Patho Exam II Remed
1/39
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
40 Terms
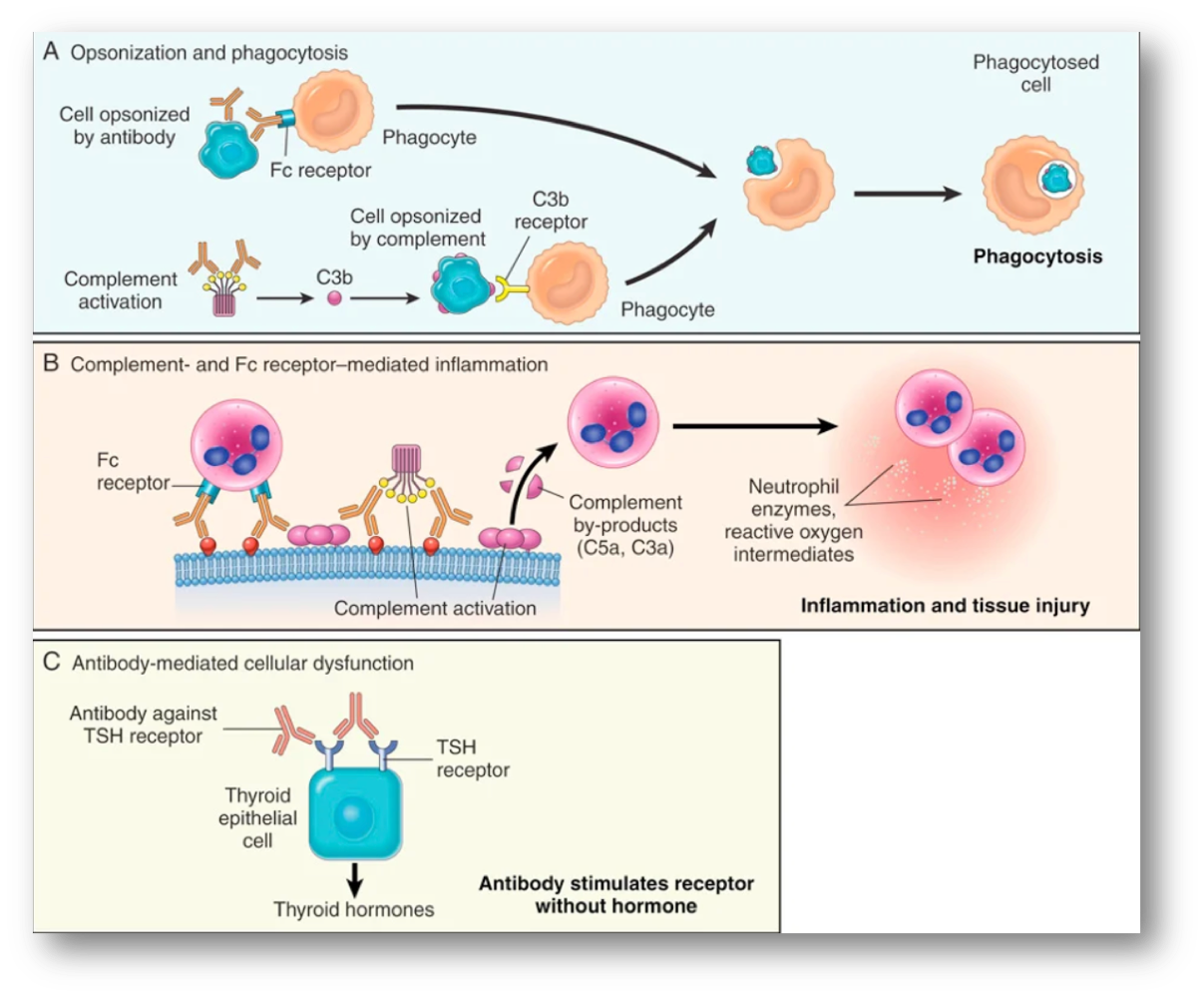
Hypersensitivity type II
all antibodies but IgE, mechanisms of antibody mediated diseases → opsonization + phagocytosis (ex hemolysis + anemia. erythroblastosis fetalis), inflammation: 1. activation of complement system (ex Good Pasture syndrome-antibodies attack basement membrane in lung + kidneys) 2. molecular mimicry (ex rheumatic fever), antibody mediated cellular dysfunction: impair/ dysregulate important functions w/out directly causing cell injury or inflammation ( ex antibodies against TSH receptor over activate thyroid cells→ Graves disease)
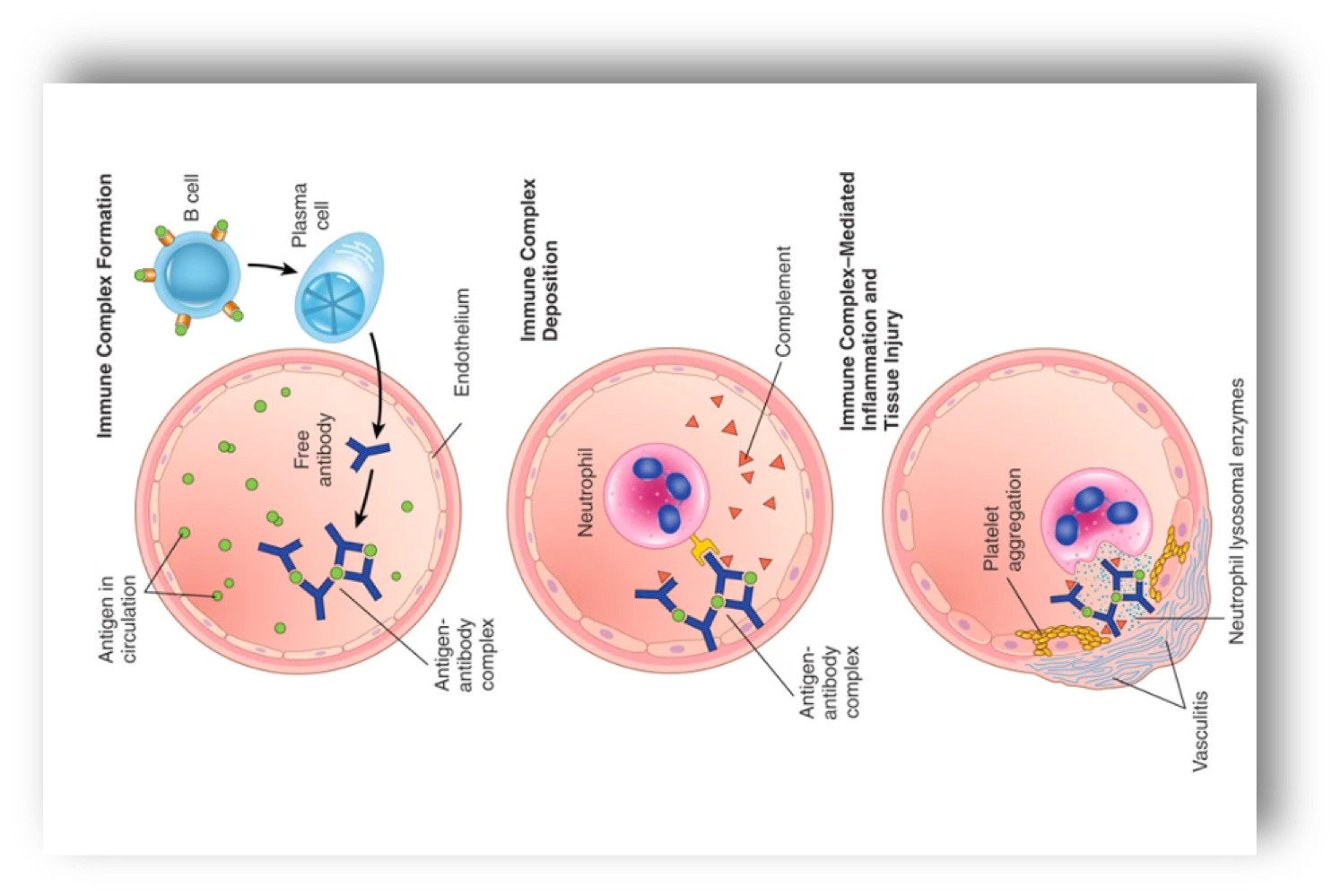
Hypersensitivity type III
mechanism of immune complex-mediated diseases→ immune complex forms: antibodies are secreted in blood, they react with antigens and antigen-antibody complex forms, immune complex deposits: antigen-antibody complexes are deposited in various tissues, immune complex-mediated inflammation and tissue injury: clinical manifestations occur about 10 days after antigen gets into blood
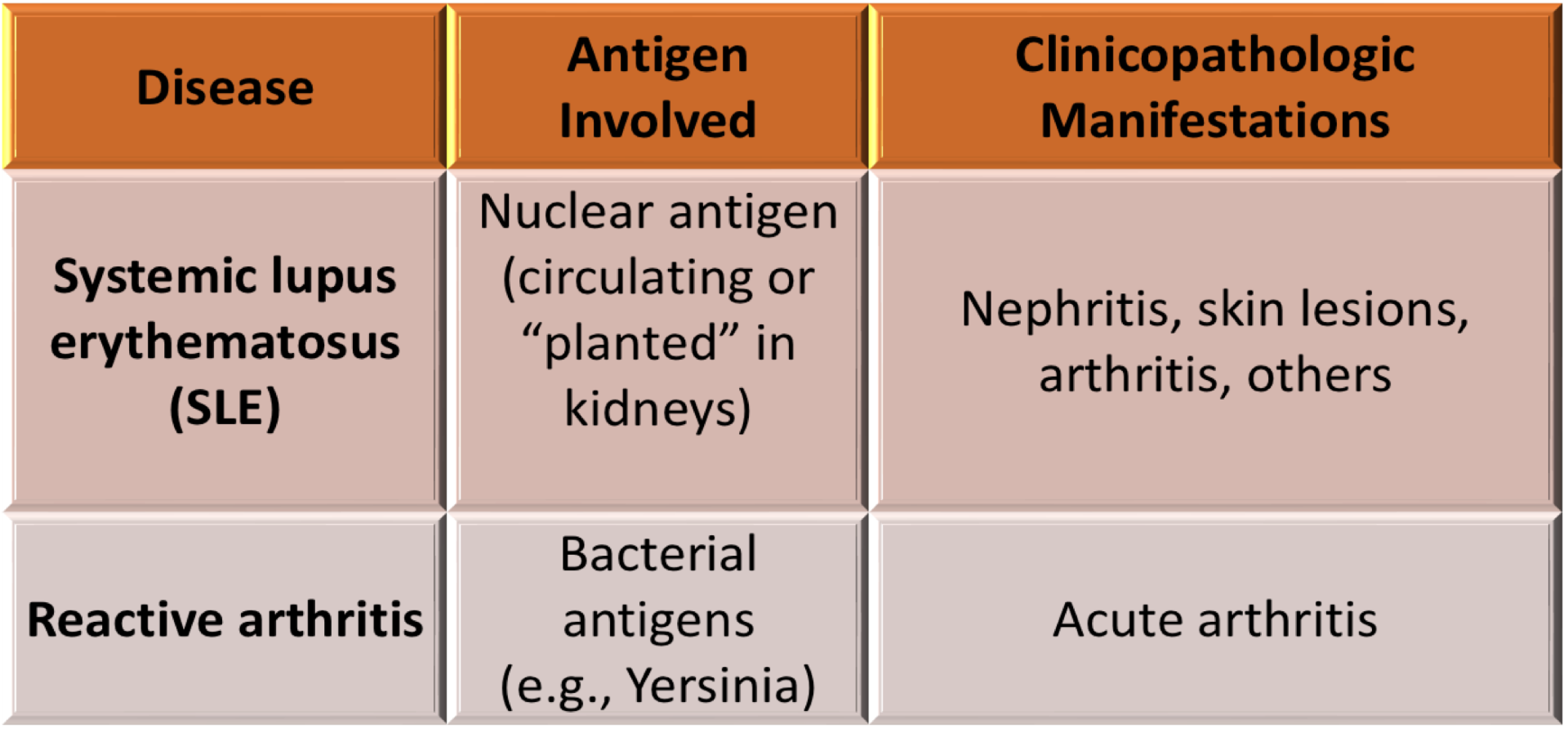
immune complex mediated clinical examples
Hypersensitivity type IV
