VI - Protein Purification & Characterization
1/45
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
46 Terms
General steps to study a protein
separation of protein of interest from: the proteome (entire set of expressed proteins), all other biomolecules
purification
characterization
Methods to disrupt a cell
Method used depends on sensitivity of cells/protein
repeated freezing/thawing; sonication; homogenization by high pressure (French press); homogenization by grinding (beads); permeabilization by detergents
Differential centrifugation
Particles separate by mass. Homogenate centrifuged at diff speeds to give a series of pellets each containing diff cellular materials and supernatant of soluble proteins
✧ heavy material pelleted before lighter material
✧ all fractions must be assayed to find where protein of interest is
Density gradient centrifugation
Sucrose density gradient with least dense at the top. Sample is carefully layered on top. Based on their densities, proteins migrate until density matches density gradient. Fractions are collected by a pinhole in the tube
✧ related to each protein’s sedimentation coefficient (how far it moves in a gradient)
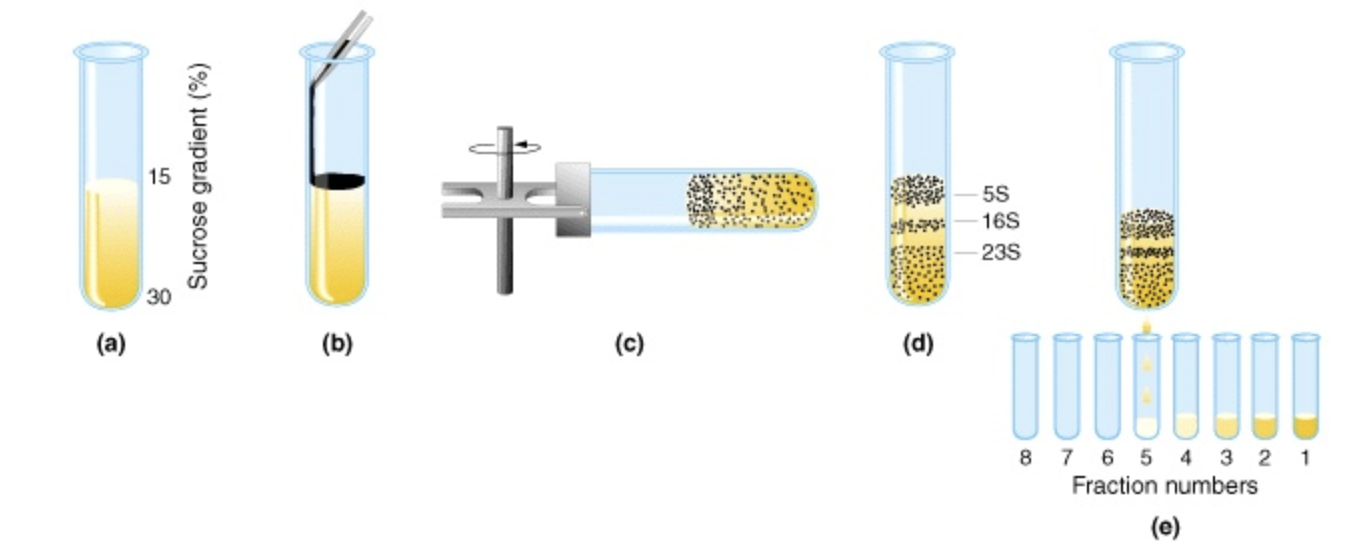
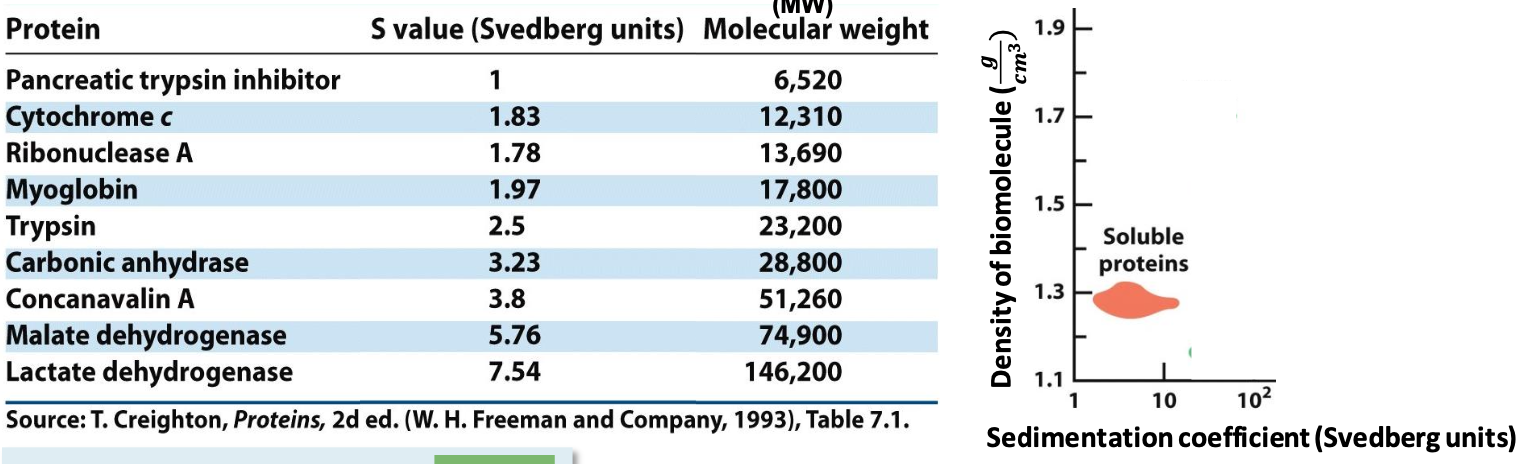
Sedimentation coefficient (S)
Soluble proteins have specific S value (∼2-20 Svedberg).
✦ the smaller the value, the less it moves in the gradients
✦ more massive proteins sediment faster than less massive ones of the same size/density

What properties can you use to separate proteins?
solubility (‘salting out’ w/ (NH₄)₂SO₄)
size (dialysis, gel filtration chromatography/size exclusion chromatography)
charge (ion exchange chromatography)
specific binding affinity
hydrophobicity
Salting out or (NH₄)₂SO₄ precipitation
Salting out: fractionation technique based on lowered solubility of some molecules at high [salt]. Most proteins aren’t soluble in high salt
Salting in: increase in protein solubility with the addition of low [salt]. Salt competes for intermolecular electrostatic interactions
✦ H₂O solvates salt instead of protein, non polar areas of protein interact with nonpolar area on another
✦ centrifugation to aggregate and dialysis to remove excess salt
