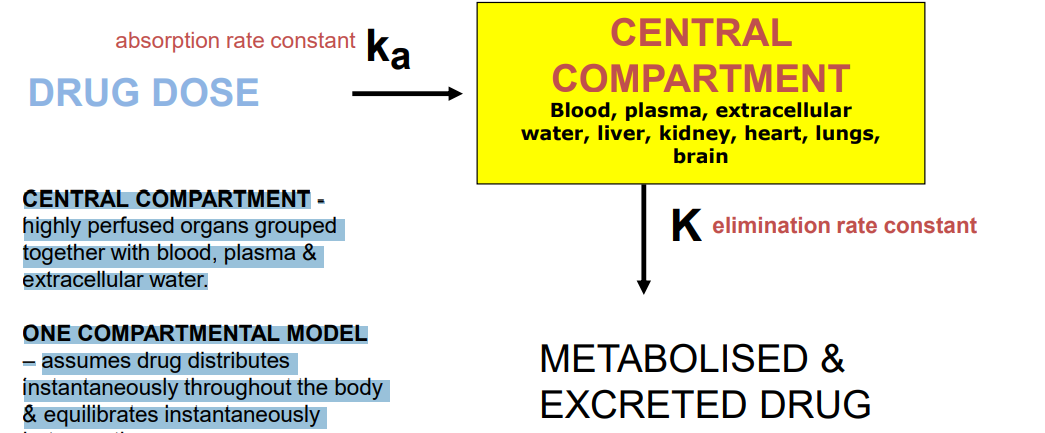
One compartment model
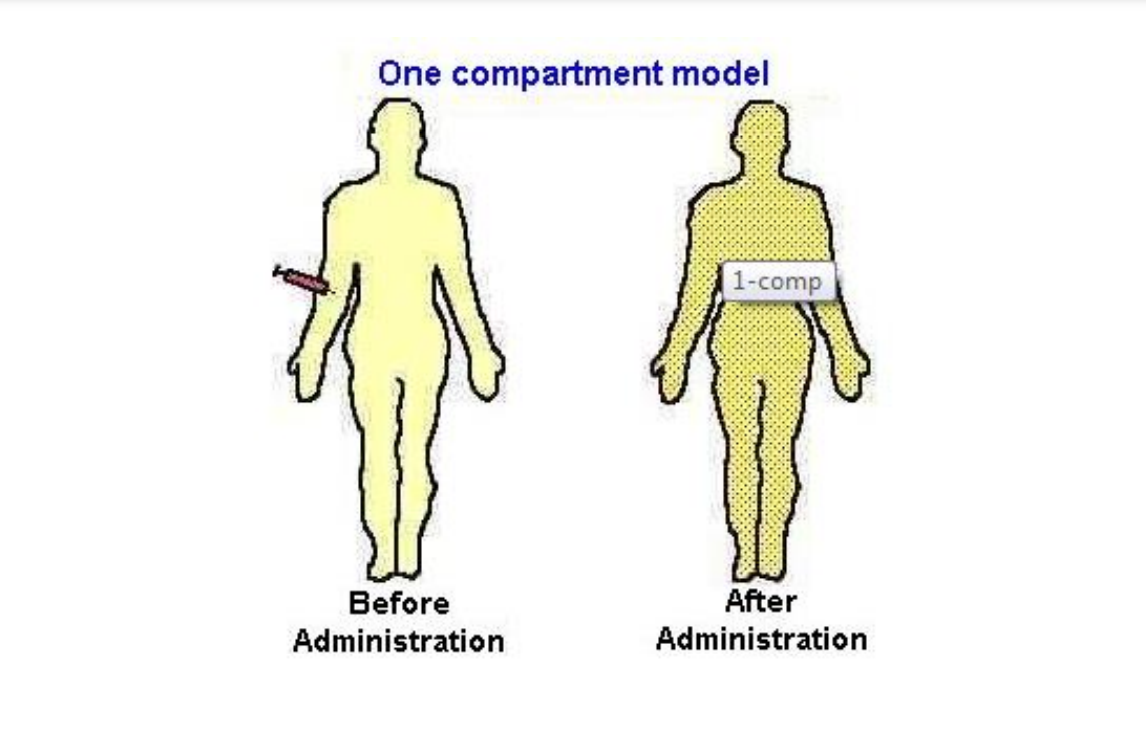
CENTRAL COMPARTMENT - highly perfused organs grouped together with blood, plasma & extracellular water. ONE COMPARTMENTAL MODEL – assumes drug distributes instantaneously throughout the body & equilibrates instantaneously between tissues. - changes in the plasma concentration quantitatively reflect changes in the tissues conc
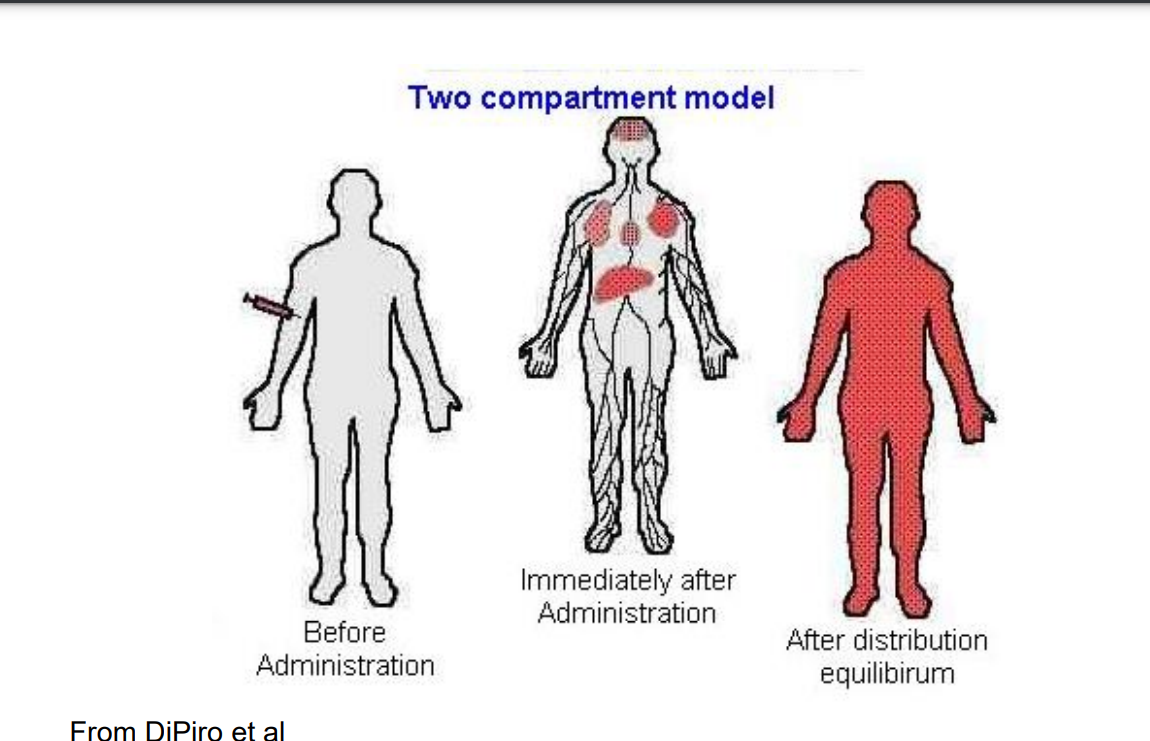
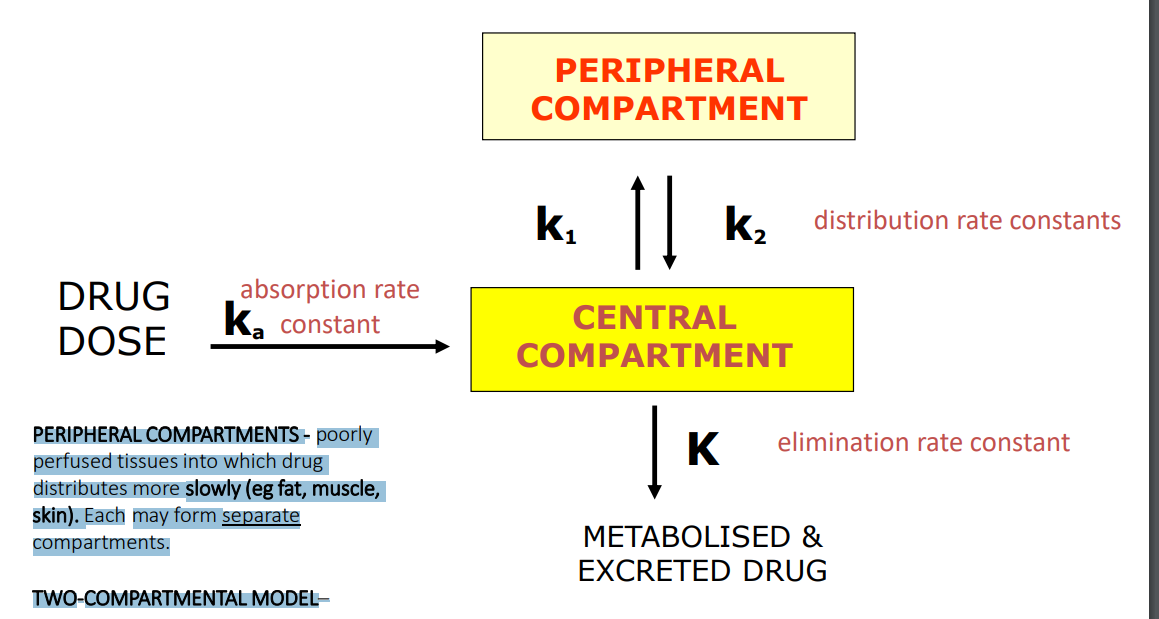
Two-Compartmental Model
PERIPHERAL COMPARTMENTS - poorly perfused tissues into which drug distributes more slowly (eg fat, muscle, skin). Each may form separate compartments. TWO-COMPARTMENTAL MODEL– assumes drug does not achieve instantaneous distribution, i.e. equilibrium, between compartments
Kinetics of drug elimination
To consider the processes of ADME the rates of these processes have to be considered • The rate of a reaction or process is the velocity at which it proceeds • This can be described as either zero-order or first-order
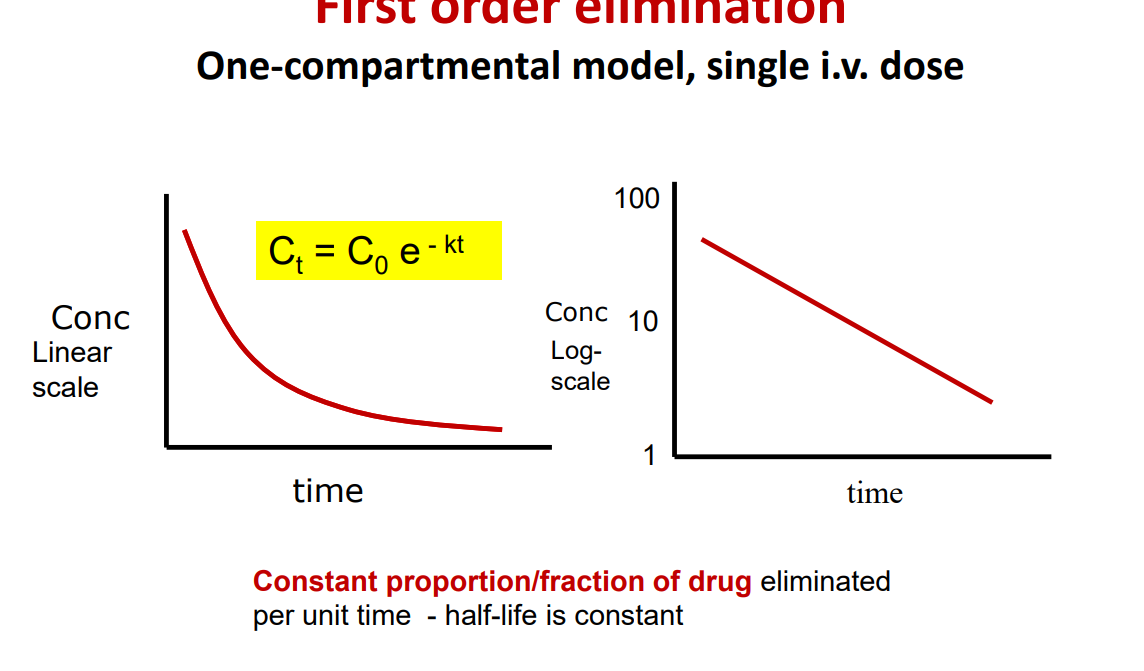