no antibodies, delayed-type hypersensitivity (DTH), mechanisms of T cell mediated diseases→ CD4+ T cell mediated mechanism inflammation: DTH, classical T-cell mediated hypersensitivity is rxn of Th1 effector cells that secrete IFN-gamma, IFN-gamma activated macrophages produce substances that destroy microbes and damage tissues and mediators that promote inflammation, activated Th17 cells secrete cytokines that recruit neutrophils and monocytes, CD8+ T-cell mediated cytotoxicity: directly kill tissue cells by granzymes and perforins
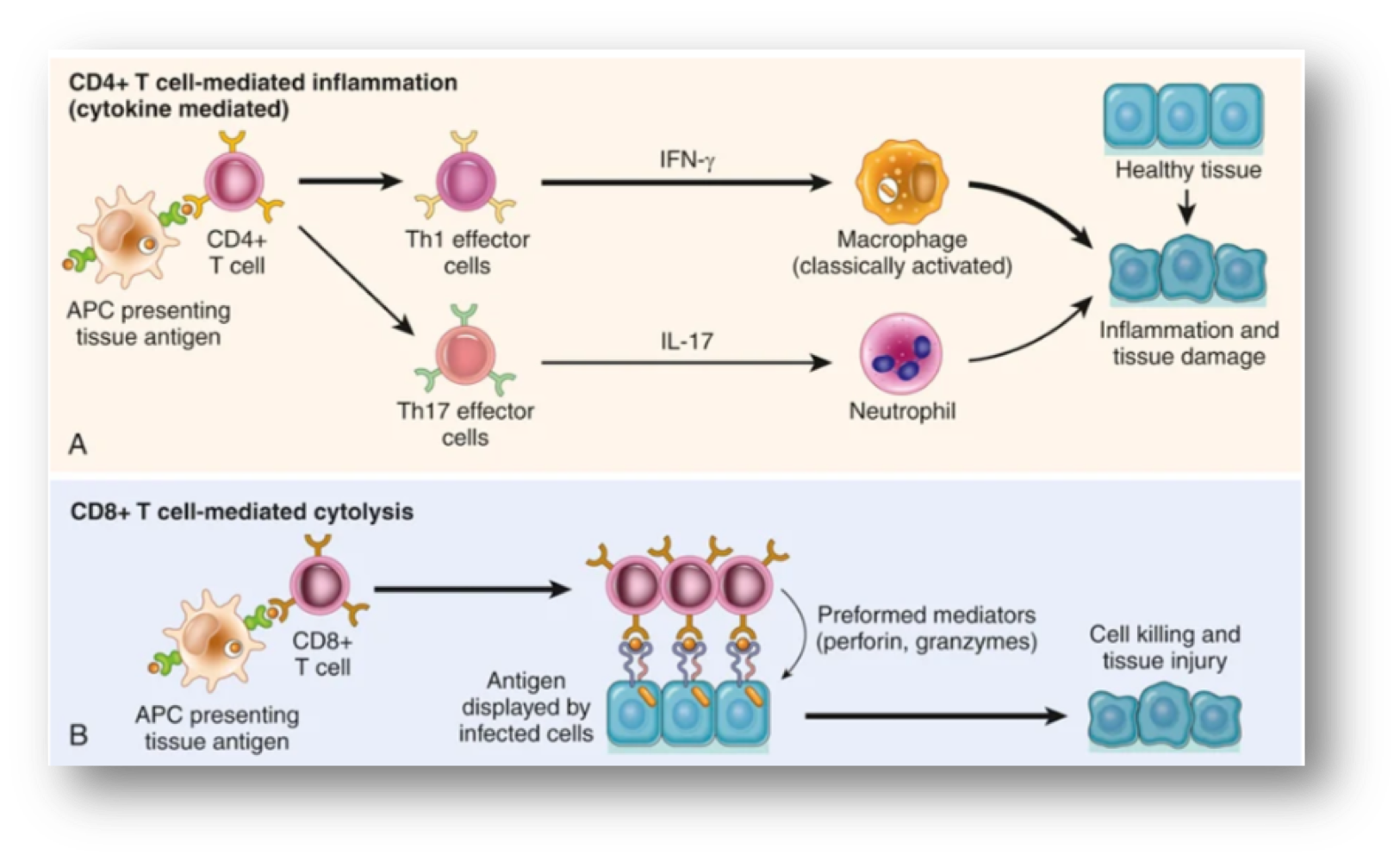
clinical connection to PPD
classic ex of DTH is the tuberculin rxn (PPD skin test) that causes by intracutaneous injection of purified protein derivative (PPD or tuberculin) which was protein antigen of Mycobacterium tuberculosis bacillus, in previously exposed pt, rxn takes 8-12 hours, reach peak @24-72 hours and slowly subside
T-cell mediated diseases, pt 1
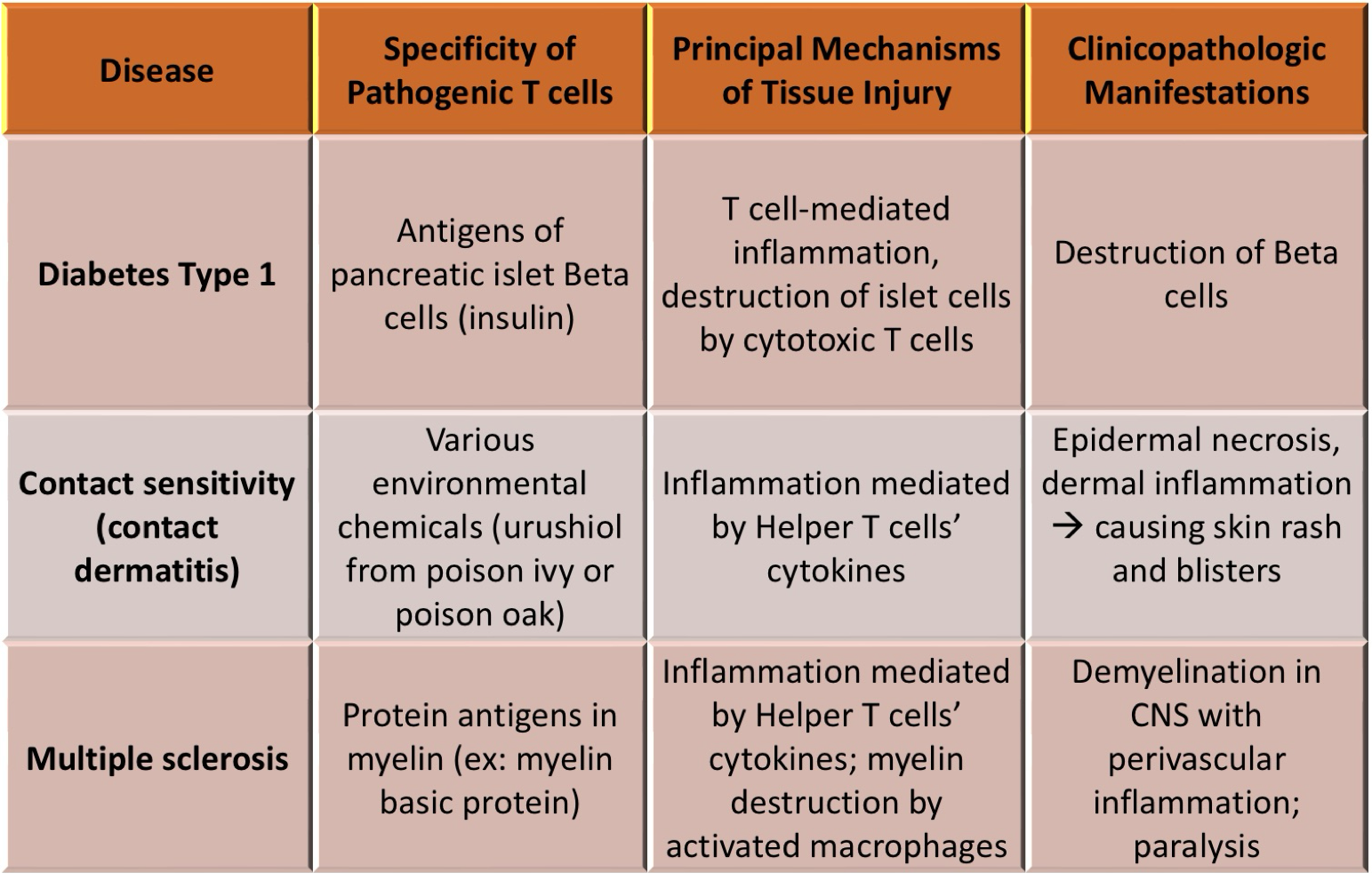
T-cell mediated diseases, pt 2
roles of infections in autoimmunity
A. expressed co stimulators on antigen presenting cells cause self tolerance failure→ self reactive lymphocytes cause autoimmunity, B. molecular mimicry where the microbes mimic self antigen that might cause autoimmune disease by T cell activation and production of cross reacting antibodies