![<p><strong><u>Salting out</u></strong>: fractionation technique based on lowered solubility of some molecules at high [salt]. Most proteins aren’t soluble in high salt</p><p><strong><u>Salting in</u></strong>: increase in protein solubility with the addition of low [salt]. Salt competes for intermolecular electrostatic interactions</p><p>✦ H₂O solvates salt instead of protein, non polar areas of protein interact with nonpolar area on another</p><p>✦ centrifugation to aggregate and dialysis to remove excess salt</p>](https://knowt-user-attachments.s3.amazonaws.com/b53f9e4f-6c7f-4d61-8bae-5edcefaa48c9.png)
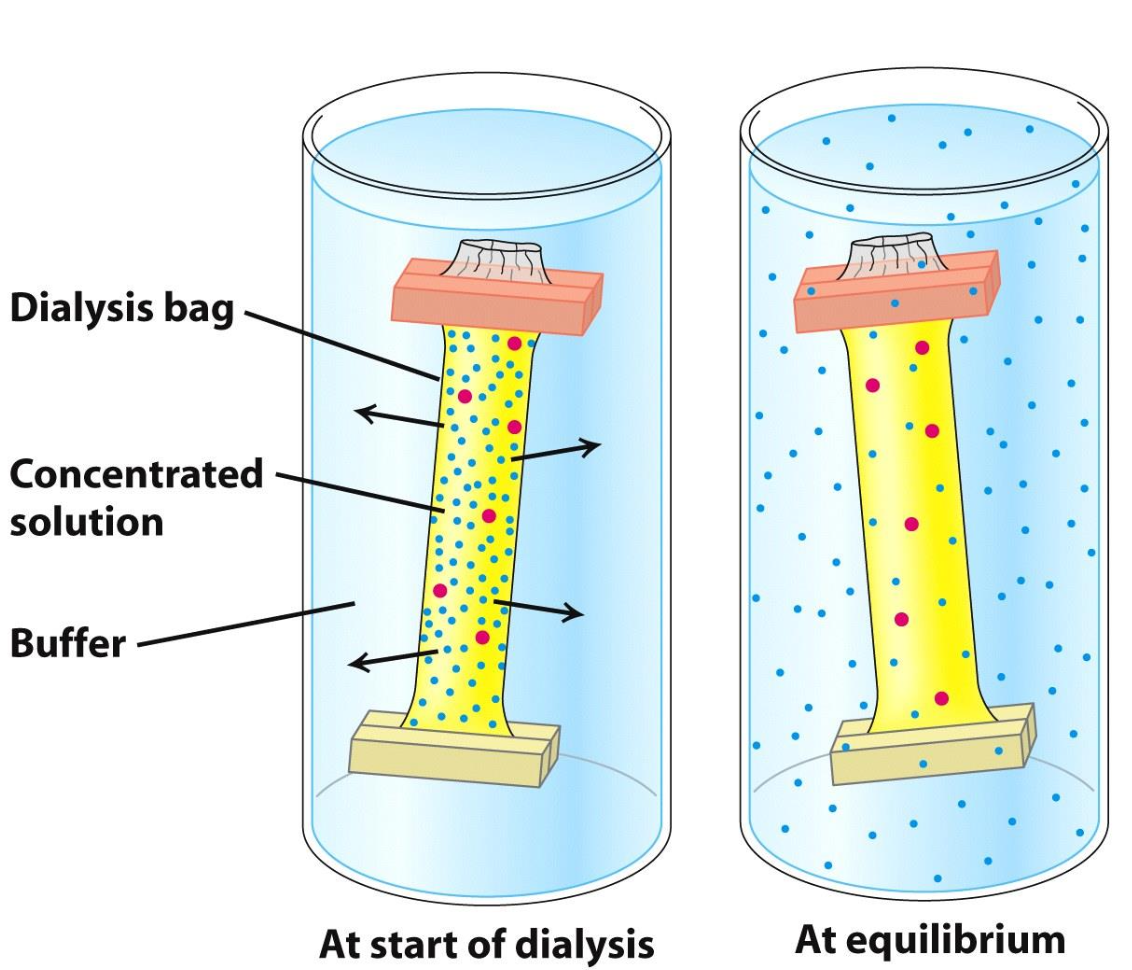
Dialysis
Protein solution is placed in a bag w/ pores allowing protein to diffuse and salt to equilibrate w/ surrounding solution
✦ molecules smaller than pores of bag move from high concentration (in bag) to an area of low concentration (surrounding sol)
✦ blue molecules found in = concentrations inside and out
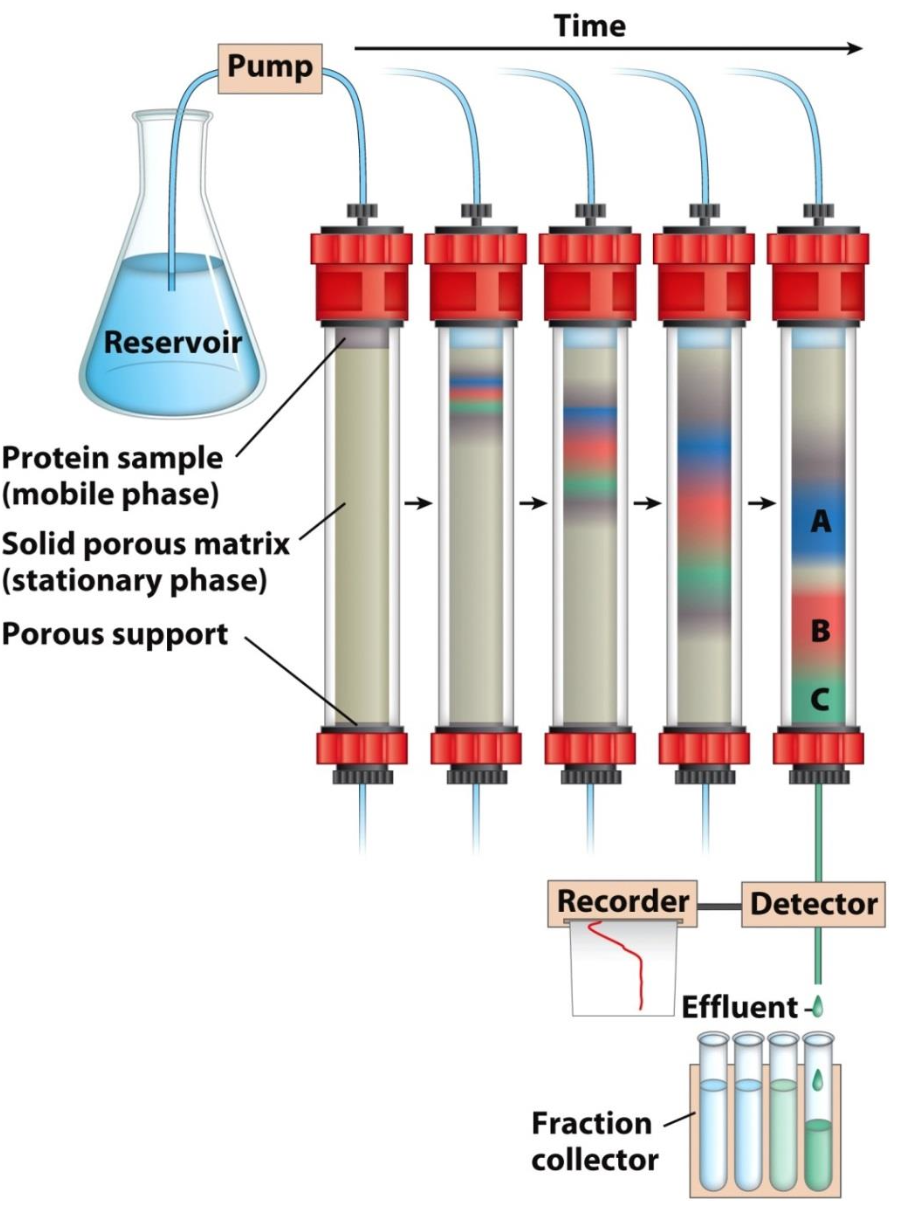
Principles of column chromatography
Contains solid stationary phase (matrix), mobile phase (flows through the matrix), and sample in the native state
✧ As sample migrates thru matrix, proteins slowed down to different degrees because diff interactions
✧ More interactions w/ matrix = slower migration
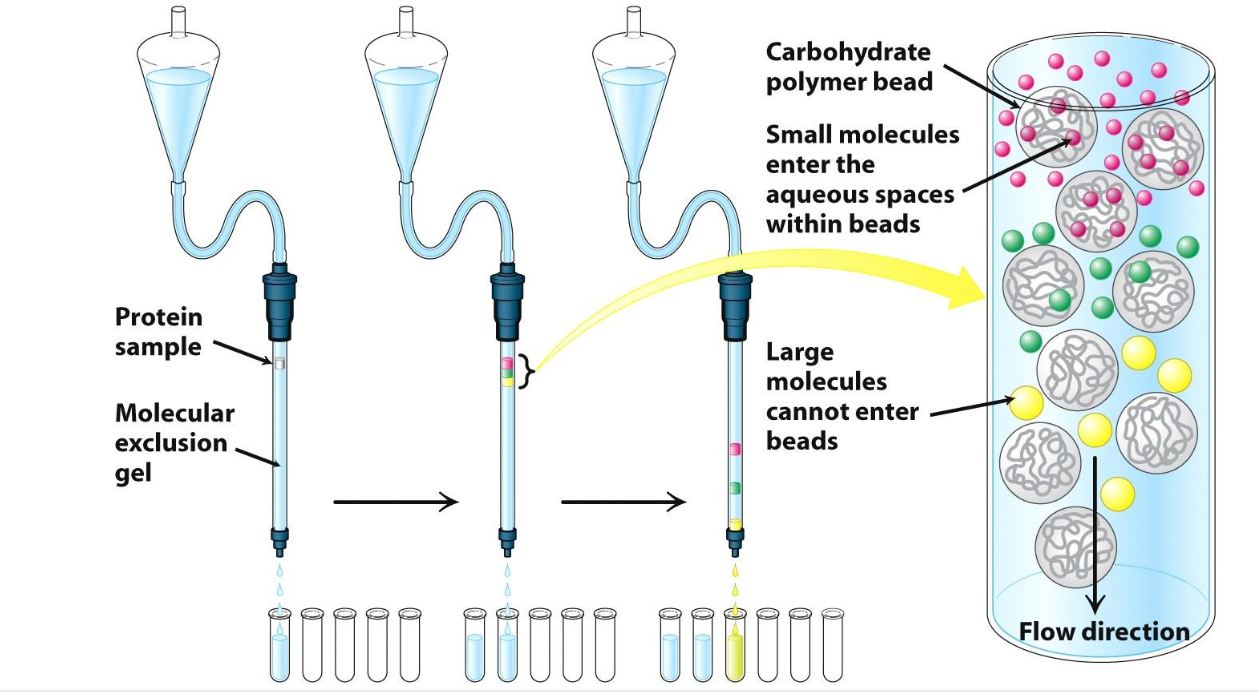
Gel filtration/Size exclusion chromatography
Separated based size
Protein that is larger than the beads’ pores = flows quickly thru the column
Protein that is small than the beads’ pores = flows thru the beads and exits column last
Ion exchange chromatography (Elution)
Separates proteins by charge
Proteins with the same charge of the beads flow quickly thru column.
The higher the opposite net charge, the tighter the binding = the slower the elution
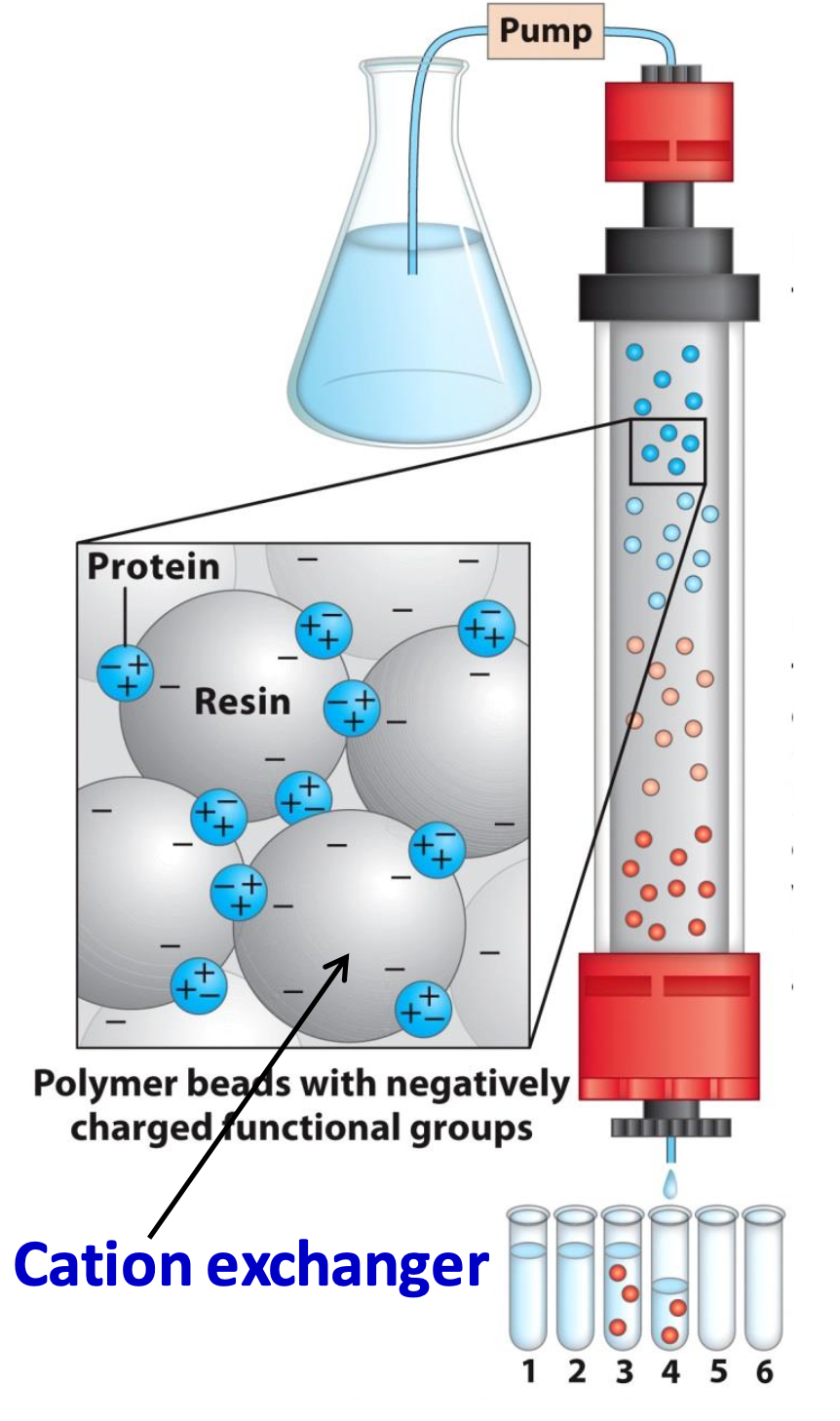
Cation exchanger
Binds only net +ve proteins (opposite for anion exchangers)
✦ increasing [salt] separates the tightly bound proteins. those that need higher [salt] elute last
✦ for cation exchangers, increasing pH = decrease of positive charge on the protein
![<p>Binds only net +ve proteins (opposite for anion exchangers)</p><p>✦ increasing [salt] separates the tightly bound proteins. those that need higher [salt] elute last</p><p>✦ for cation exchangers, <strong>increasing pH </strong>= decrease of positive charge on the protein</p>](https://knowt-user-attachments.s3.amazonaws.com/50f25e0e-4d8a-4f8c-89ed-9dd87daac406.png)
