Experiment 7 (week 2) - purification of lactate dehydrogenase from chicken breast tissue by affinity chromatography
1/84
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
85 Terms
Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE)
relies upon the same general principles that you are already familar with, but due to the high structural variation of proteins, there are additional technical details that this type of electrophoresis depends on.
the migration of a charged particle in solution under the influence of an electric field can be represented by the equation
v = velocity of the charged particle
E = strength of the electric field to which the charged particle is exposed.
q = net charge of the particle
f = frictional coefficient of the particle as it moves through the solution.
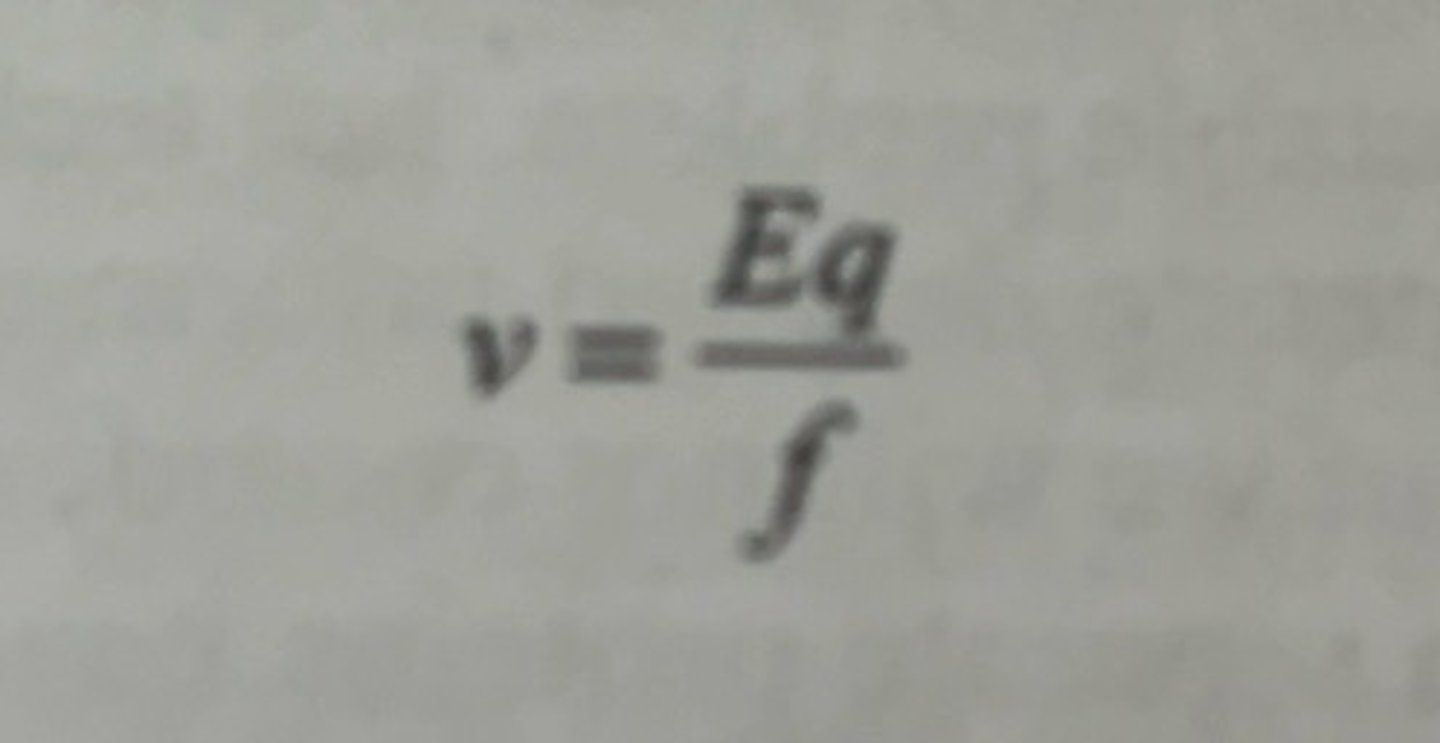
uniformity of DNA structure
DNA molecules of different sizes had similar q/f ratios and therefore would behave similarly in solution.