First order elimination
Most drugs at therapeutic doses are eliminated by a first order process • A constant fraction of drug is metabolised per unit of time. • But the amount of drug eliminated in a set amount of time is directly proportional to the amount of drug in the body (ie the dose)
• If you double the dose you will double the plasma concentration
• Accumulation does not occur (at normal doses) • However, if you continue to increase the amount of drug administered then all drugs will change from showing a first-order process to a zero-order process, for example in an overdose situation
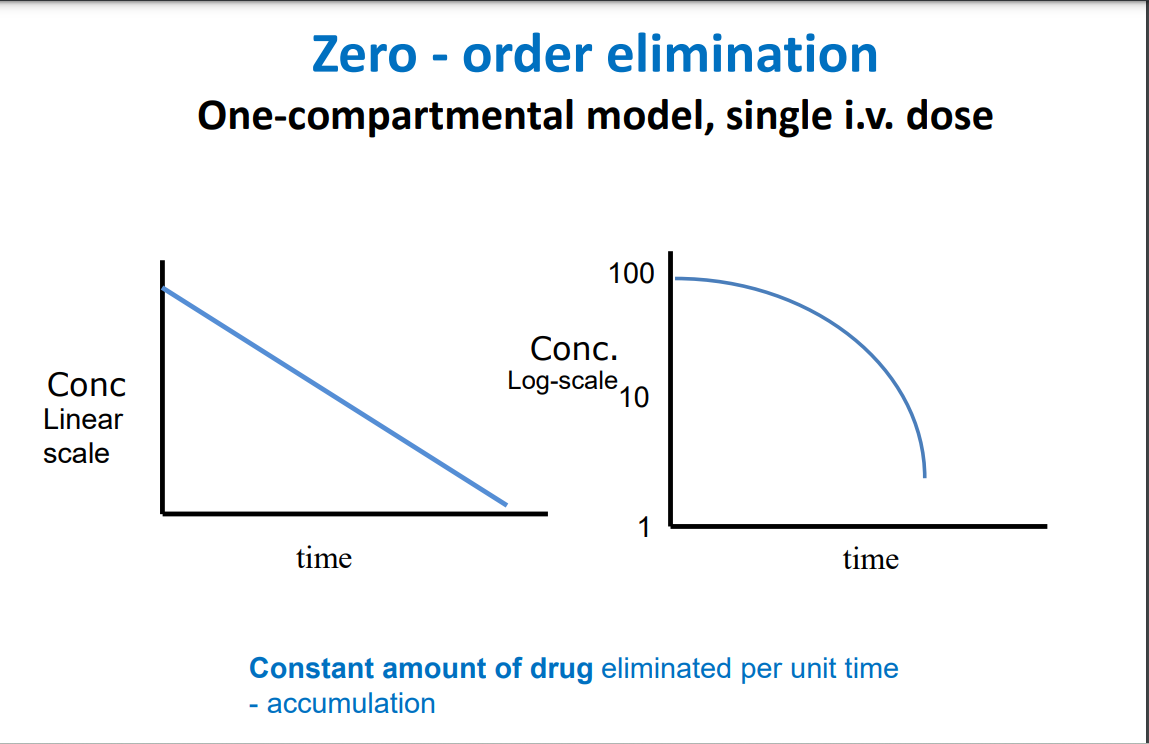
Zero order elimination
Amount of drug eliminated for each time interval is constant, regardless of the amount of drug in the body
• Some drugs at high doses display zero-order elimination • Body’s ability to eliminate drug has reached maximum capacity and accumulation occurs
• An example is the elimination of alcohol • Change in plasma conc with increasing dose is less predictable especially at higher end of dose range