Systemic Lupus Erythematosus (SLE)
systemic inflammatory autoimmune disorders, characteristics → autoantibodies, mainly antinuclear antibodies (ANAs), causes: deposition of immune complexes and binding of antibodies to cells and tissues → injury, clinical features: anti-double-strand DNA (dsDNA), anti-Smith (Sm)→ its against Smith (Sm) antigen, which is a core protein of small ribonucleoprotein particles
T-cell activation and exhaustion in viral infection
acute viral infection: virus-specific CD8+ T cells proliferate and differentiate into effector cytotoxic T cells + memory cells→ viral clearance
chronic viral infection: CD8+ T cells initially respond, but begin to express inhibitory receptors → viral persistence
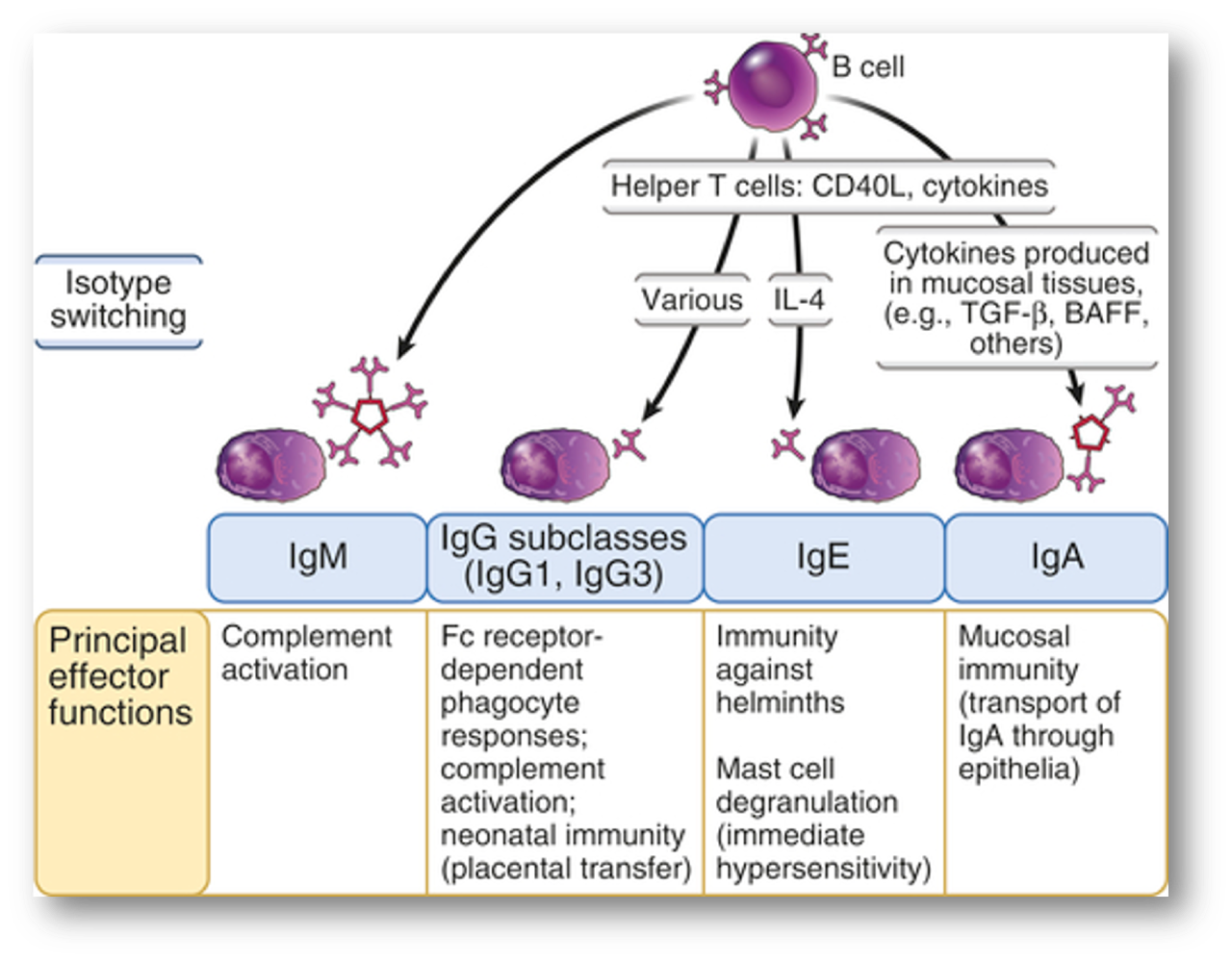
Immunoglobulin (Ig) isotype switching
heart rate (HR)
number of heart beats in 1 min, normal value 60-100 bpm
end-diastolic volume (EDV)
is the filled volume of the ventricle prior to contraction
end-systolic volume (ESV)
the residual volume of blood remaining in the ventricle after ejection
stroke volume (SV)
the volume of blood pumped out by the left ventricle in one contraction
relationship btwn heart factors
SV=EDV-ESV
preload
ventricular volume @end of diastole, approximated by LVEDV