-this is very different for proteins
what are proteins
proteins are polymers of 20 different amino acids, and some amino acids have ionizable side chains, some of which will be deprotonated depending on the pH of the solution into which the protein is dissolved.
what does the charge of the protein depend on
depends upon the actual amino acids that are present, and the pH of the solution
-the effect of this is that proteins always have a charge, and the net charge of the protein (q) is the sum of al the various charges of these ionizable side chains at any particular pH.
-since different proteins have different amino acid compositions, different proteins will have a different charge at a particular pH which means of course that the numerator will be different for different proteins.
variety of charge
-heterogeneity
protein shapes
a protein exists in a variety of shapes and sizes
-some proteins are spherical, other proteins can exist as rods, but the vast majority of proteins have very complex shapes and sizes meaning that the frictional coefficient is highly variable.
native structure
inherent charge, shape and size of a protein
variability in migration
there is potential variety of protein native structure so there is variable migration velocity of proteins during electrophoresis because q and f parameters are different for every protein
electrophoresis scenario
you have a protein named A with a charge of -5 with an imaginary frictional coefficient of 10
-you also have a second protein, B, with a charge of -10 but it is much bigger than protein A and has a frictional coefficient of 20.
-if you tried to separate these 2 proteins by electrophoresis, they would have the same velocity because the ratio of their charge to size/shape (q/f) is the same and both proteins are subject to the same electric field.
complicated electrophoresis scenario
-you have a protein C that has a net charge of -2 and a frictional coefficient of 10.
-you have protein D that has a net charge of -20 and frictional coefficient of 20.
-you have protein E that has a net charge of -8 and a frictional coefficient of 7.
-if you ran protein A, B, C, D and E, their relative electrophoretic velocity would be E>D>A=B>C
-this example reveals that if you electrophoresed these proteins and you knew nothing about them apart from how fast they migrated, you could not draw conclusions about their relative size and charges.
the challenge of protein electrophoresis
-proteins can also have positive charges, which would mean that they run in an opposite direction to the negatively charged proteins; these positively charged proteins would of course exhibit the same structural complexity as the negatively charged proteins.
solution
techniques have been developed that specifically exploit this variety to generate detailed information about the protein composition in cells, namely 2-D electrophoresis.
-we can also use SDS PAGE.
SDS PAGE of proteins
electrophoretic method that separates proteins from each other on the basis of the length of their polypeptide chains.
-the most commonly used technique in a biochemistry lab for the evaluation of protein purity and for obtaining some basic structural information about a protein.
what does SDS only worry about
separates proteins on the basis of size alone
what are the 2 reasons why SDS PAGE can separate proteins on the basis of size
1. during the preparation of the proteins for loading on the gel they all are structurally altered to give them the same q/f ratio
2. they are electrophoresed through a sieving porous structural support which we are calling gels.
how is a sample prepared using SDS
-when a protein sample is prepared for SDS PAGE, it is heated to about 95 degrees C for 5 minutes in loading buffer that contains beta-mercaptoethanol and sodium dodecyl sulfate (SDS).
-the 3D structure of the protein is destroyed (denatured).
why does the structure of the protein get denatured
the combination of high temperature and the reduction of disulfide bonds between cysteine residues by the beta-mercaptoethanol.
how does SDS work
-SDS associates with the peptide bonds of the protein at a constant ratio of approximately 1.4 g of SDS for every 1 g of protein.
charge of SDS
SDS carries a negative charge
charge of denatured protein
it is now covered with negative charges that greatly exceed the original charge on the protein.