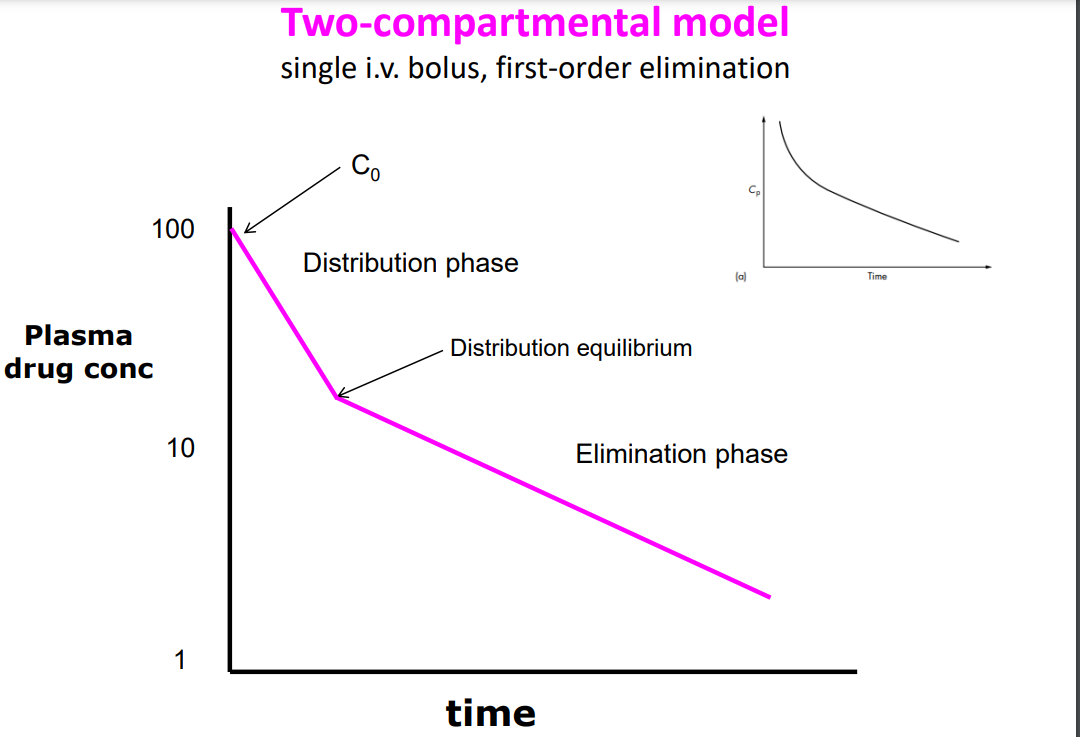
C0 – injection just completed, drug density in central compartment (plasma) is at its highest. Drug distribution and elimination have just begun • Distribution phase - rapid decline in the plasma drug concentration owing to elimination from the central compartment and distribution to the peripheral compartment. Distribution is the main factor determining conc in central compartment • Distribution equilibrium is achieved between the central and peripheral compartments (ie conc approx. equal in both), and main determinant of drug disappearance from central compartment is elimination (rather than distribution) • Elimination phase – drug is being drained from both compartments out of body at around the same rate • The rate processes are described by first-order reactions
Volume of Distribution (V)
Volume of fluid in which total amount of drug in body would need to be distributed to give a concentration equal to the concentration measured in plasma
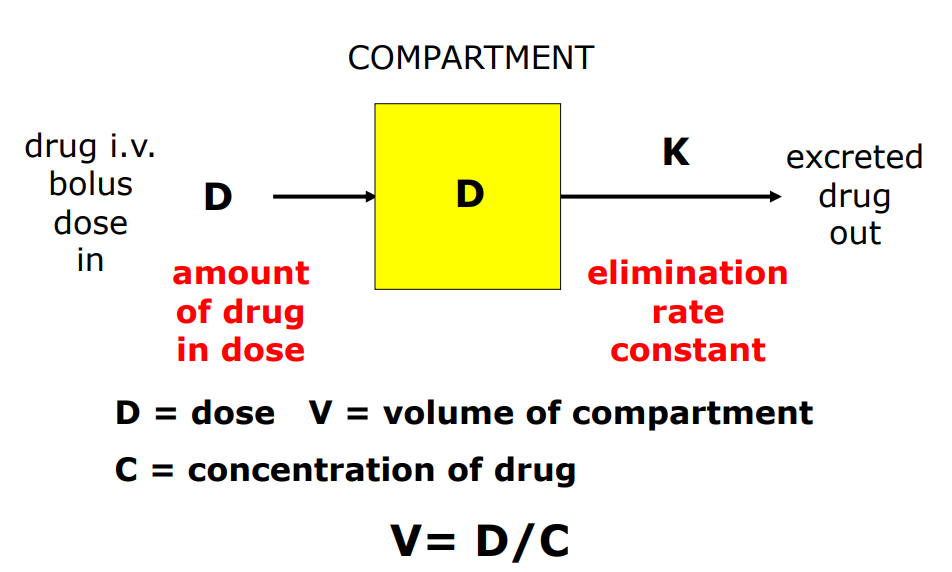
Calculation of V One-Compartment Model i.v.