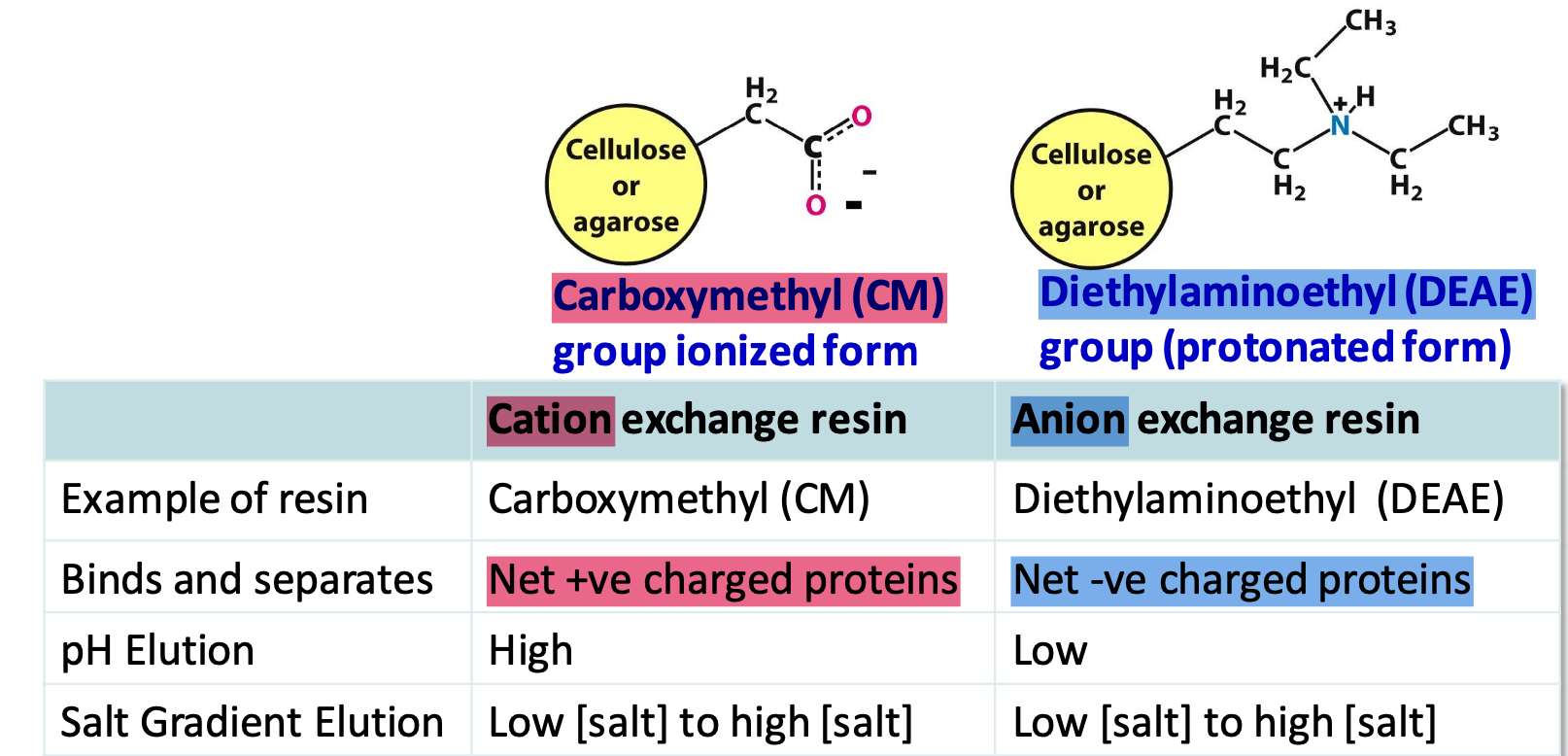
Ion exchange resins
Carboxymethyl (CM) cation and Diethylaminoethyl (DEAE) anion
✦ pH < pI → net +ve charge (binds to cation exchangers)
✦ buffers can control charge on protein and elution
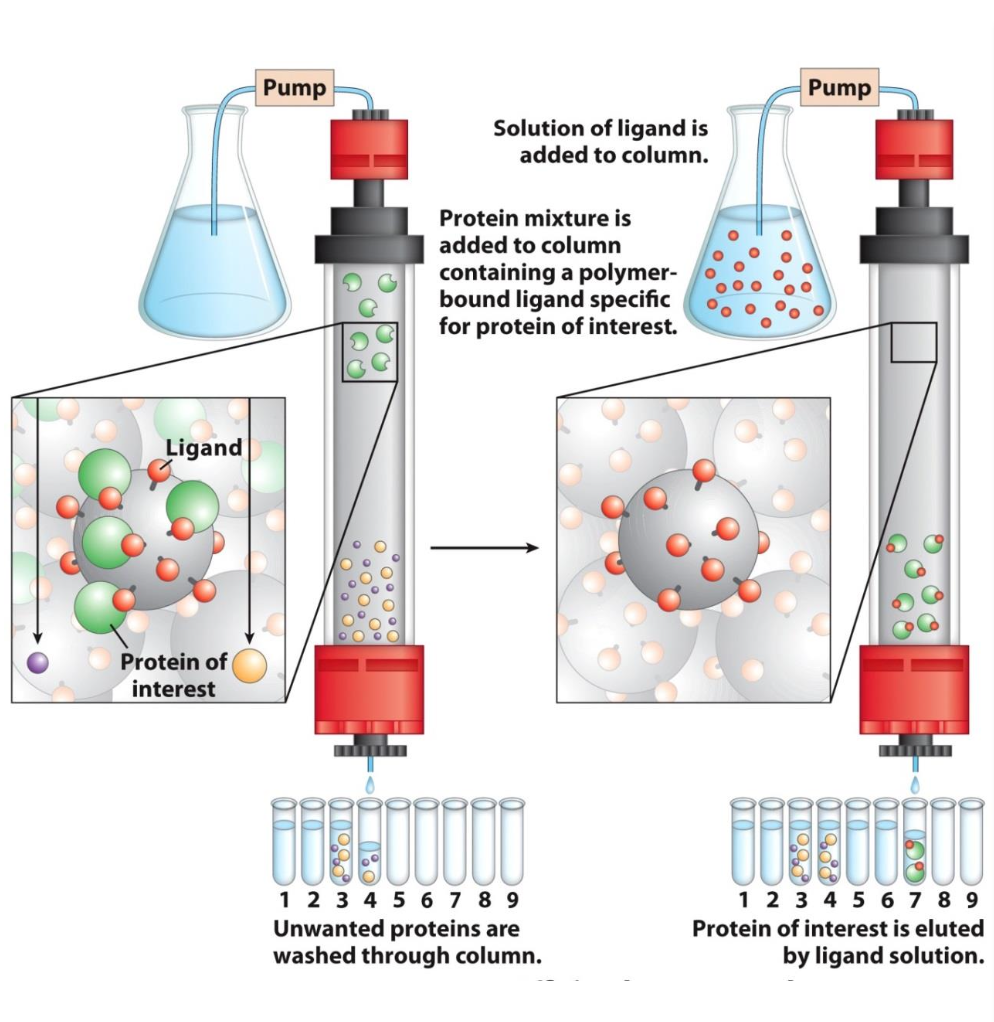
Affinity chromatography
Separate based on specific binding interactions
✦ beads have a proteins’ specific ligand covalently attached to them.
✦ only proteins w/ ligand affinity will be retained in column = elutes last
✦ excess ligand being passed thru the column will release bound proteins
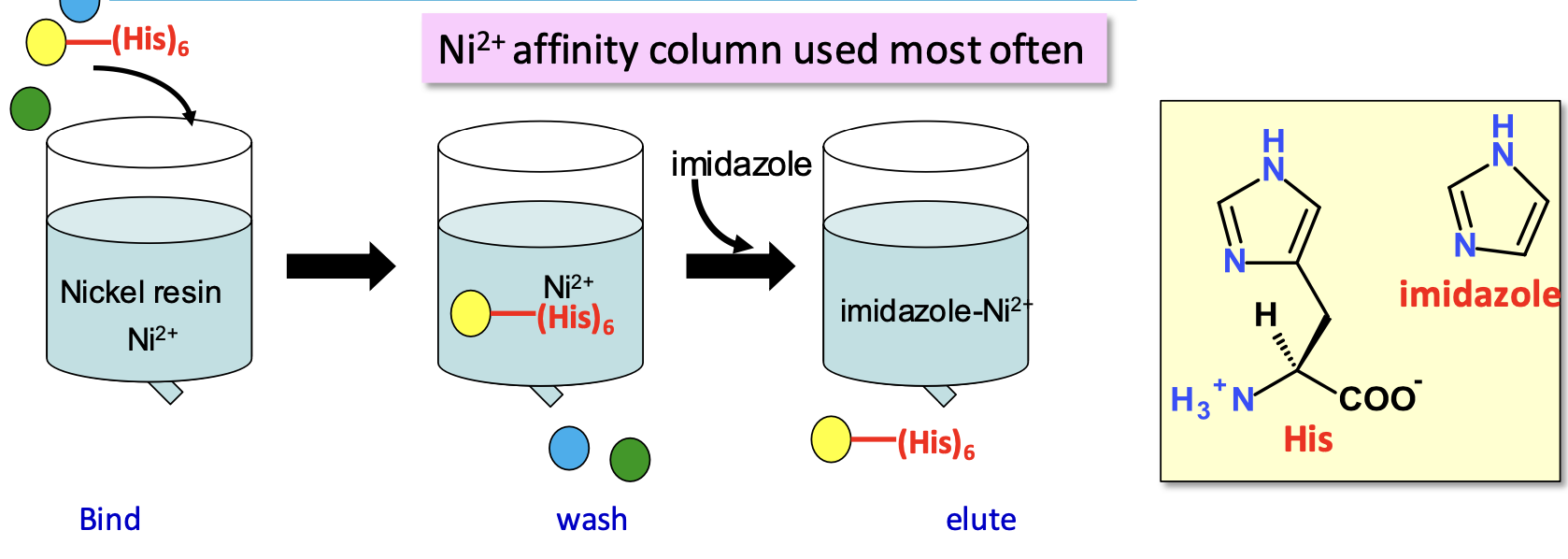
Separation by affinity (His tags)
Adding a tag onto your gene of interest at either N or C terminal
✦ Ni resins purify cloned proteins w/ poly 6 x His tag: the column has Ni (covalently bound to resin) which chelates His tag. Imidazole (has high Ni²⁺ affinity) elutes protein
Hydrophobic interaction chromatography
Commonly a C18 column (carbon chain attached to silica). A protein w/ exposed hydrophobic groups will bind to hydrophobic column. Elution with increasing hydrophobicity
✦ high [salt]: H₂O solvates salt leaving protein’s phobic areas exposed → stronger interactions
✦ protein eluted when [salt] decreases (resolvates protein)
![<p>Commonly a C18 column (carbon chain attached to silica). A protein w/ exposed hydrophobic groups will bind to hydrophobic column.<strong> Elution with increasing hydrophobicity</strong></p><p>✦<strong> high [salt]</strong>: H₂O solvates salt leaving protein’s phobic areas exposed → <strong>stronger interactions </strong></p><p>✦ protein eluted when [salt] decreases (resolvates protein)</p>](https://knowt-user-attachments.s3.amazonaws.com/ef5da124-44c2-43e7-90fa-fac089147015.png)
High-Pressure Liquid Chromatography (HPLC) and Fast Protein Liquid Chromatography (FPLC)
HPLC used for small molecules and on finer packed columns which provide more sites of interaction for molecule
✦ Greater resolving power but slower flow rates w/out applied pressure
FPLC adapted for proteins, uses buffers as a mobile phase
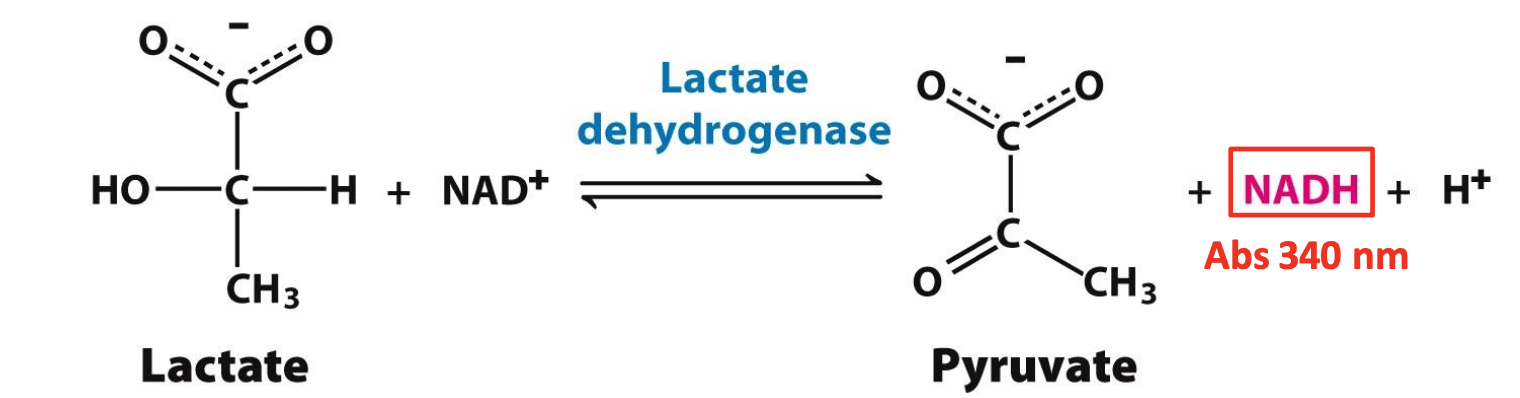
Working assay
Used to identify fractions collected & find protein amongst them. Needs to monitor a unique property of the protein.