structure of SDS
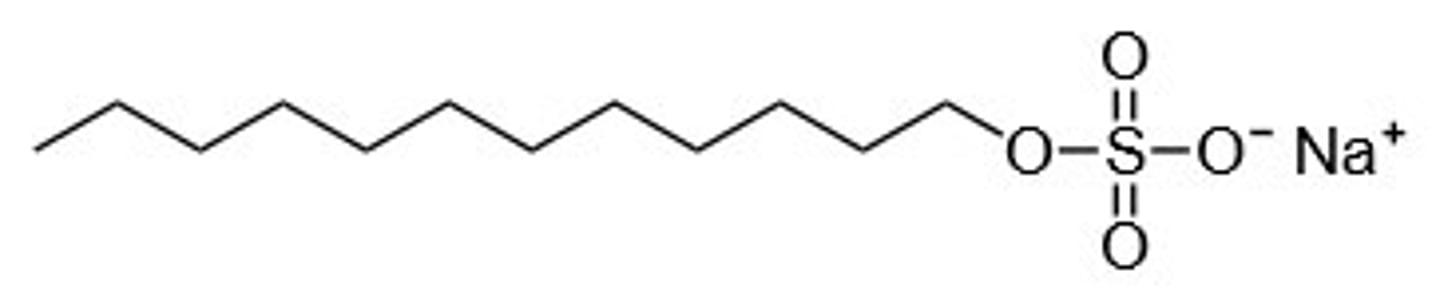
what is the combined effect of SDS
-all the proteins now roughly look like long rods coated with negative charge.
-proteins of different sizes will now be rods of different lengths, but all will have similar q/f values (a structural scenario you already saw with DNA).
what is added to the loading buffer
the loading buffer will also contain either glycerol or sucrose to give the loading buffer greater density so that when it is applied to the wells of the gel, the sample sinks directly to the bottom of the well.
Gel for SDS-PAGE
-Polyacrylamide gels
-based upon acrylamide.
how are polyacrylamide gels formed
-formed by the cross-linking (polymerization) of acrylamide with bisacrylamide in a 37:1 ratio in the presence of the initiator catalyst system of ammonium persulfate and TEMED.
TEMED
-it is the catalyst.
-it causes the formation of free radicals from the ammonium persulfate which induces the polymerization of the acrylamide molecules into long chains with the occasional bisacrylamide forming a cross-link joining 2 polyacrylamide polymers.
what does TEMED form
a mesh network
-it generates a gel that is characterized as having numerous pores of varying sizes
when does the average pore size decrease
with higher concentration of acrylamide.
when the concentration of acrylamide exceeds 4%
the average pore size is sufficiently reduced such that solvent molecules can still easily pass through the gel unimpeded, but larger molecules like denatured proteins can only migrate through large pores.
distribution of pore sizes
-since there is a distribution of pore sizes, smaller proteins will have a greater number of pores that they can migrate through compared to the larger proteins.
what happens when you apply a protein sample to the top of the gel and activate the voltage
the negatively charged proteins will begin to migrate towards the positively charged bottom of the gel.
what size molecule migrates faster
since the smaller proteins are able to more easily find a pore that they can fit through, they migrate faster down the gel than the larger proteins
for SDS PAGE, what is it frequently used with
a discontinuous gel
discontinuous gel
-the bottom portion of the gel is called the separating or resolving gel
-a stacking gel sits atop the separating gel and functions primarily to focus the protein sample into a tight band before the separation of size that occurs in the separating gel.
acrylamide concentration of the resolving gel
6-15%
-depends on application
acrylamide concentration of stacking gel
4%
positive pole
the bottom of the gel
-the negatively charged modified protein molecules will migrate down their lanes from directly beneath the wells into which they were loaded.