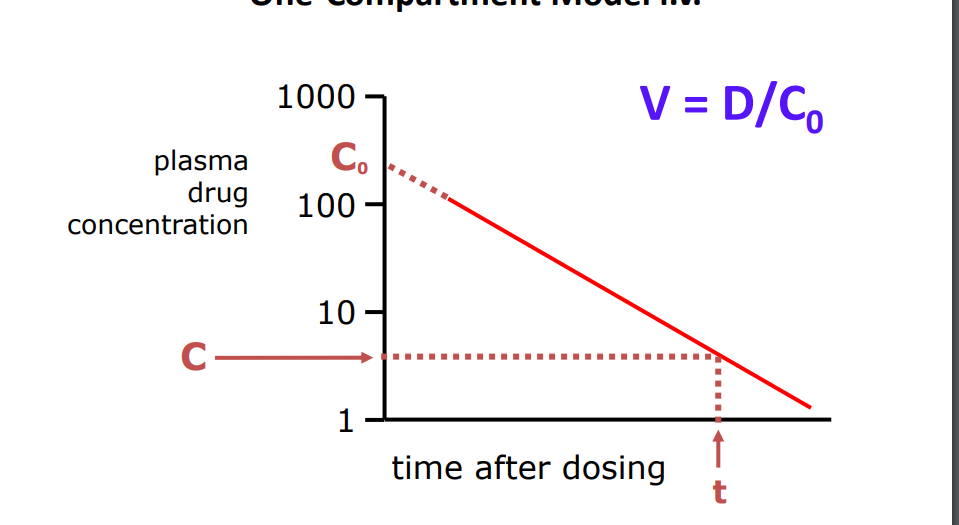
Magnitude of V
Drug in plasma vs keeping drug in tissue:
• Small V – drug held in plasma: water solubility, plasma protein binding, large RMM
• Large V – drug in tissue: lipid solubility, binding to tissue proteins, smaller RMM
• Lowest ~ 3L (plasma volume) • Usually constant for a drug • Units: L (L/Kg)
Use of V
where the drug is distributed in the body
• the physicochemical properties of the drug
• the amount of drug in the body at a particular time, if the conc of the drug is sampled
• calculation of loading dose of a drug
Patient factors affecting V
Liver, renal, cardiac impairment
• Reduced blood perfusion to tissues
• Changes in plasma protein binding
• Odema, ascites
• Old age
• Pregnancy
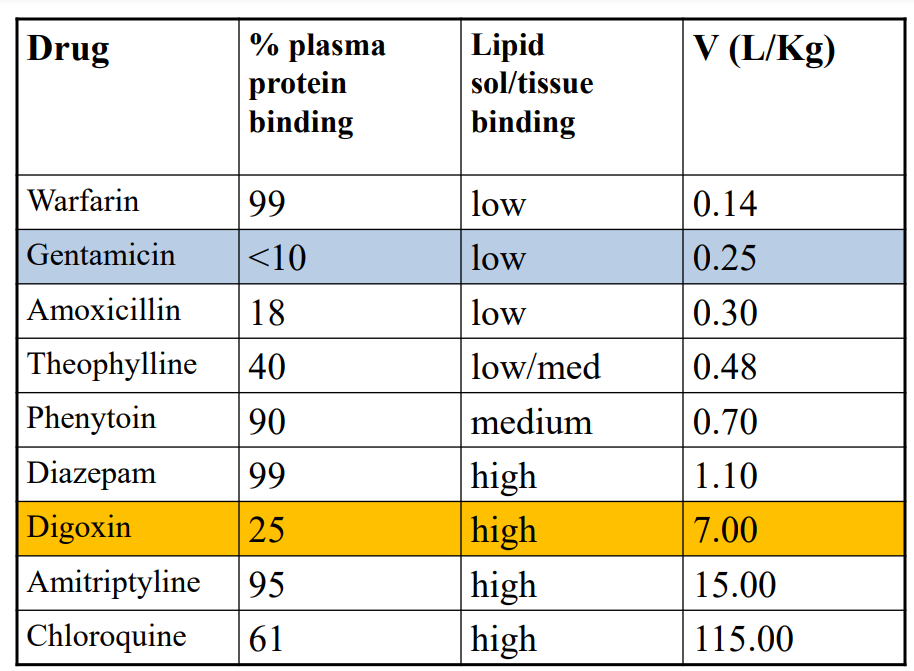
Digoxin (7L/Kg) Large volume of distribution: • Relatively small molecule • With low binding to plasma proteins - mostly albumin, averages 20 to 30% • And high affinity for skeletal & cardiac muscles, intestines and kidney.
