Week 7-12 Structure and Reactivity ILOs
1/44
Earn XP
Description and Tags
Only relevant ones
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
45 Terms
Explain the existence of isomers in internal alkenes, in terms of orbital overlap in pi systems
Multiple structural isomers exist in alkenes because the pi (double) bond can be between different carbons.
Use the Cahn-Ingold-Prelog priority rules to assign the absolute stereochemistry (E/Z) of alkenes and analogous systems (e.g. oximes, imines)
Higher atomic number has higher priority
If equal priority, tiebreak by listing the next attached atoms in atomic order
Multiple bonds: list element twice
note: basically highest atomic weight/ total atomic number wins but this is not technically correct
Explain the origin of molecular chirality
Molecular chirality occurs when a carbon center bonded to 4 different atoms or groups of atoms are nonsuperposable on their mirror images: cannot align perfectly with their mirror image through rotation or translation and the two molecules are enantiomers.
Use the Cahn-Ingold-Prelog priority rules to assign the absolute stereochemistry (R/S) of stereogenic centres
Highest atomic number has highest priority
Assign numbers 1-4. Rotate molecule so that 4th priority is facing back (into the page). If 1→2→3 is clockwise, R, if anticlockwise, S
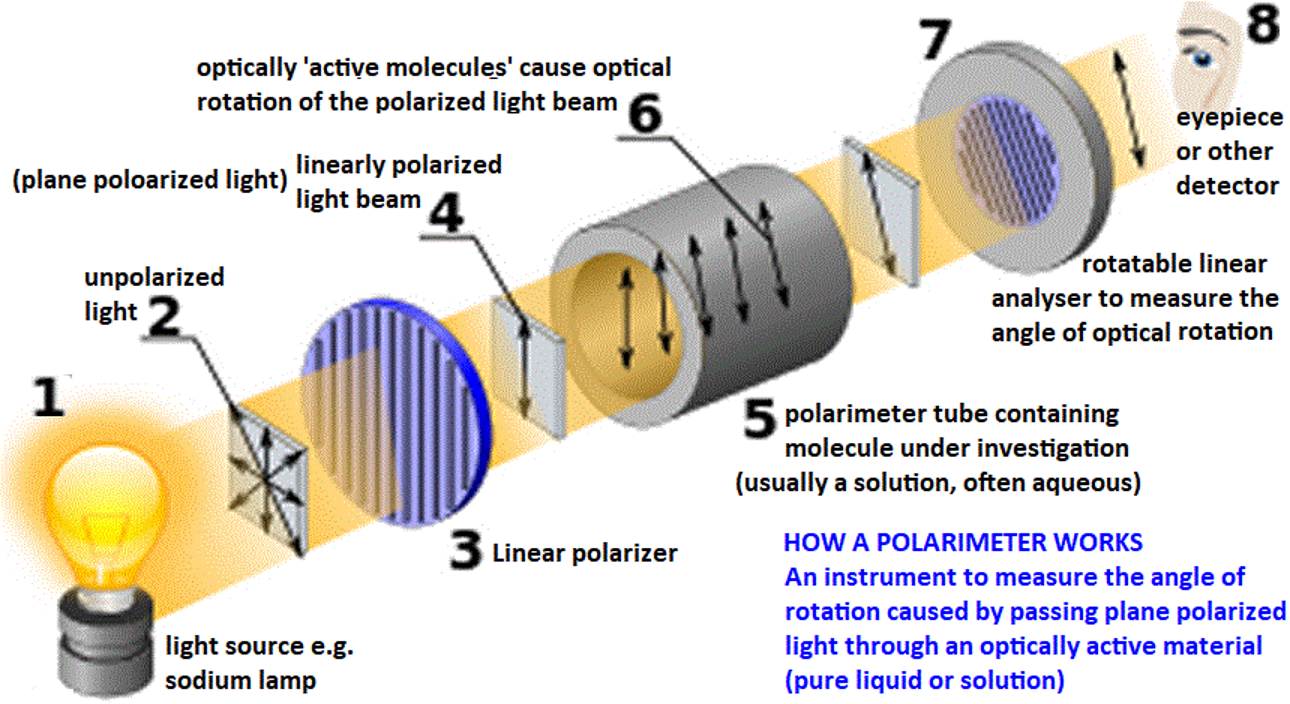
Explain how optical activity is measured using a polarimeter
light emitted through a light source, e.g. sodium lamp
unpolarized light passed through a linear polarizer to get linearly polarized light beam
polarized light passed through polarimeter tube containing the molecule under investigation (usually solution)
light hits a rotatable linear analyser to measure the angle of optical rotation
Calculate the specific rotation of a compound using polarimetry data
obtain observed rotation (alpha)
measure path length (l) of sample cell in dm
determine the concentration of the sample in g mL^-1
use the formula angle of rotation (alpha) depending on wavelength and temperature = angle of rotation/(path length dm x concentration g mL-1)
Determine the enantiomeric composition of a sample of a chiral compound, and describe this using appropriate stereochemical terminology
Pass through polarimeter and calculate angle of rotation. Refer to data tables to assign which enantiomer it is, R (clockwise) or S (anti clockwise). If the angle of rotation is 0 but you know it is nonsuperposable on its mirror image, it is a racemic mixture with an equal 1:1 ratio of each enantiomer.