✧ LDH can be verified by measuring enzymatic activity = generation of NADH during conversion, NADH has a specific absorbance of 340 nm. The more ∆Abs/t, the more NADH/t
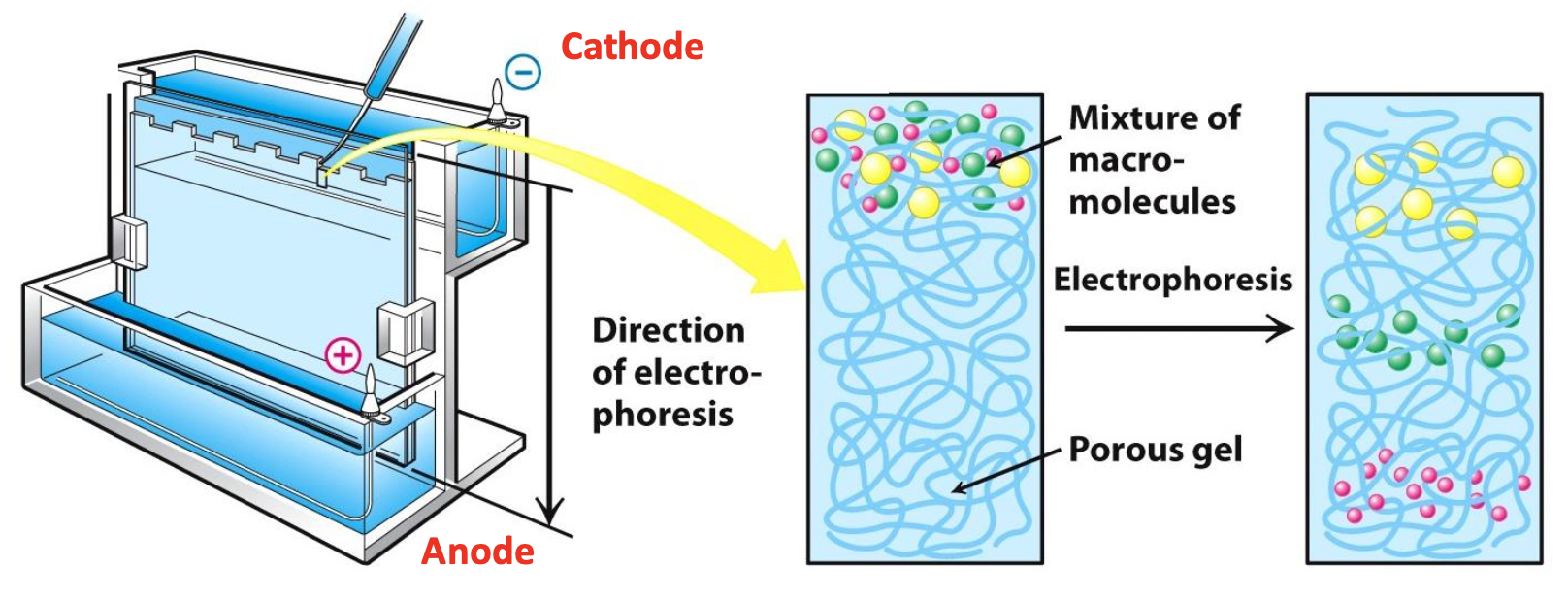
Gel electrophoresis
Migration of a molecule w/ a net charge in an electric field (on gel)

Polyacrylamide gel & sample preparation
Gel is a 3D mesh generated from acrylamide monomers cross-linked w/ methylenebisacrylamide
Disulfides are reduced w/ 𝛽-ME or DTT → S-S exchange
Protein is denatured with SDS detergent → disrupts almost all non-covalent interactions
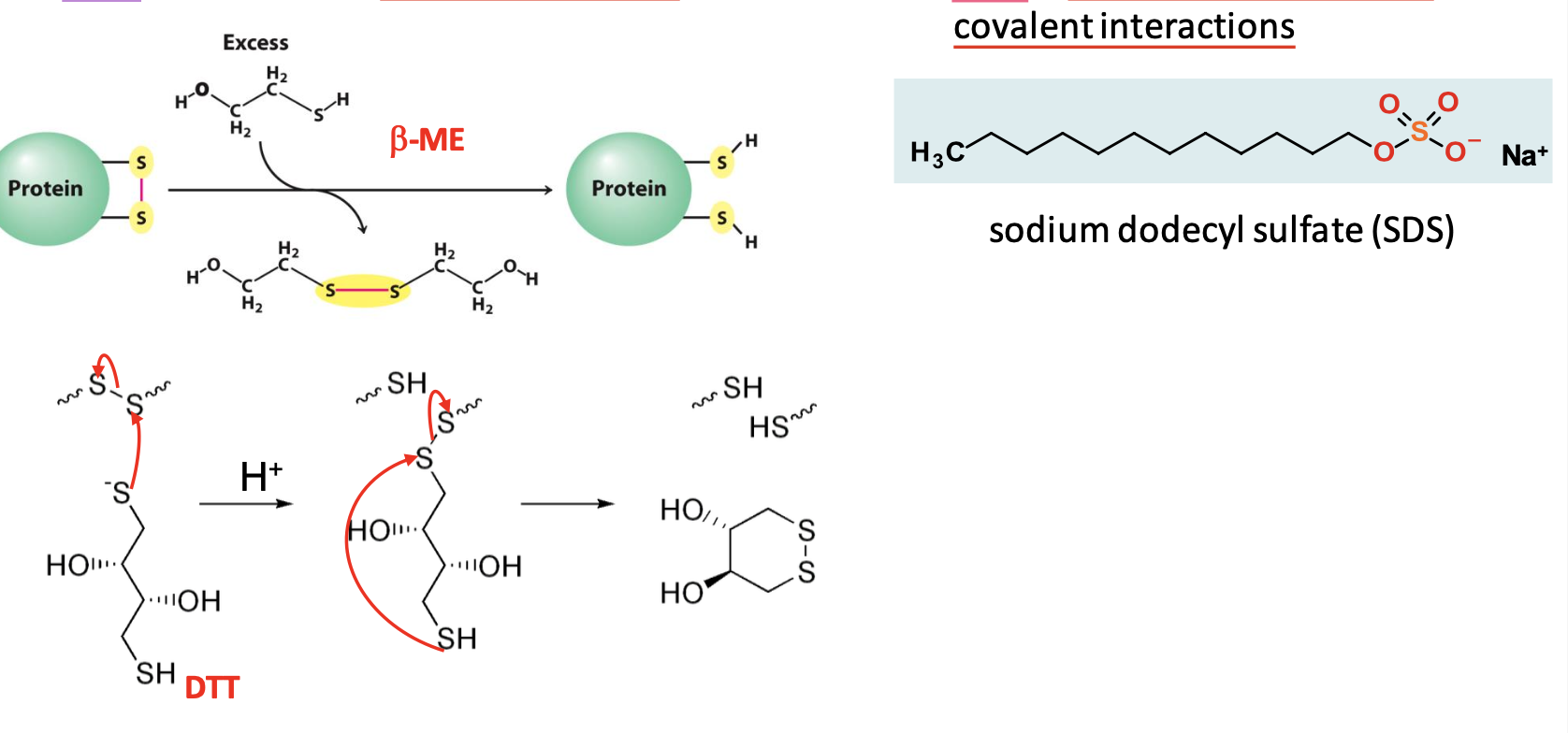
Effect of Sodium Dodecyl Sulfate (SDS)
One mole of SDS binds for every 2 AAs. An excess of SDS = a net -ve charge.
Net charge is now proportional to mass of denatured protein = SDS standardizes charge, density, shape of proteins
SDS Polyacrylamide Gel Electrophoresis (SDS-PAGE)
Sample is loaded into wells at the top of the plate. Electrical voltage causes -ve proteins to migrate towards +ve electrode
✧ The size of protein relative to size of the pores in gel determines mobility
✧ Smaller proteins migrate farther into the gel
Coommassie-blue staining
Used to help visualize proteins after electrophoresis. It binds to the protein itself, not the gel. It can detect 0.1 µg of a single chain protein.
✧ Protein of interest gets enriched w/ each purification technique.