Gentamicin (0.25L/kg) Small volume of distribution: • Smaller molecule • Does not bind to plasma proteins • But highly ionised; does not cross cell membranes easily
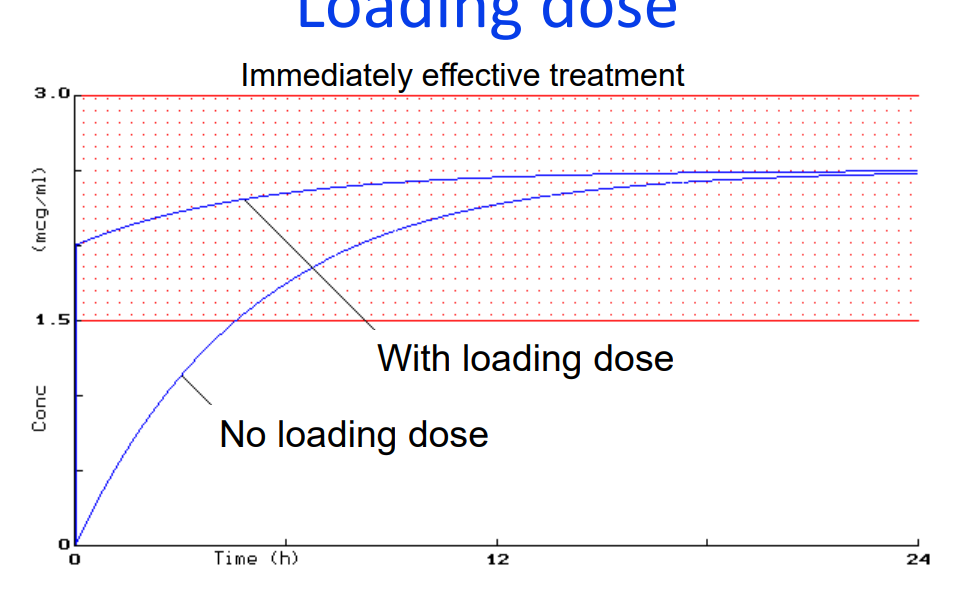
Calculation of Loading Dose (LD)
• What is a “Loading dose?
➢LD is the initial
➢usually higher dose given
➢so that therapeutic concentrations are achieved quickly
In what conditions might a LD be given?
➢ if the drug has a long elimination half life, eg digoxin, amiodarone
➢ in acute conditions e.g status asthmaticus, status epilepticus
➢ in life-threatening arrhythmias, e.g rapid digitalisation.
• Look up the LD of digoxin and amiodarone in the BNF
Calculating loading dose

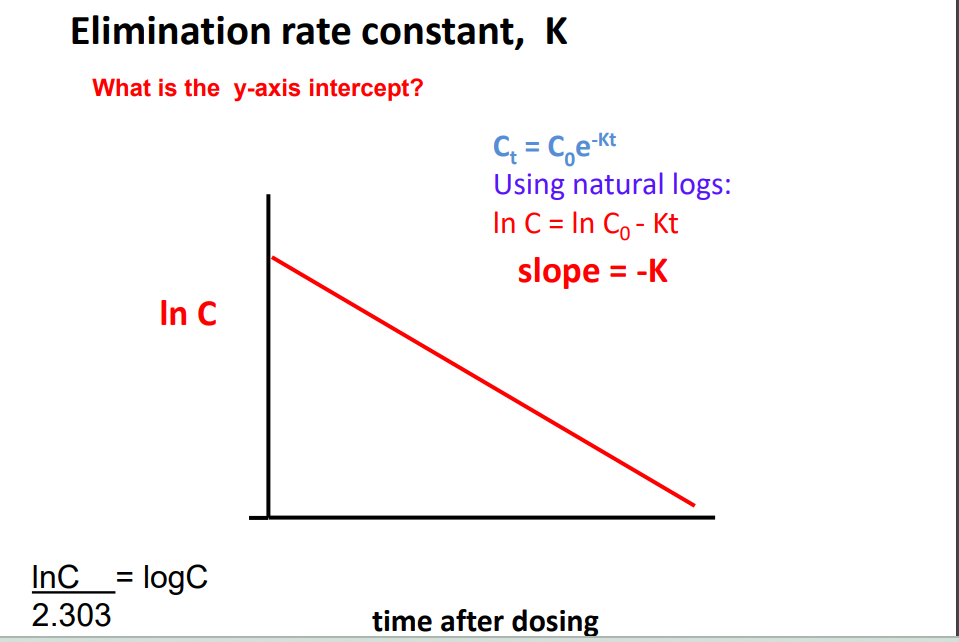
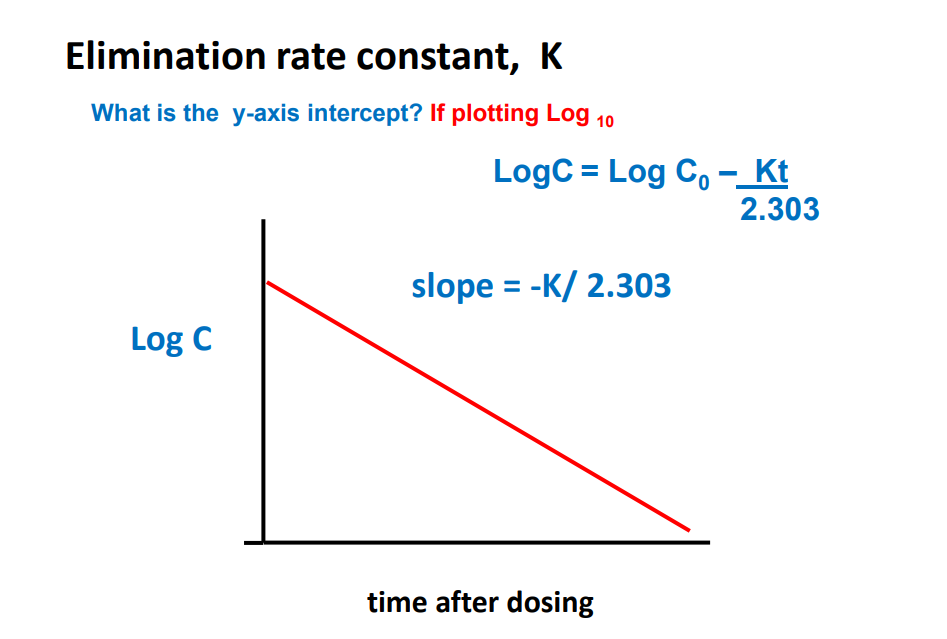
Elimination Rate Constant K
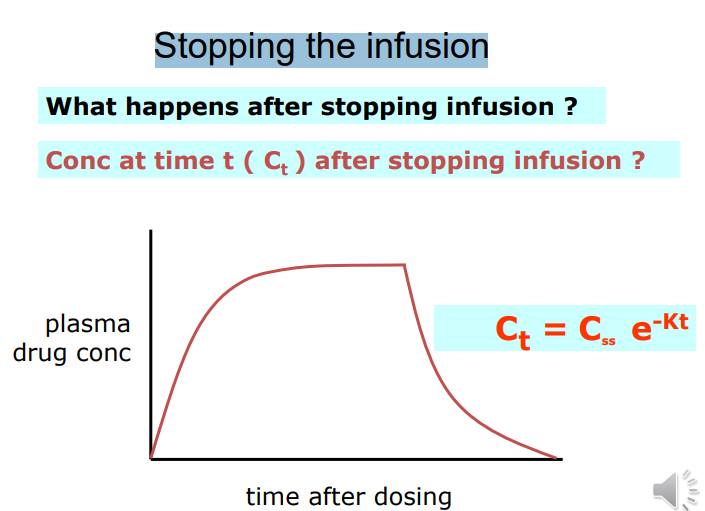
Ct = C0 e -Kt K is the elimination rate constant • K is fraction of drug dose eliminated from compartment per unit time (units = min -1 or h-1 ) • 1 st order elimination • Amount drug eliminated decreases as plasma conc. decreases • But fraction of drug eliminated is constant • Elimination rate = K x A • A = total amount drug in body • e.g. Procainamide, K = 0.25 h-1
Clinical importance of K
Comparing groups of drugs, larger K means faster rate of drug elimination from body
• Drugs with rapid elimination have a short duration of effect after single dose For multiple administrations
: • drugs eliminated rapidly will need to be given more frequently
• drugs eliminated slowly should be given less frequently
Half - life t1/2
t1/2 is time taken for plasma drug conc to fall by half of what it was at beginning of measurement period (Units = mins or h)
• It is another index of rate of drug elimination, along with elimination rate constant (K) and clearance (CL)

Steady state
Rate of drug going in = rate of drug going out
• Steady state is when the amount of drug administered over a dosing interval = amount of drug being eliminated over the same period
• For 1st order elimination, steady state will always be reached after repeated administration at the same dosing interval
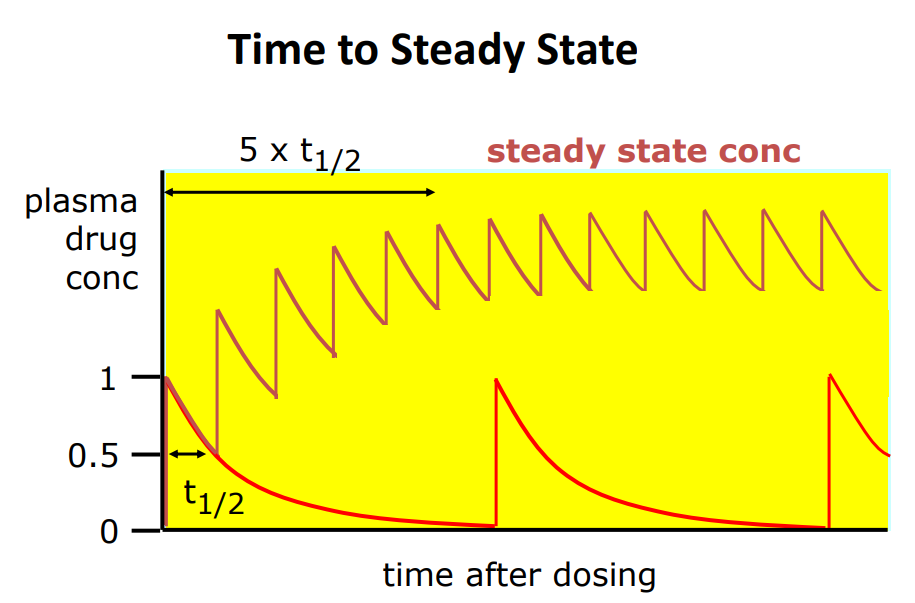