Identify and draw diastereomers of a given molecular structure
Diastereomers - stereoisomers (same structural formula but different arrangement in space) that are not enantiomers/not mirror images
Calculate the maximum number of possible stereoisomers of a given compound
2^n where n is the number of chiral centres in the molecule
Identify meso compounds
Optically inactive member of a set of stereoisomers - achiral molecule with multiple chiral centres, usually symmetrical.
Explain the principles of resolving chiral compounds using enantiopure derivatising agents or chiral stationary phase chromatography
Resolution - mix the racemic compound and a single enantiomer of a resolving agent (enantiopure deriving agents) to give seperable diastereomer products
Chromatography - use a chiral stationary phase and one enantiomer will elute before the other
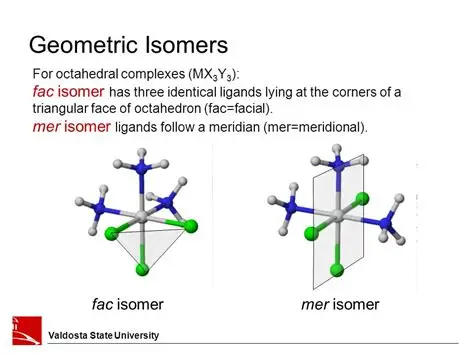
Use cis/trans and fac/mer nomenclature to describe the geometries of square planar and octahedral transition metal complexes
mer isomers follow a meridian - in a line/plane (mer=meridian)
fac isomers have 3 identical ligands in a corner (fac=face)
cis isomers if 2 identical ligands next to each other
trans isomers if 2 identical ligands, one above and one below in a straight line
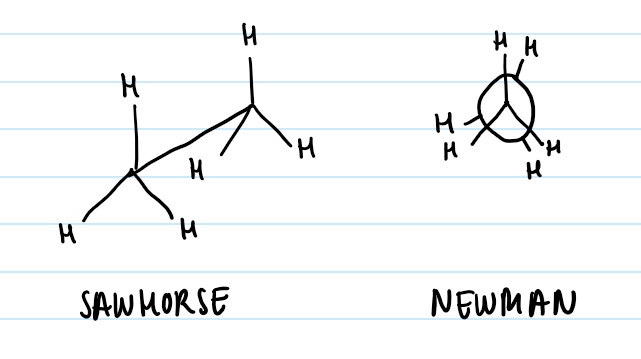
Dihedral angle: angle between front substituent and back substituent
Staggered: Front substituents are equally between back substituents when looking from the front
Eclipsed: Front substituents are directly in front of back substituents
Torsional strain - occurs when molecules are in eclipsed conformations due to electron-electron repulsion between eclipsing substituents
Steric strain - occurs when substituents are close together (within their van der waals radii) and there is electron-electron repulsion between the groups
Torsional and steric strain increase the energies of conformations.