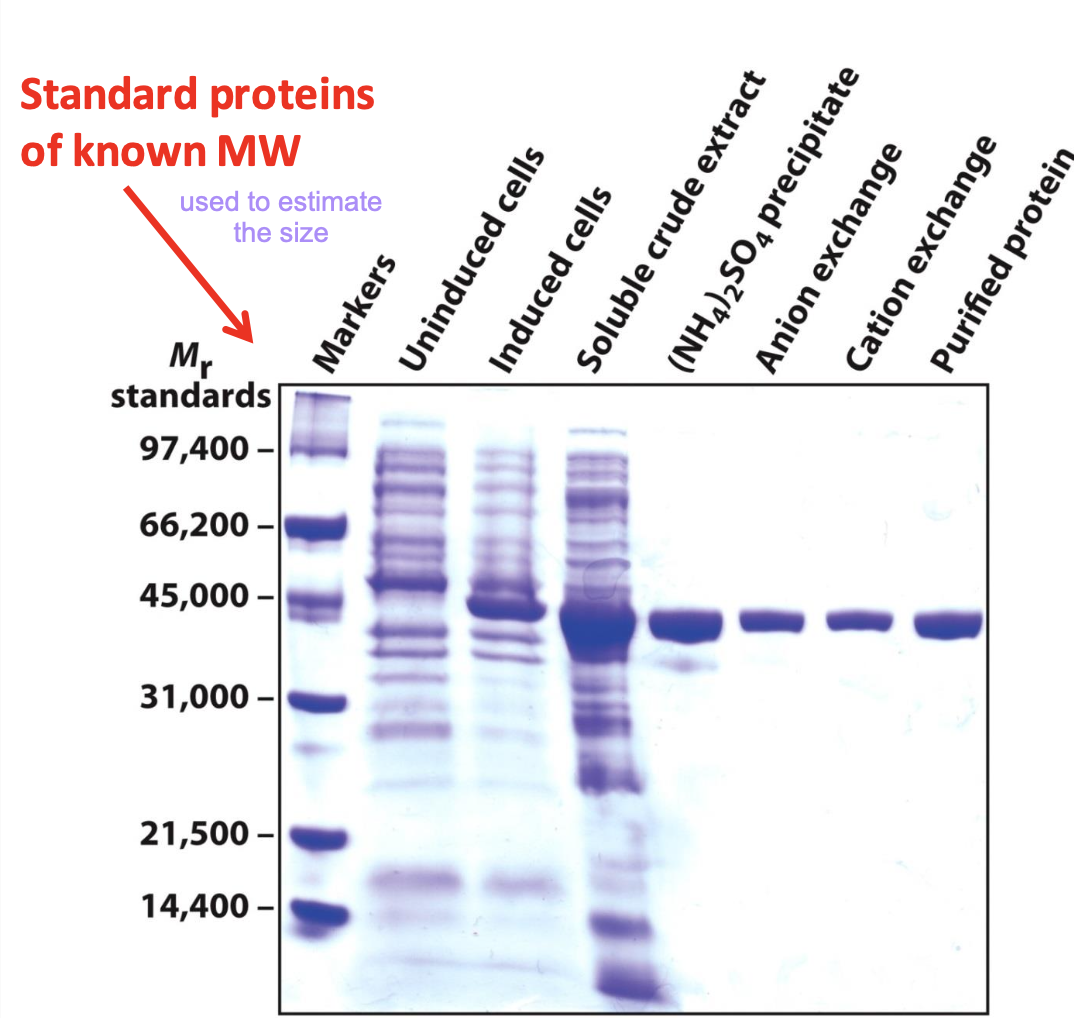
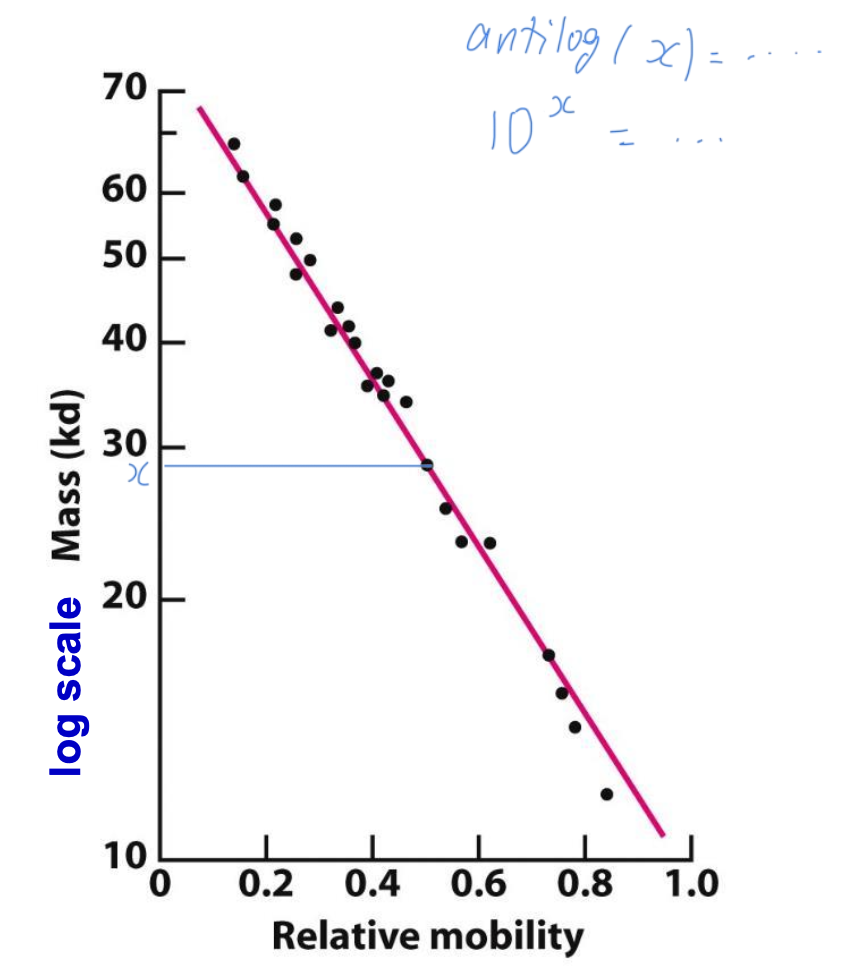
Estimating protein size with SDS-PAGE
Electrophoretic mobility of many proteins is inversely proportional to the logarithm of their mass
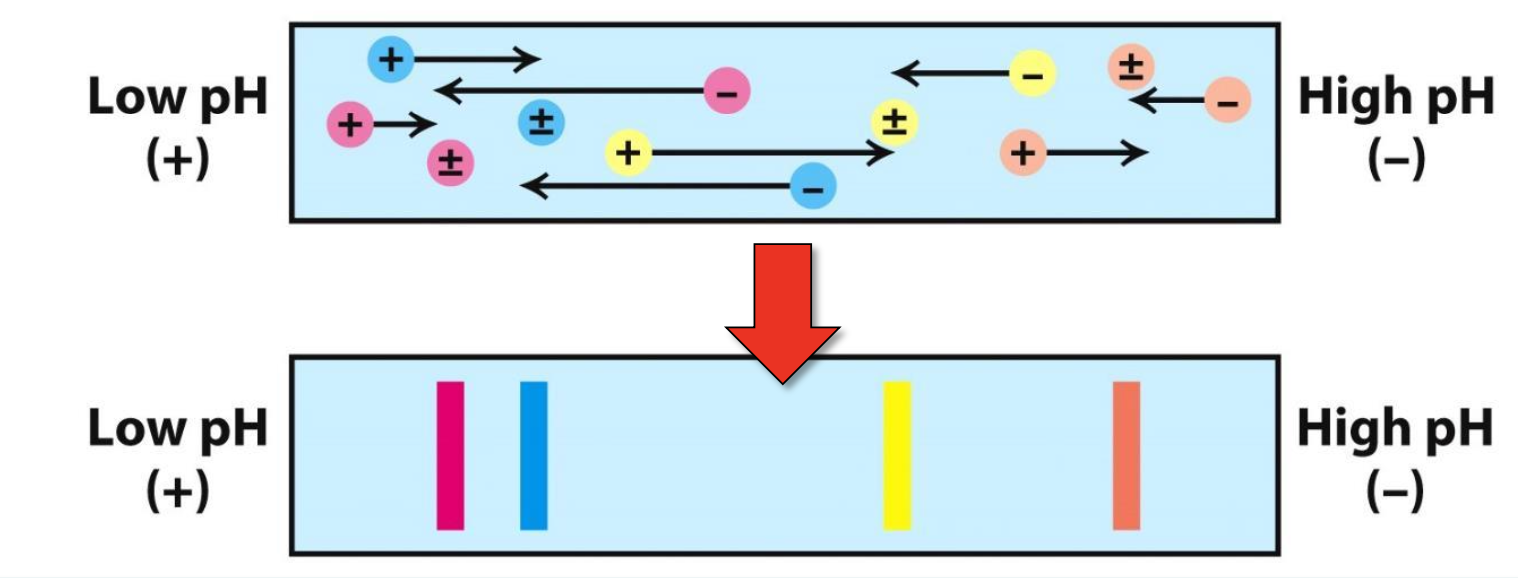
Isoelectric focusing (IEF)
With an electric field, proteins migrate on the pH gradient gel until pH=pI (net neutral protein)
When, pH < pI → net +vely charged protein that moves toward negative electrode
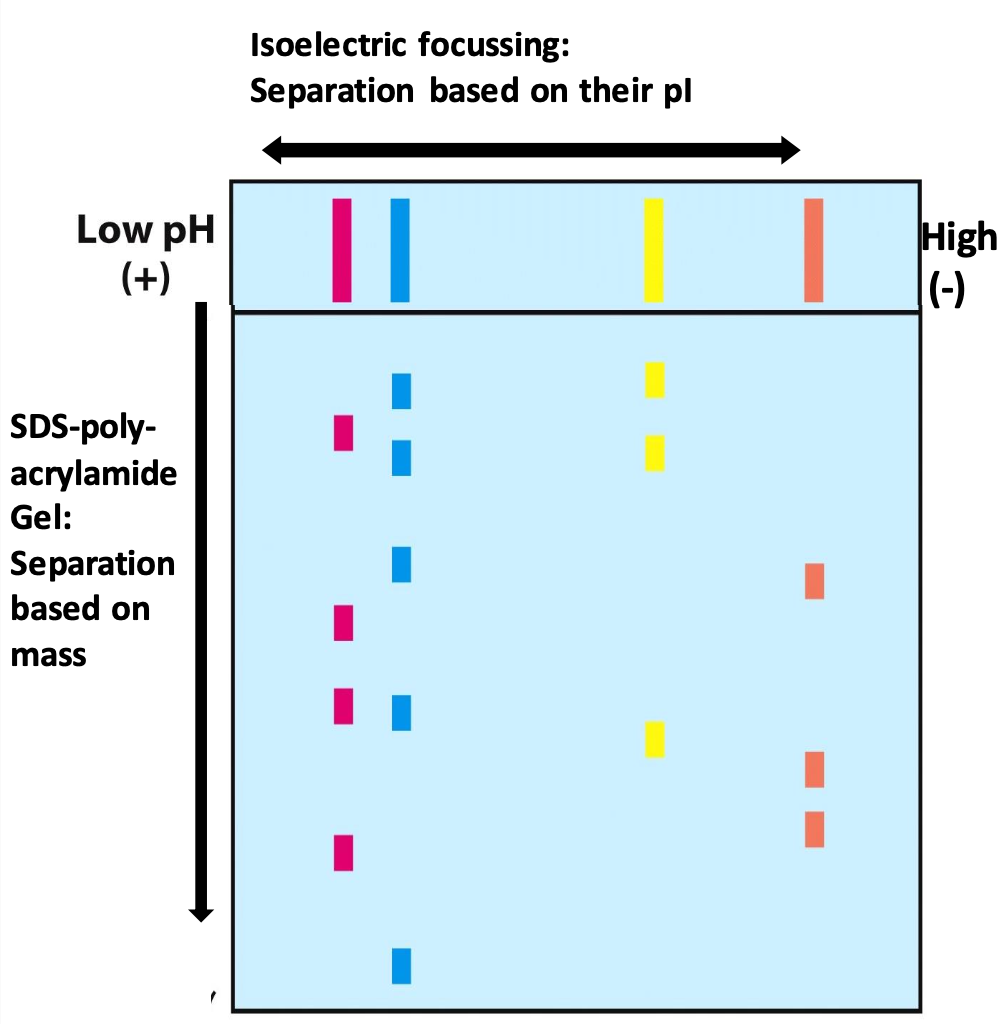
2D gel electrophoresis
Allows for efficient separation of a large # of proteins
After IEF, lane treated with DTT and SDS to denature each band. Electrophoresis is performed perpendicular to direction of original separation (proteins of same pI are now separated by mass)
Specific activity
Used to monitor purification: specific activity = enzymatic activity / total [protein]
The goal is to increase specific activity w/ each purification procedure. When enzyme is pure, specific activity should be constant
Purification scheme formulas
Tot protein (mg) = [protein sample] * tot fraction vol
Tot activity = enzyme activity/ vol * tot fraction vol
Yield (%) = tot activity in any given step/ tot activity in 1st step * 100
Purification level = specific act at any level/ specific activity in 1st step
![<p>Tot protein (mg) = [protein sample] * tot fraction vol</p><p>Tot activity = enzyme activity/ vol * tot fraction vol</p><p>Yield (%) = tot activity in any given step/ tot activity in 1st step * 100</p><p>Purification level = specific act at any level/ specific activity in 1st step</p>](https://knowt-user-attachments.s3.amazonaws.com/a5e22914-17f1-45c2-9c55-3b6cf57339e5.png)
Immunological techniques for purification
The specific reaction of an antibody with its antigen is the basis of these techniques to identify/quantify samples.