Time to steady state varies from drug to drug
• High K, short half-life, steady state reached quicker than lower K and longer half-life
• eg time to Css (h): aspirin 1.3, digoxin 120-148, vancomycin 25-30, ranitidine 10.5
• Is a loading dose needed? - for a drug with a long half life and if there is a need to reach steady state quickly then yes
Factors affecting Half-Life t1/2
T1/2 = 0.693 x V Cl
• Why does V affect t1/2 ?
• Elimination processes (metabolism & excretion) work only on drug in plasma – large V - little of drug in plasma and more in body – small V - most of drug in plasma
• What about effect of Cl ?
Disease can decrease both V & Cl
Disease can decrease both V & Cl
• Hepatic or renal failure reduces drug clearance
• Because V & CL have opposing effects on t1/2 , a decrease in both might still result in no change in t1/2
• Half-life not good indicator of changes in CL
Proton-pump inhibitors (PPIs)
Esomeprazole, omeprazole, lansoprazole, pantoprazole, rabeprazole
• Inhibit gastric acid secretion by inhibition of the gastric proton pump - very effective Short elimination half lives ~ 1h ➢ quick onset of action ➢ taken ~30 mins before breakfast - so peak blood concs coincide with maximal gastric pump activation ➢ only about ~ 70% of proton pump enzymes are inhibited ➢ ~ 66% inhibition of maximal acid output (o.d admin) ➢ effects last about 24h - pump inhibition is permanent
Takes ~2-3 days to achieve steady state inhibition of acid secretion • Increasing the dose doesn’t have much further effect once optimal dose is reached • However, increasing frequency of administration (b.d.) increases effect to ~ 80% inhibition of maximal acid output • Bedtime dose doesn’t add to nocturnal acid breakthrough • To improve acid inhibition - new PPIs with longer plasma half-lives
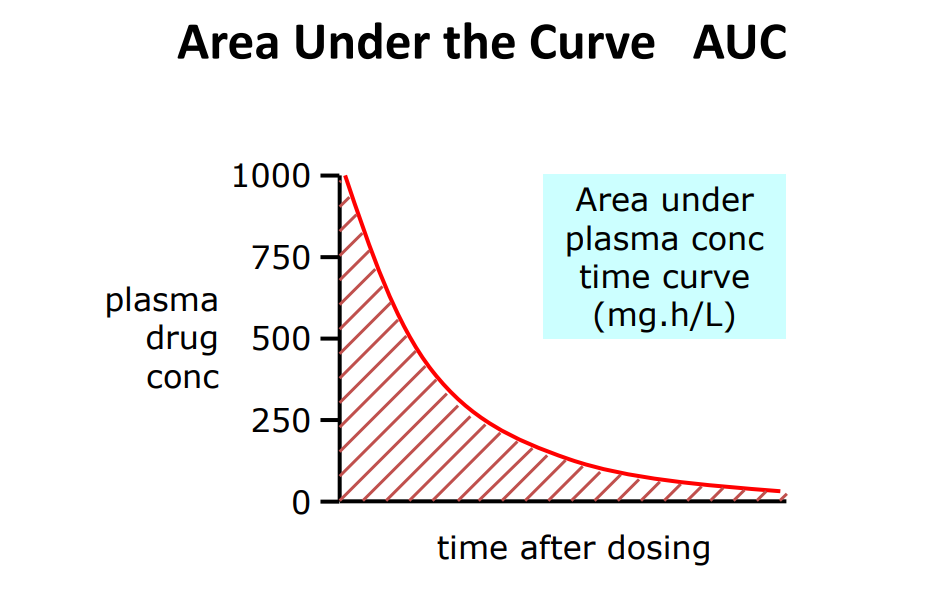
AUC
A measure of drug exposure
• AUC gives a measure how much & for how long a drug stays in a body
• AUC is used extensively in the calculation of bioavailability of different dosage forms
• AUC values can be used to determine other pharmacokinetic parameters, such as Clearance.