afterload
ventricular wall tension during contraction, depends on the atrial bp and vascular tone
afterload relationship
INCREASE afterload= INCREASE cardiac workload= DECREASE cardiac output
ejection fraction (EF)
the % of blood that is pumped out of a filled ventricle with each heart beat, usually measured only in the left ventricle, EF=SV/EDV x 100, heart %= less than or equal 40% (red), about 41-49% (yellow), about 50-70% (green)
atrial septal defect (ASD)
abnormal, fixed openings in atrial septum that allows communication of blood btwn left and right atrium, most common congenital cardiac anomalies in adults, usually asymptomatic until adulthood, pathophysiology of ASD depends on → pulmonary and systemic vascular resistances, compliance (if stiff or noncompliant, bp increase) of right and left ventricles, size of defect, L→R (L= high pr, R=low pr), blood goes to path of least resistance, clinical features= can be asymptomatic, a murmur is often heard as result of flow through ASD, pulmonary hypertension→ tachypnea and respiratory symptoms (dyspnea), ASD closure reverses the blood flow abnormalities and prevents complications
ventricular septal defect (VSD)
incomplete closure of ventricular septum causes blood to communicate btwn the left and right ventricles, overall most common form of congenital anomaly, clinical features= small VSDs are asymptomatic, most VSDs that clinically manifest in kids are associated with other congenital cardiac anomalies such as Tetralogy of Fallot, large defects generally cause significant L→R shunting, causing right ventricular hypertrophy (increase workload) and pulmonary hypertension
patent ductus arteriosus (PDA)
ductus arteriosus (DA) allows blood flow btwn the aorta and pulmonary artery during fetal development, thus bypassing the lungs, the ductus normally closes within 1-3 days of life, persistent DA is called PDA and can cause L→R shunting, clinical symptoms + implications= a characteristic continuous murmur can often be heard on physical exam, described as “machine like” murmur, as a result of L→R shunt from aorta into pulmonary arteries→ increase pulmonary hypertension
Tetralogy of Fallot (ToF)
R→L, cyanotic, one of the most common congenital heart lesions that need attention in 1st year of life, major pathologic feature= 1. stenosis of the pulmonary artery, determining factor, valve or artery, 2. VSD, 3. deviation of the aorta to the right (overriding aorta), 4. right ventricular hypertrophy, clinical features= symptom severity is related to the extent of pulmonary artery stenosis