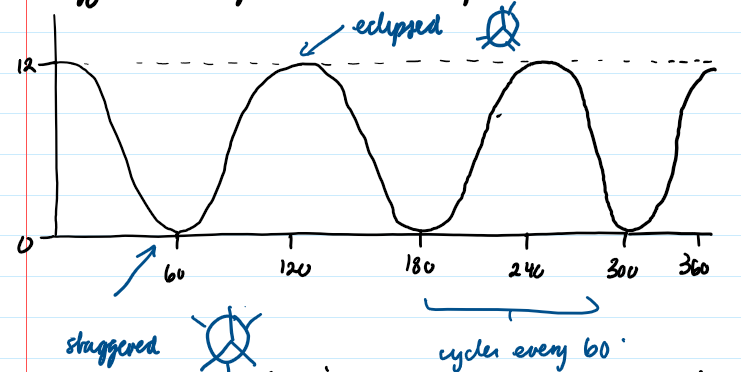
Construct and interpret torsional energy profiles for small molecules
Shows the energy of a molecule as a function of rotation around a bond (see image: torsional energy profile for ethane)
Rationalise the geometries of cycloalkanes (C3-C6) on the basis of angle and torsional strain
Angle strain: increase in energy of a molecule due to bond angle values deviating from ideal, hybridised bond angles. For example, sp³ hybridised angle is 109.5 but is 60 in cyclopropane so cyclopropane has angle strain
Axial - substituents are above and below the plane of carbon atoms
Equatorial - substituents are in the planes of the carbon atoms
Substituted cyclohexane chair conformers usually have substituents in the equatorial position as this reduces steric strain that would occur with Hs in the axial positiion
BL acid - proton (H+) donator
BL base - proton (H+) acceptor
pH = -log[H+]
pKa = -log[Ka] where Ka is the acid dissociation constant
pKb = -log[Kb] where Kb is the base dissociation constant
pkaH = -log[KaH] where KaH is the acid dissociation constant of the conjugate acid of a base (reverse reaction for Kb)
carboxylic acid pKa ~ 5
alcohol pKa 16-18
amine pKa 38-40
phenol pKa ~ 10
water pKa ~ 14-15
Ka = 10-pKa
KaH = 10-pKaH
Across a period - acidity increases and basicity decreases due to increase in electronegativity and effective nuclear charge (Zeff) so it is easier for atom to donate protons.
Down a group - acidity decreases and basicity increases as increasing atomic size means Zeff decreases, harder to release a proton and easier to accept a proton
Explain how inductive (+I, −I) effects stabilise charged species,
Inductive - through sigma framework as some atoms more electronegative, leading to polarized bond and redistribution of electron density
-I occurs when electron withdrawing groups pull electron density away from adjacent atoms, increasing the positive charge on those atoms (e.g. halogens)
+I occurs when electron donating groups push electron density towards adjacent atoms, reducing positive charge (e.g. methyl groups)
Define and explanation hyperconjugation as part of inductive stabilisation
Hyperconjugation - delocalisation of electrons from a sigma bond (usually C-H or C-C) to an adjacent empty or partially filled p orbital or pi system when a filled orbital overlaps with empty p or pi system, usually stabilising carbocations and alkenes.
Explain how resonance/mesomeric (+M, −M) effects stabilise charged species,
Resonance/mesomeric - through pi framework of the molecule through delocalisation and spreading the charge across multiple atoms
+M electron donating groups, stabilise cations
-M electron withdrawing groups, stabilise anions
Strong acids have weak conjugate bases and strong bases have weak conjugate acids.
Inductive effects - electron withdrawing or donating groups caused by difference in electronegativity, can stabilise conjugate bases, leading to stronger acids. E.g. halogen is electron withdrawing group and increases acidity because pulls e.d. away from oxygen atom in water and stabilising hydroxide ion
Resonance effects - delocalisation of electron pairs through resonance stabilise charges on conjugate base, making it more stable and more likely to donate a proton, making stronger acid. Resonance of double bond can stabilise hydronium ion, making it a stronger acid than water.
Nucleophiles - high electron density, attracted to regions of positive charge or cations. Usually donate electrons
Electrophiles - electron deficient, attracted to regions of high electron density or anions. Usually accept electrons
Create and interpret reaction coordinate diagrams for simple reactions
Energy on y axis, reaction coordinate/progress on x axis
Transition states - maximums on reaction coordinate diagrams. Barriers between reaction intermediates with bonds partially broken or formed with a very short lifetime.