One way of calculating: • AUC = C0/K Assumes one-compartmental model, 1st order elimination and i.v. admin
AUC = Dose/CL • CL = Dose/ AUC • Model-independent • Units mg.hr/L • Other ways
Bioavailability
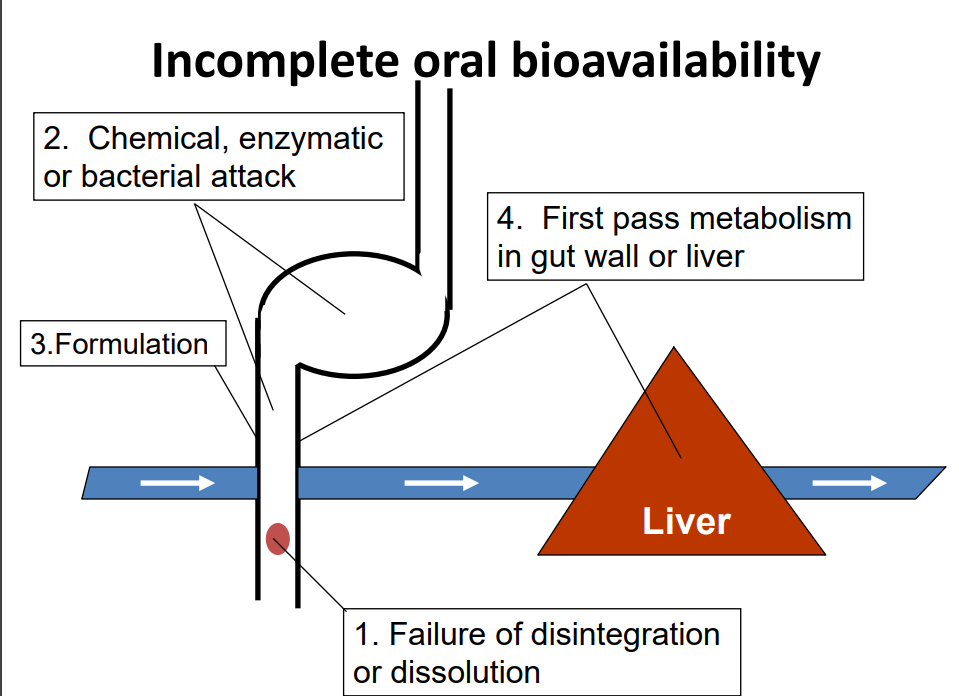
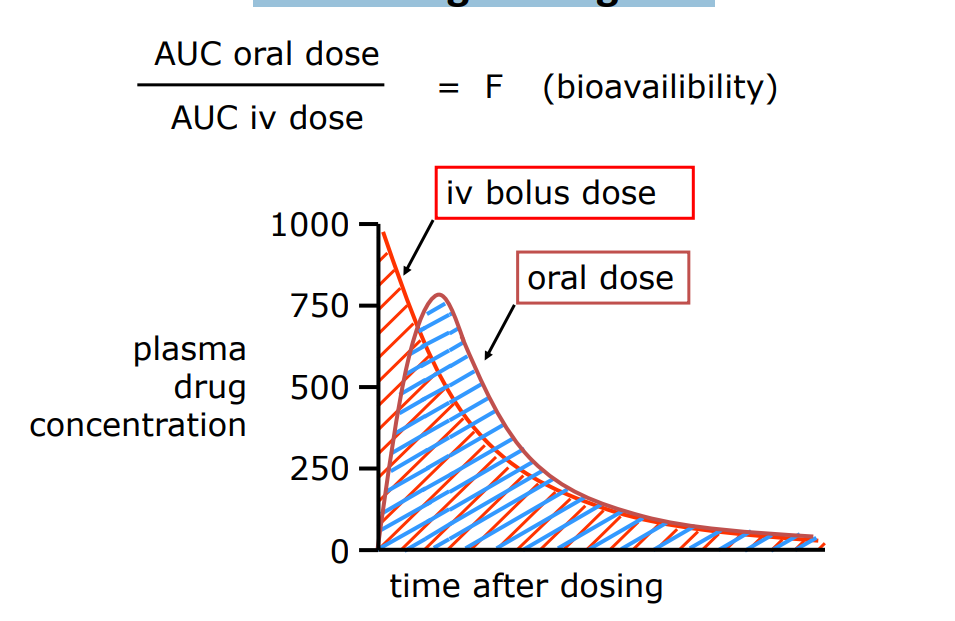
Bioavailability is the fraction of dose reaching the systemic circulation in a chemically unaltered form. No units
Calculating F using AUC
Clearance
Removal of drug from a volume of plasma in a given unit of time (units = L h-1 or ml min-1 ) • Model-independent parameter • CLt = CLr + CLm + Cb + CLother • Most drugs eliminated by combination of hepatic clearance (metabolism) and renal clearance (excretion without metabolism)
For 1 st order elimination, as plasma conc. falls, rate of elimination also falls • At steady state: Rate administration = Rate of elimination Rate of elimination = CL x C CL = Rate of elimination C
Factors altering clearance
Body weight • BSA • Cardiac output • Drug-drug interactions • Extraction Ratio • Genetics • Hepatic and renal function • Plasma protein binding
With 1st order elimination process, drug clearance remains constant at all doses. Rate of elimination = CL x C Rate of elimination = K X A And C = A/V Therefore CL = K x V A = total amount of drug in the body
Decrease in drug clearance means slower rate of elimination • Dosage adjustment may be required in eliminating organ dysfunction
Estimating Renal Clearance
Elimination of drug by renal excretion • Declines with age
• Impaired renal function/CKD
• Reduced excretion
• Nephrotoxic drugs
• Dosage adjustment
Estimating renal function: eGFR useful for: • assessing severity of Chronic Kidney Disease (CKD).
• initial assessment of change in renal function.