Antibody = protein w/ 4 polypeptide chains held together by S-S that is generated in response to an antigen, for which they have specific binding sites for
Antigen = foreign substance that contains an epitope, a particular structural feature that is recognized by a specific antibody. An antigen and its binding site on the antibody are complementary
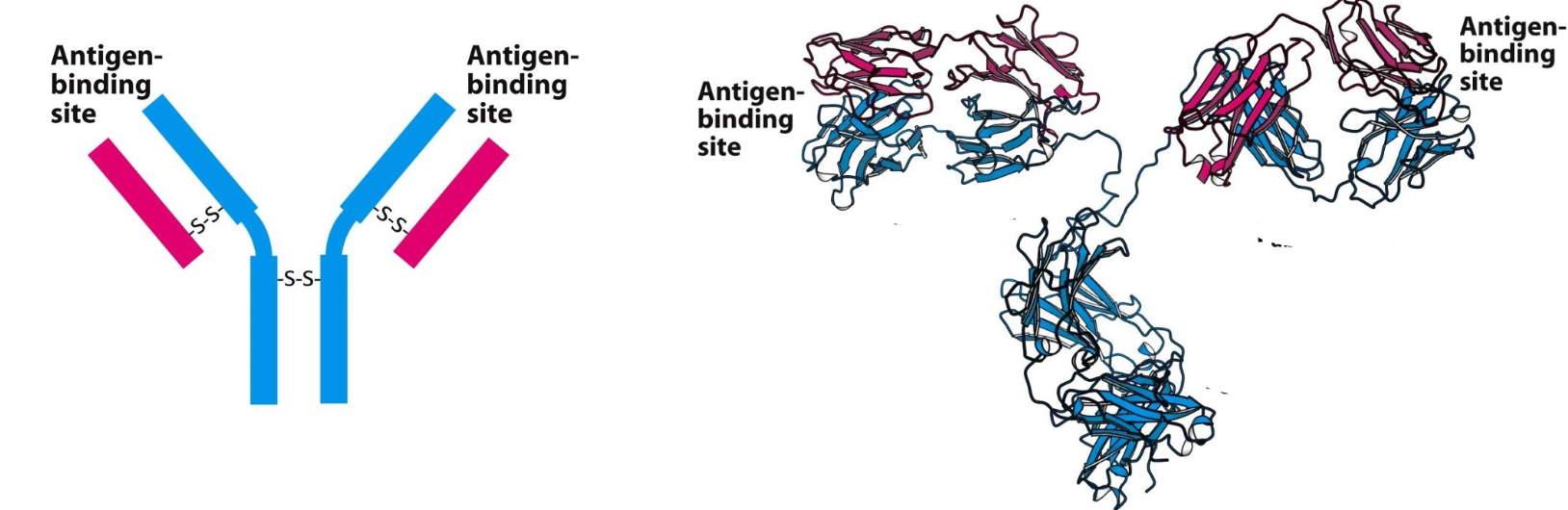
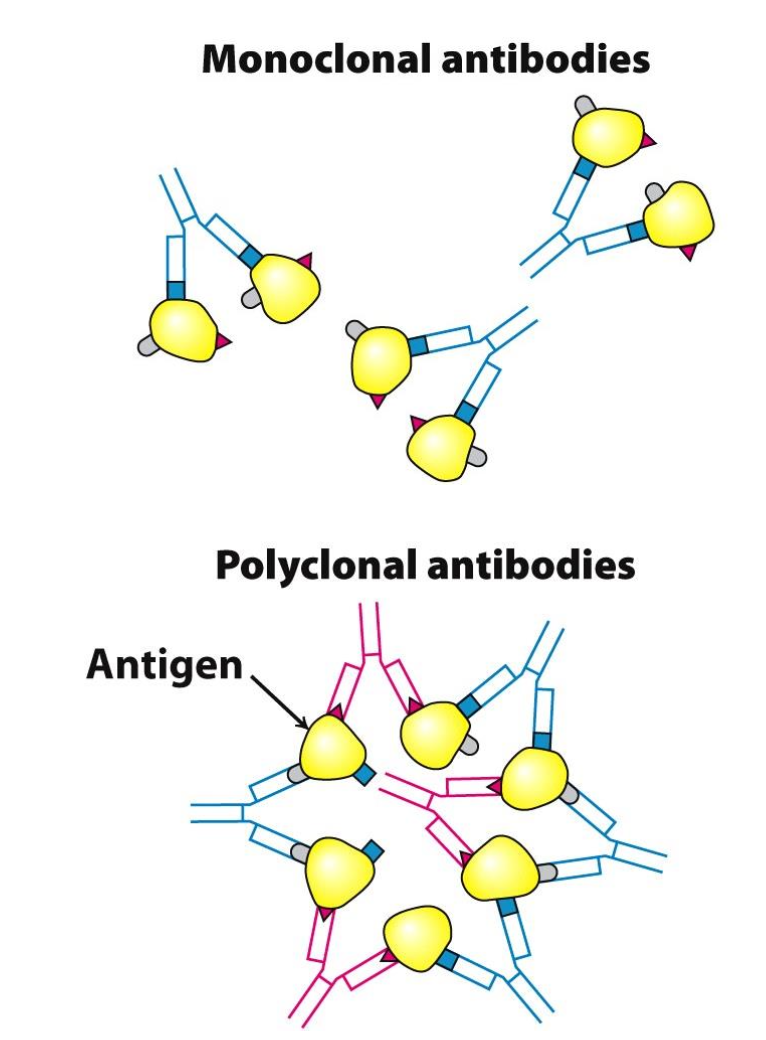
Types of antibodies
✦ monoclonal antibodies: all identical, recognize only ONE epitope on a given antigen
✦ polyclonal antibodies: heterogeneous mixture of antibodies, each one specific for an epitope
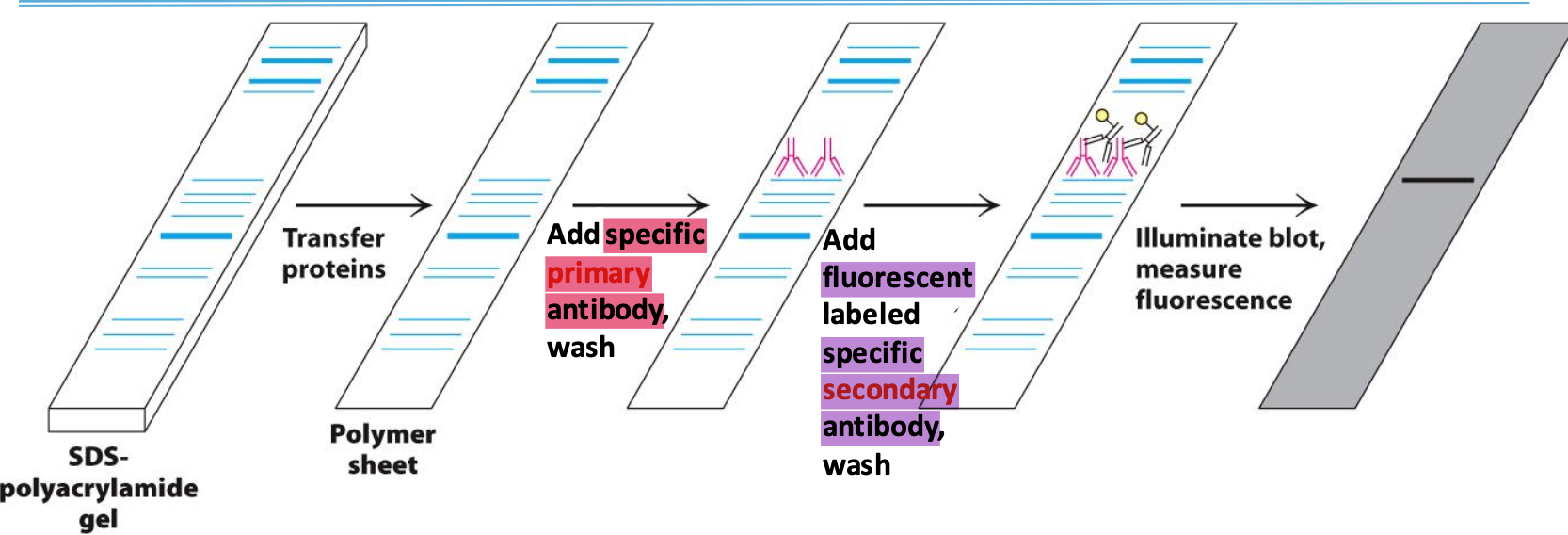
Western blotting
Used to confirm the presence of a protein
Proteins on SDS are transferred to polymer sheet. A primary antibody specific to the protein is added. To detect the antibody, a secondary, fluorescent-labelled, antibody complexes the primary and is excited by light to be visualized.