compensatory mechanism of HF
signs and symptoms of hf are caused by compensatory (good short term but bad long term) mechanisms: 1. Frank-Sterling mechanism: increase EDV→ increase CO, more secretion → more force 2. activation of neurohormonal symptoms: SNS→ increase hr, myocardial contractility, ADH→ increase in systemic vascular resistance, RAAS→ systemic vasoconstriction and renal sodium retention, 3. Myocardial adaptions: cardiac hypertrophy is the compensatory response of the myocardium to increase mechanical work, in pressure overload states (ex hypertension or vascular stenosis) the ventricular wall thickens increase without increase in size of chambers, in volume overload states (wx vascular regurgitation or shunts), the ventricle tends to dilate and the resulting wall thickens can be increase, normal, or decrease
left-sided HF (CHF)
or congestive, systolic dysfunction (decrease in myocardial contractility), etiologies → ischemic heart diseases (IHD), systemic hypertension, mitral or aortic valve disease, clinical features= are caused by decrease systemic perfusion and increase back pressures within pulmonary circulation, categorizing pt w/ left sided hf based on left ventricular ejection fraction (LVEF): HFpEF, HFrEF, and HFmEF
HFpEF
abnormal diastolic function, LV (stiff, have hard time relaxing) end-diastolic volume and SV are preserved, decrease in LV compliance (Diastolic flexibility ) → increase in LV diastolic pressure → often with LV hypertrophy, LVEF is preserved ( greater than or equal to 50%)
HFrEF
abnormal systolic function, LVEF less than or equal to 40%
HFmEF
hf with mid-range, LFEF=40-49%
right sided HF
diastolic dysfunction (increase in LV stiffness), etiologies= the consequence of left sided HF, since any pressure increase in pulmonary circulation inevitably cause an increase burden on right side of heart, clinical features= pure (rare, usually caused LVHF) right sided HF is not typically associated with respiratory symptoms, its manifestation is mainly related to systemic and portal venous congestion → increase jugular vein pressure, hepatic and splenic enlargement, peripheral edema and ascites (abnormal cavity filled with fluid)
prothrombin time (PT) assay
assesses function of protons in extrinsic and common pathways (tissue factor, factor VII, X, V, II and fibrinogen), INR= international neutralized ratio
partial thromboplastin time (PTT/ aPTT) assay
screens function of proteins in intrinsic and common pathways (factor XII, XI, IX, VIII, X, V, II and fibrinogen)