Creatinine clearance:
• Cockcroft & Gault formula
• estimates renal function
• dosage adjustment
Creatinine Clearance
CrCl = F x (140 – age(yr) x weight (kg)/ Serum creatinine (μmol/L)
F = 1.04 (females) or 1.23 (males)

Dosage adjustment in renal impairment
Loading dose – adjustment generally unnecessary as accumulation is unlikely after one dose
• Maintenance dose – reduce dose/or increase time between doses
• If half-life is prolonged then time to steady state will also be increased •
Hepatic clearance
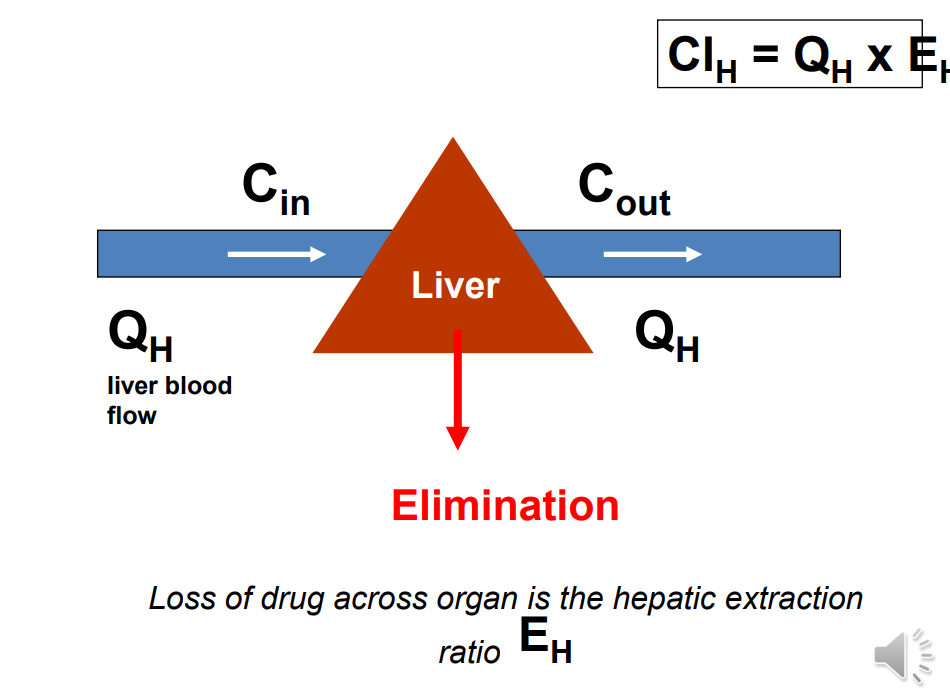
Hepatic Extraction Ratio ( EH )
Fraction of drug removed from blood during one pass through the liver. • EH = Cin – Cout/ Cin
EH is determined by: • Hepatic blood flow • Plasma protein binding of drug • Intrinsic clearance
First pass effect
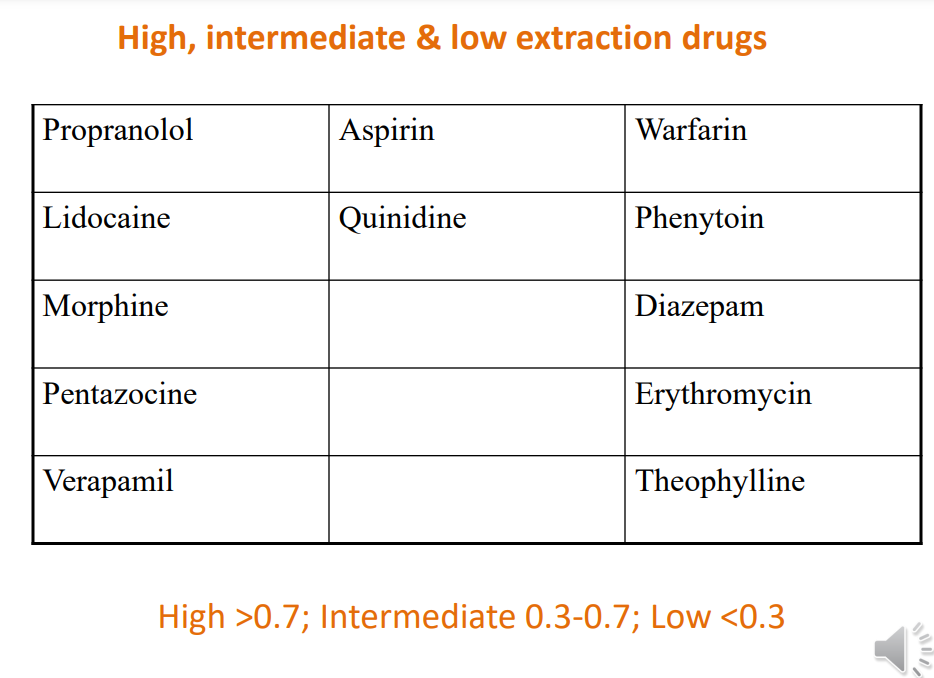
High EH drugs, e.g. propranolol (EH = > 0.9) • After oral administration • Most of the drug is removed by one pass through liver • i.e. significant amount of drug is metabolized before reaching systemic circulation • So amount of drug reaching the systemic circulation is considerably less than the dose given • See BNF – e.g. propranolol, lidocaine, diazepam
Some consequences of EH values

Assessing liver impairment
Liver function tests (LFTs)
• ALT and AST levels (aminotransferases)
• Alkaline phosphatase (ALP)
• Gamma GT (GGT), Bilirubin, Albumin
• Clotting studies
• Indicators of liver disease
• Multifactorial nature of hepatic clearance makes finding direct relationships between impairment and changes in PK is more challenging
• Dosage adjustment
Continuous Intravenous Infusion
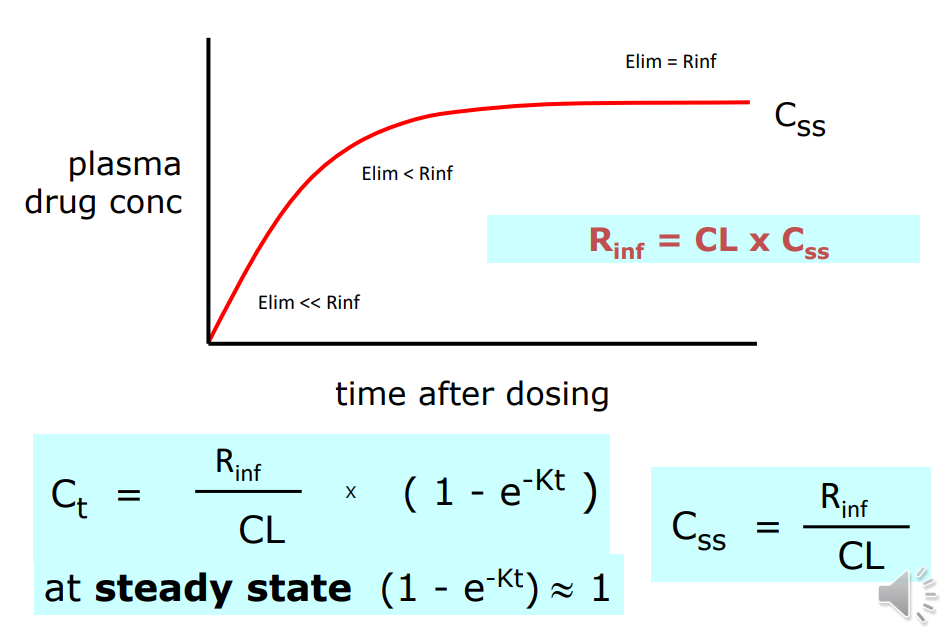
Predicting Css

stopping infusion