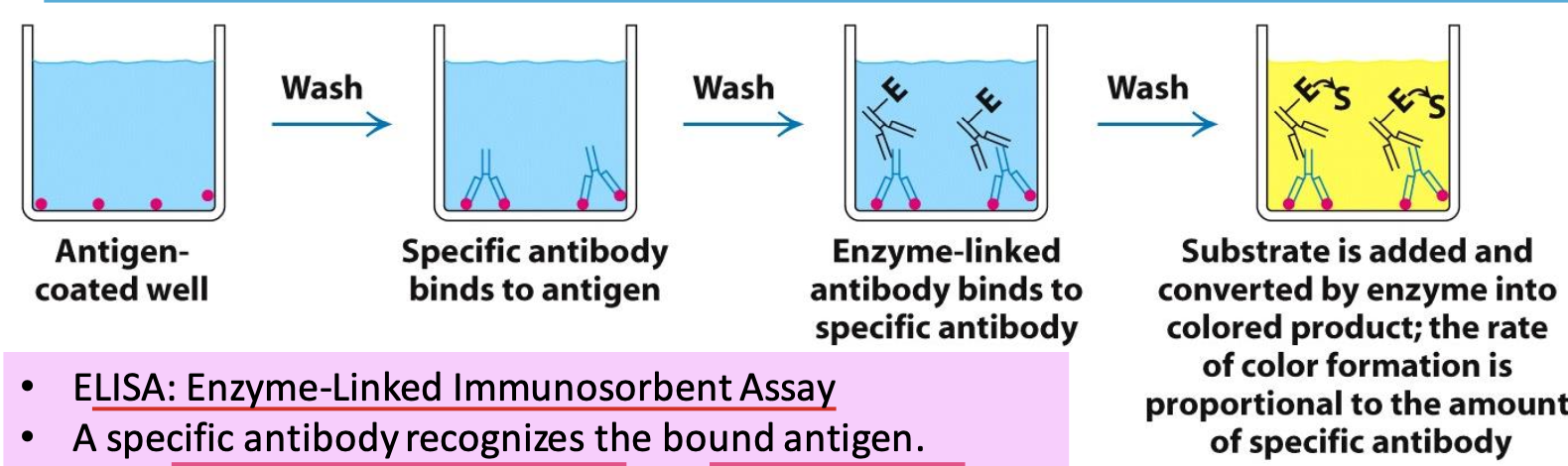
Indirect ELISA
ELISA = Enzyme-Linked Immunosorbent Assay
A specific antibody will recognize the antigens in well. If this antibody is present, an enzyme-linked antibody will bind. Adding the enzyme’s substrate will catalyse the conversion of clear → colored product. The rate of color formation ∝ amount of specific antibody
✧ the more antigen, the stronger the color. can usually detect >1ng of protein
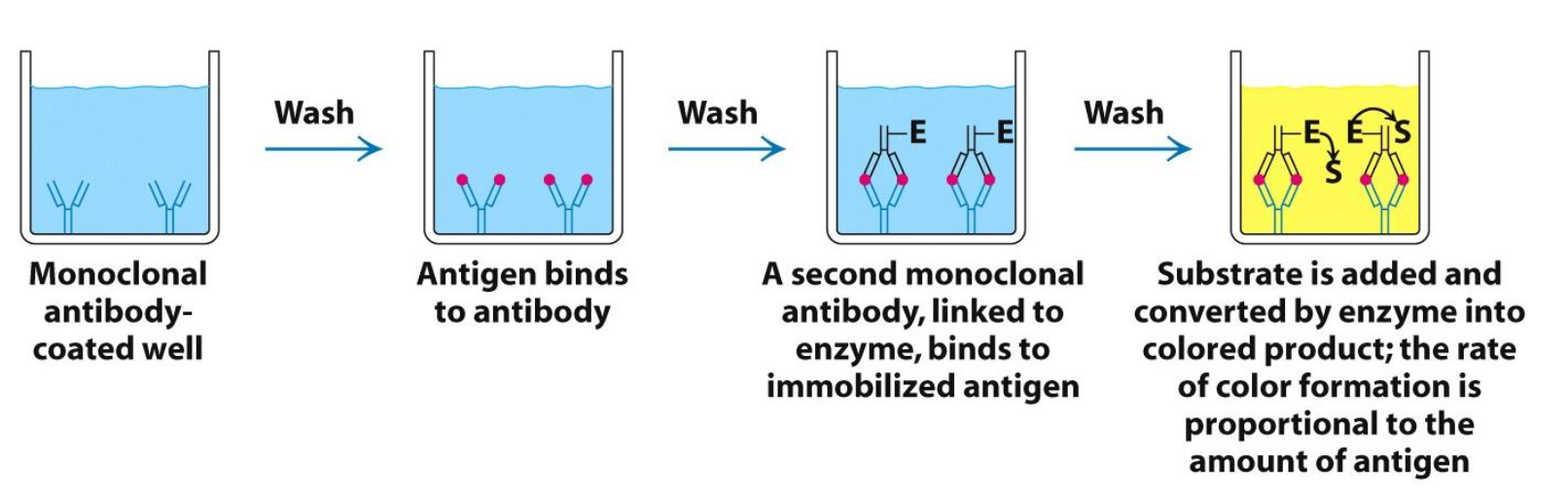
‘Sandwich’ ELISA
Instead of starting w/ antigen coated well, the well is coated w/ monoclonal antibody which attaches antigen and is “sandwiched” by another monoclonal antibody linked to an enzyme. The amount of colored product per given time ∝ amount of antigen bound
X-ray crystallography
Used to determine 3D arrangement of atoms in a crystalline solid. When in contact w/ protein, some rays can diffract off atoms. The diffraction patterns tell us about structure.
X-rays are used since their wavelength is approx. the length of a covalent bond
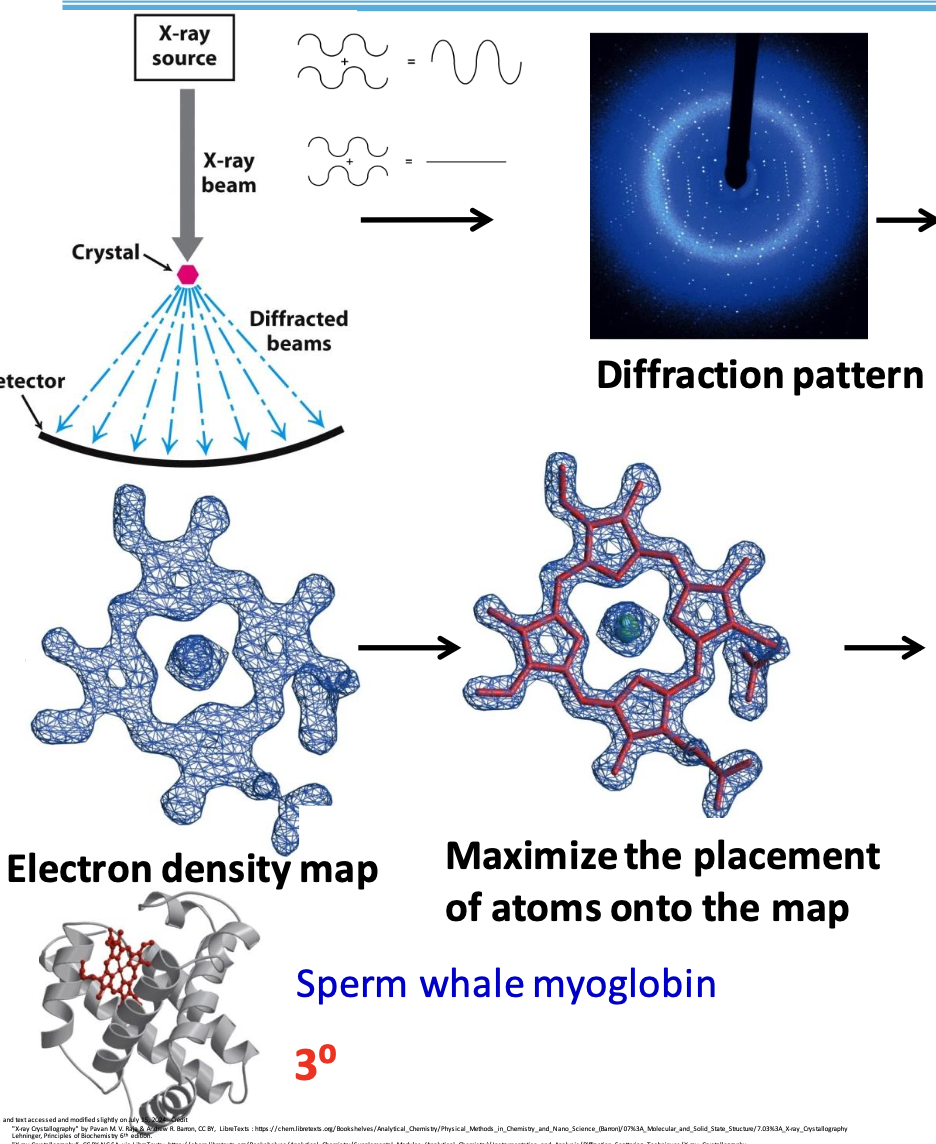
Three main principles of x-ray crystallography
✦ the more electrons an atom has, the greater the amplitude of the scattered wave
✦ scattered waves will either reinforce each other (dark spot) or cancel out (no spot)
✦ atomic arrangement reflected in the way the waves combine
Limitations of X-ray crystallography
☆ requires concentrated sample
☆ works as long as the protein can crystallize (which isn’t always guaranteed)
☆ crystal form hinders protein’s natural movement
☆ challenging to analyse membrane proteins
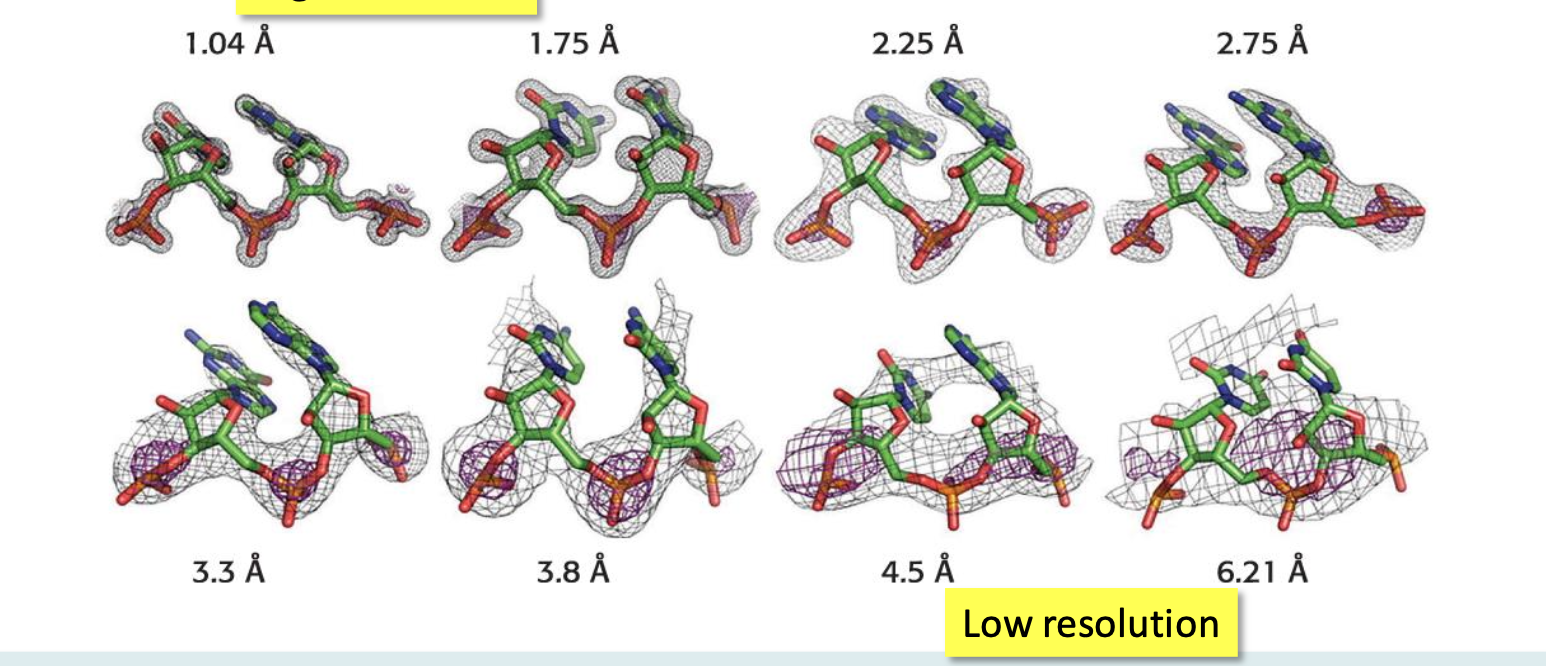
Resolution
Refers to the quality of the electron density map. The better the resolution, the better we can interpret the map
6.0 Å → can see polypeptide chain and few structural details
1.0 Å → can see the position of individual atoms
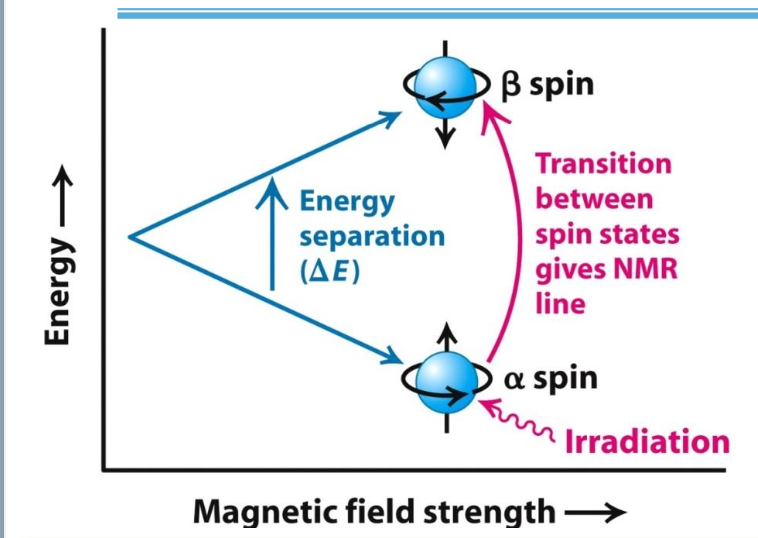
Nuclear Magnetic Resonance (NMR)
✧ Some atomic nuclei (e.g. ¹H) are naturally magnetic w/ 2 spin states: α (aligned with the magnetic field) and β (not aligned).