thrombin time (TT)
measures final step of coagulation (Conversion of fibrinogen to fibrin)
antithrombotic properties of endothelium
1.inhibiting platelets activation in basal state= shielding (hiding) platelets from vWF (von Willlebrand factor) and collagen, synthesizing NO→ inhibits platelet aggregation, 2. anti-coagulant effects on basal state= thrombomodulin binds thrombin→ inhibits thrombin and activates protein C, 3. fibrinolytic effects= synthesizing t-PA (tissue type plasminogen activator)
pathways of systemic edema in diseases
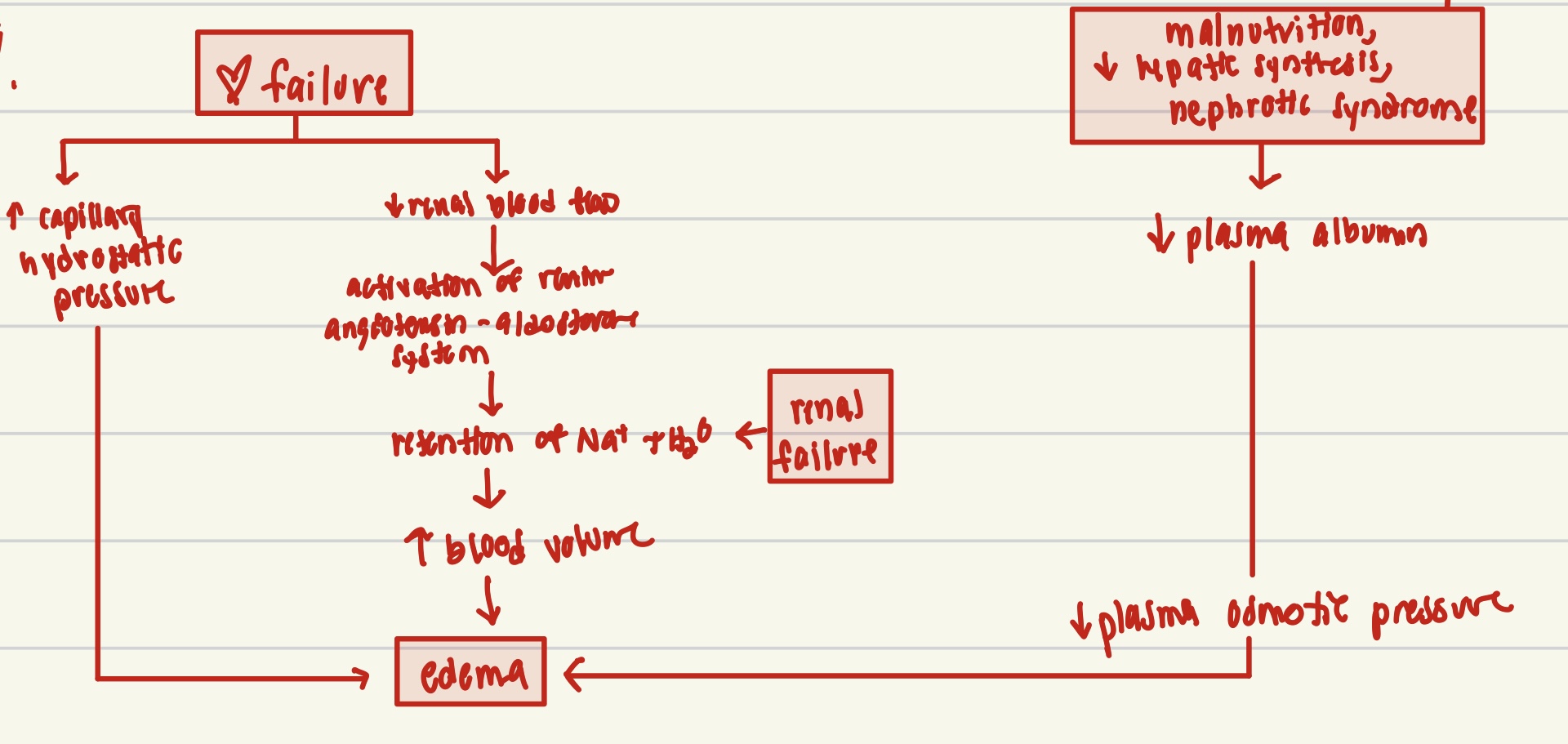
hyperemia
too much blood in arterial, arteriolar dilation leads to increase blood flow, affected tissues turn red (erythema)
congestion
reduced outflow of blood from tissue (venous end), it can be systemic, such as heart failure, or localized such as isolated venous obstruction
acute DIC
acute exposure to large amounts of pro coagulant substances decompensated DIC, bleeding predominates (no coagulation, can’t be stopped), might be caused: sepsis and trauma
chronic DIC
intermittent/ continuous exposure to smaller amounts of pro-coagulant substances, compensated DC, thrombosis predominated, could be caused by advanced cancer
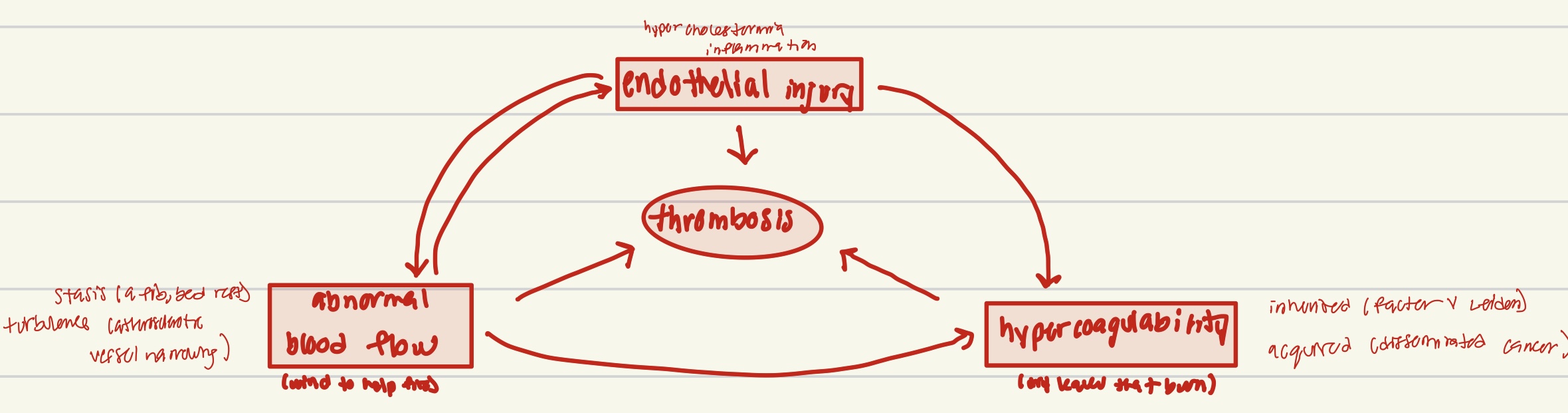
Virchow Triad