Reaction intermediates - minima on reaction coordinate diagrams. Stable species with finite lifetimes that form during a chemical reaction and are later used so do not appear in overall reaction equation
Heterolytic bond fission in H-Br with Br getting both electrons. Pi electrons bond to H+ from H-Br to form a carbocation. Br- acts as nucleophile and forms bond with the positive C in the carbocation to form a bromoalkane
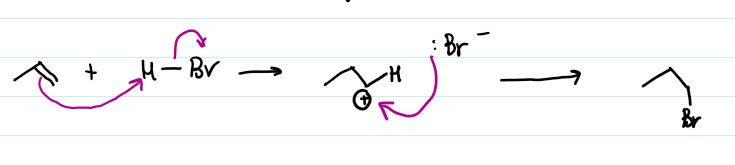
H added to site where H most present because this mechanism forms greater substituted carbocations which are more stable due to hyperconjugation and inductive effects from alkyl groups. Primary carbenium ions are less stable as not stabilised by resonance or hyperconjugation. This causes the energy on the reaction coordinate diagram to increase, as well as the energy of the transition state before the carbenium ion intermediate (same factors that destablise carbenium ion apply to transition state - build up of positive charge in same place).
Predict the stereochemical outcome of electrophilic addition reactions of alkenes
If a stereogenic center is formed in the reaction, the products will be racemic as the carbenium ion is sp² hybridised and planar, equal probability of attack on either side of the planar carbenium ion
Draw and justify reaction mechanisms for nucleophilic addition to carbonyl compounds, as exemplified by cyanohydrin formation and nucleophilic attack by borohydride and organometallic species such as organolithiums and Grignard reagents,
Cyanohydrins - alcohol bonded to CN on same group as OH
Nucleophilic attack to carbonyl compounds (mainly aldehydes or ketones) proceeds by:
Nucleophile HOMO (lone pair on C in -CN) begins to interact with carbonyl LUMO which is the pi* orbital
Filling the pi* orbital causes the pi bond to break (BO is now 0)
Electrons from the pi bond end up as a negative charge on the more electronegative atom - which is O
in forming cyanohydrins and nucleophilic attack by borohydride:
protonation of - charge on O by bonding with H+ molecule
in nucleophilic attack by borohydride:
Instead of breaking B-H bond in BH4- to form H- (which is non-nucleophilic as small and charge dense), B-H bond is transferred to the carbonyl compound with the H effectively taking electrons with it
in nucleophilic attack by organometals or grignard reagents (R-MgX where X halide):
Bond between the metal and carbon is polarised towards carbon, interacts with the carbonyl LUMO, bond forms between carbon from metal-carbon bond and carbon in carbonyl compound
Prochiral carbonyl compounds - achiral carbonyl compounds that can be converted to chiral carbonyl compounds with changing only one atom.
Carbonyl carbon atom is sp² hybridised and planar so nucleophile can attack from top or bottom side, forming a racemic mixture.
Draw and justify reaction mechanisms for nucleophilic substitution at acyl carbons (exemplified by the synthesis of esters from acid chlorides or anhydrides).
Nucleophilic substitution at acyl carbons with alcohol to form esters occur with the nucleophile being the OR- (where R is an alkyl group) and attacking the acyl carbon centre LUMO (pi*) which breaks the pi bond and forces the electron density towards the O to form a tetrahedral intermediate. Instead of protonating the O- in addition reactions, the C=O bond reforms with O- donating electron density to the C centre to form an ester. This occurs in the presence of a base.

Explain reactivity trends in SN1 and SN2 reactions as a function of leaving group ability,
More stable the anion, the better the leaving group (lower pKaH = better leaving group) As you go down group 7 with group 7 elements as leaving groups, rate of reaction increases as anions get bigger so the - charge is stabilised. Conjugate bases of strong acids make good leaving groups.