✧ Resonance: α → β transition when nuclei exposed to EM radiation of the right frequency
✧ ∆frequency between spin states depends on the environment of each nucleus, leading to chemical shifts (measured in ppm).
✧ Shifts vary based on protein structure.
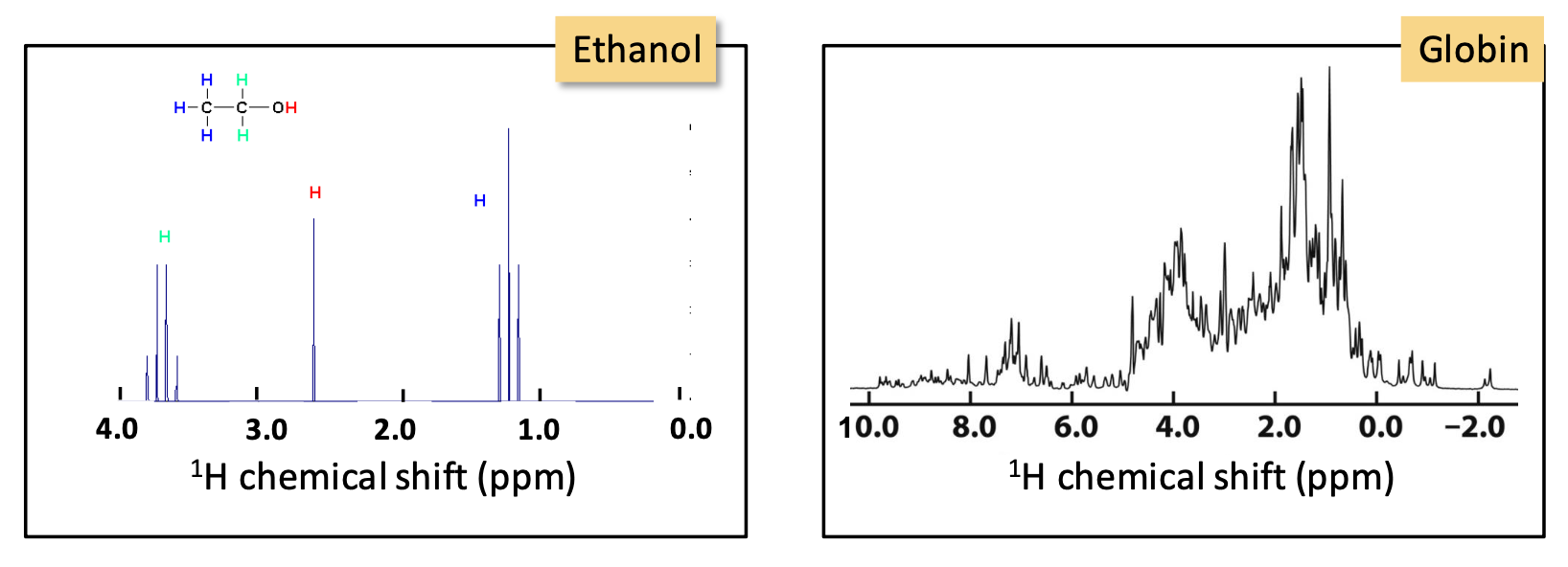
H¹ NMR
Identifies the chemical surrounding of ¹H nuclei
As the molecule becomes complicated, so does the ¹H spectra. An overlap of peaks means a better method needs to be used in order to determine which protons are in close proximity and thus, determine the protein structure → 2D NMR
Nuclear Overhauser Effect
A transfer of energy between nearby nuclei (less than 5 Å apart from each other in the 3D)
2D NMR (Nuclear Overhauser Enhancement spectroscopy)
It is used to identify pairs of atoms >5 Å of each other in 3D space. Finding where these atoms are can help plot the folds of our unknown protein
☆ ¹H peaks are shown in diagonal line
☆ Altering frequency of one nucleus will impact others in close proximity. e.g. 2 and 5 are close, shown by symmetry above/below diagonal line
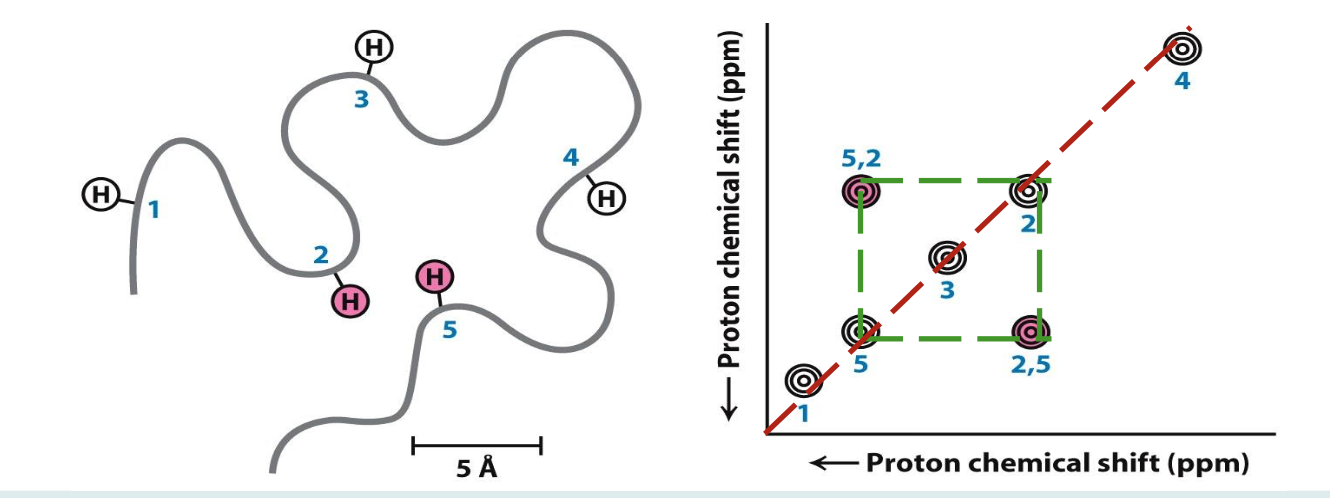
Family of structures
By knowing which protons are in close proximity (>5 Å apart) and the chemical shift of each proton, one can deduce the family of structures that the protein may belong in
NMR limitations
☆ requires concentrated sample
☆ dissolved form of protein needs to stay homogenous for duration if experiment
☆ only works for proteins smaller than 40 kDa
☆ in large proteins, requires enrichment with ¹⁵N or ¹³C for proper signal since ¹H is overly abundant
Technique for exact MW determination
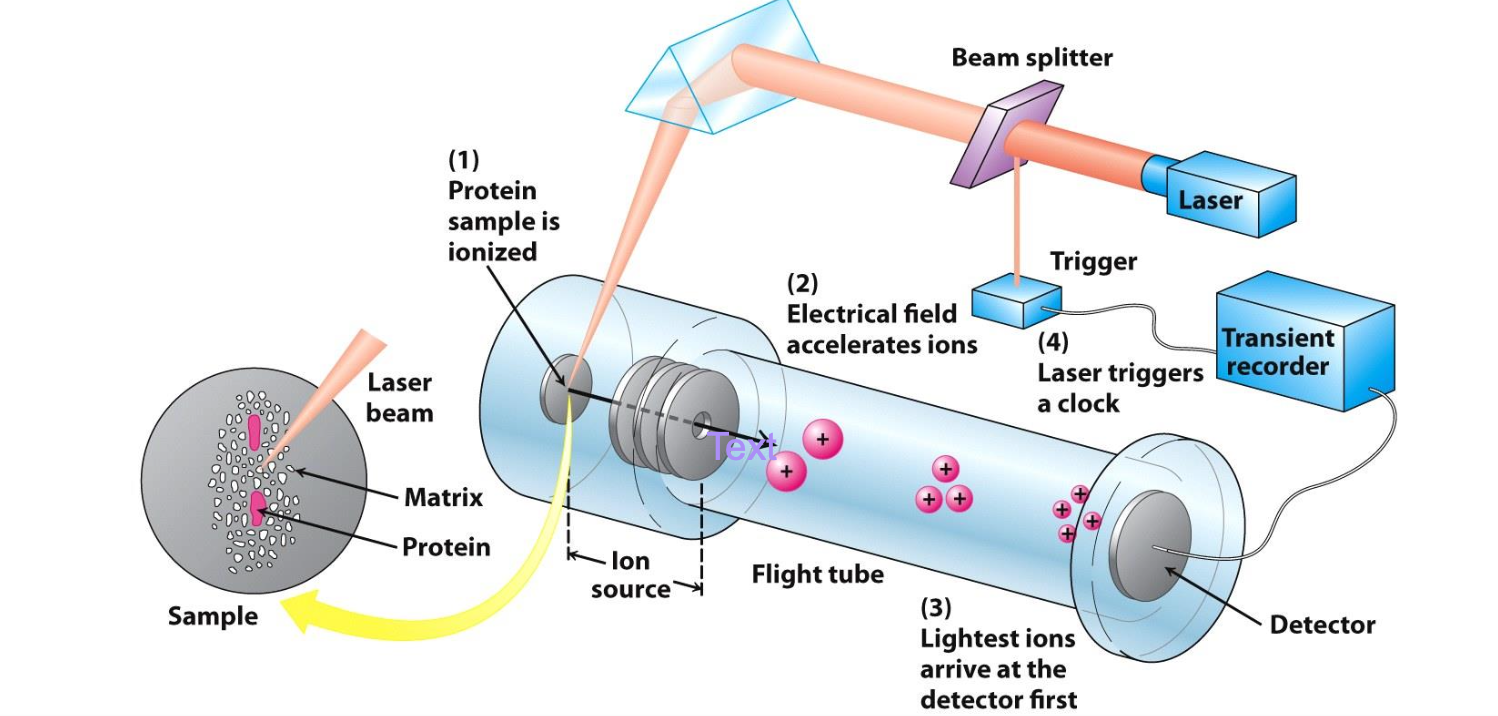
MALDI-TOF or Matrix Assisted Laser Desorption Ionization-Time Of Flight
✦ Protein of interest converted to gaseous charged ions in presence of matrix that prevents degradation. Electrostatic potentials are applied to measure mass/charge ratio
✦ Mass determined by time taken to pass thru chamber
MALDI-TOF advantages
☆ gentle on protein → won’t fractionate/fragment your sample
☆ sample doesn’t need to be 100% pure
☆ any AA changes in protein should be detected since its mass will be different
☆ useful for post-translational modifications like glycosylation, phosphorylation,…
Peptide fingerprinting
Isolating protein by SDS-PAGE, 2D gel, FPLC
Cleave into fragments (enzyme trypsin)
Identify mass (MALDI-TOF)
Search protein database and compare results obtained from virtually cleaving all proteins in database with the same enzyme used experimentally