Explain reactivity trends in SN1 and SN2 reactions as a function of nucleophile (effects of charge, resonance stabilisation, periodic trends),
negatively charged better nucleophiles because electrostatic attraction stronger, usually higher electron density to donate
higher pKaH (more basic) of nucleophile is more reactive, faster rate of reaction
resonance stabilised nucelophiles are less good nucleophiles, harder to donate an electron pair if shared about molecule
as we go down a group, tends to get more nucleophilic (e.g. S compounds better nucleophile than O compounds because larger molecule, less electronegative, hold electrons less well)
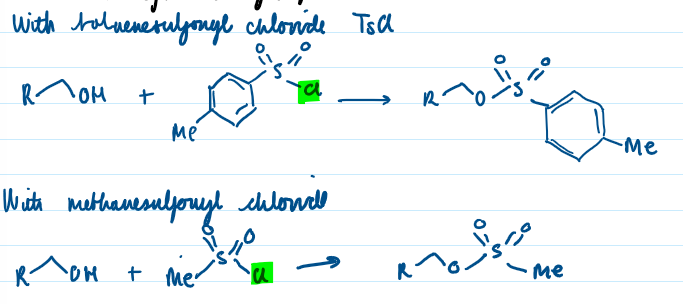
Suggest appropriate electrophiles for synthetically useful SN2 reactions, including the activation of alcohols by conversion to their sulfonate esters,
Alcohols converted to sulfonate esters which can undergo SN2 (OH on alcohol bad leaving group with high pKaH, replace the H on OH with something else) through treatement with base, reacting with sulfonyl chloride where chloride leaves the molecule and sulfonate ion bonds to the O of the OH group. Toluenesulfonyl chloride and methanesulfonyl chloride usually used. Sulfonate ion now is a better leaving group so can undergo SN2 reactions.
Tertiary compounds react by SN1, primary by SN2 due to increased stability of the tertiary carbocation during SN1 mechanisms. This stability is because of inductive effects as alkyl groups have high electron density and are electron donating sigma bonds, pushing electron density towards the positively charged C, reducing the positive charge. Stability also because of hyperconjugation: donation of electron density from adjacent C-H sigma bonds in to the empty p orbital of the carbocation (as is sp² hybridised)
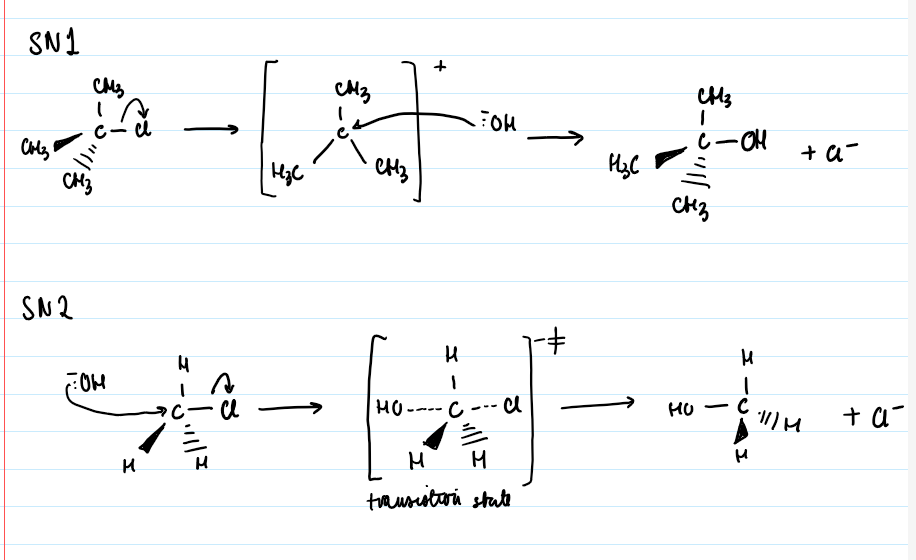
SN1 produces racemic mixtures (no optical activity) whereas SN2 reactions always result in complete inversion of configuration, same angle but opposite to starting compound when polarimetry performed.
SN1 is unimolecular, 2 steps:
Leaving group departs (RDS) forming a carbocation intermediate
Nucleophile attacks the carbocation, forming the product
SN1 mechanism has 2 transition states (maximums) and 1 intermediate (minimum - the carbocation).
SN2 is bimolecular, 1 step
Nucleophile attacks substrate simultaneously as the leaving group departs.
No intermediates, only 1 transition state where the carbon centre is partially bonded to both the nucleophile and the leaving group.