figure of discontinuous SDS PAGE
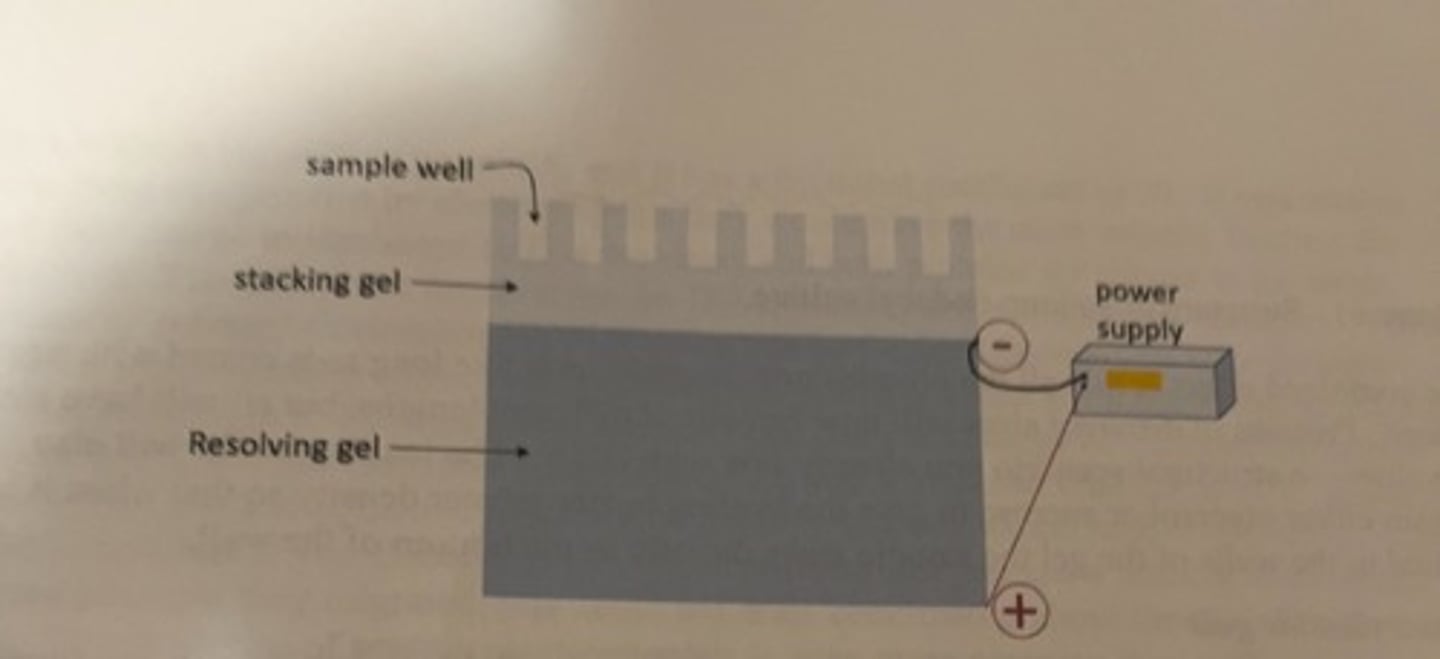
loading the gel
-the micropipettor tips are used to load.
-they have long, flexible tips that enable you to insert the tip between the glass plates and load sample directly into the well.
how much samples is loaded
-typically, one loads between 10-20 ul of a mixture of your sample in loading buffer in its designated well
if sample has low concentration of protein
you load more
visualizing the proteins on the gel
after the electrophoresis is completed, one needs to add some kind of dye in order to visualize the location of the proteins that have been separated.
what is the most common dye method
Coomassie-based stain
-recall that Coomassie is the dye that is used for the Bradford protein quantitation assay
how long is the gel stained for
10-20 minutes
how is the gel stained
-stained in a mixture of the dye, methanol and glacial acetic acid
glacial acetic acid
allows the dye to reach the protein embedded in the gel
problem with coomassie
this method effectively stains the proteins, but it also stains the gel itself blue which obscures the stain that is bound to the protein.
solution
to actually see the proteins, the gel needs to be de-stained to remove stain that has not bound to protein in the gel.
de-stain solution
-usually a glacial acetic acid/methanol mixture.
how long does de-staining take
24-48 hours depending on the de-staining conditions.
once the background stain is sufficiently removed to reveal the protein bands on the gel
some record of the results of the electrophoresis must be obtained for later analysis.
how can results be read
-in some cases, the gel itself is sandwiched between 2 layers of plastic film and stored in a lab notebook, or the gel can be imaged using either a digital camera or specialized piece of gel imaging equipment.
what will gel look like
when a sample of protein is run on this type of gel, the proteins separate on the gel vertically in lanes. each column of bands is a lane, and reveals the distribution of sizes of protein content of a sample that was originally loaded in the well in the stacking gel.
-the proteins higher on the gel are larger than those lower on the gel.
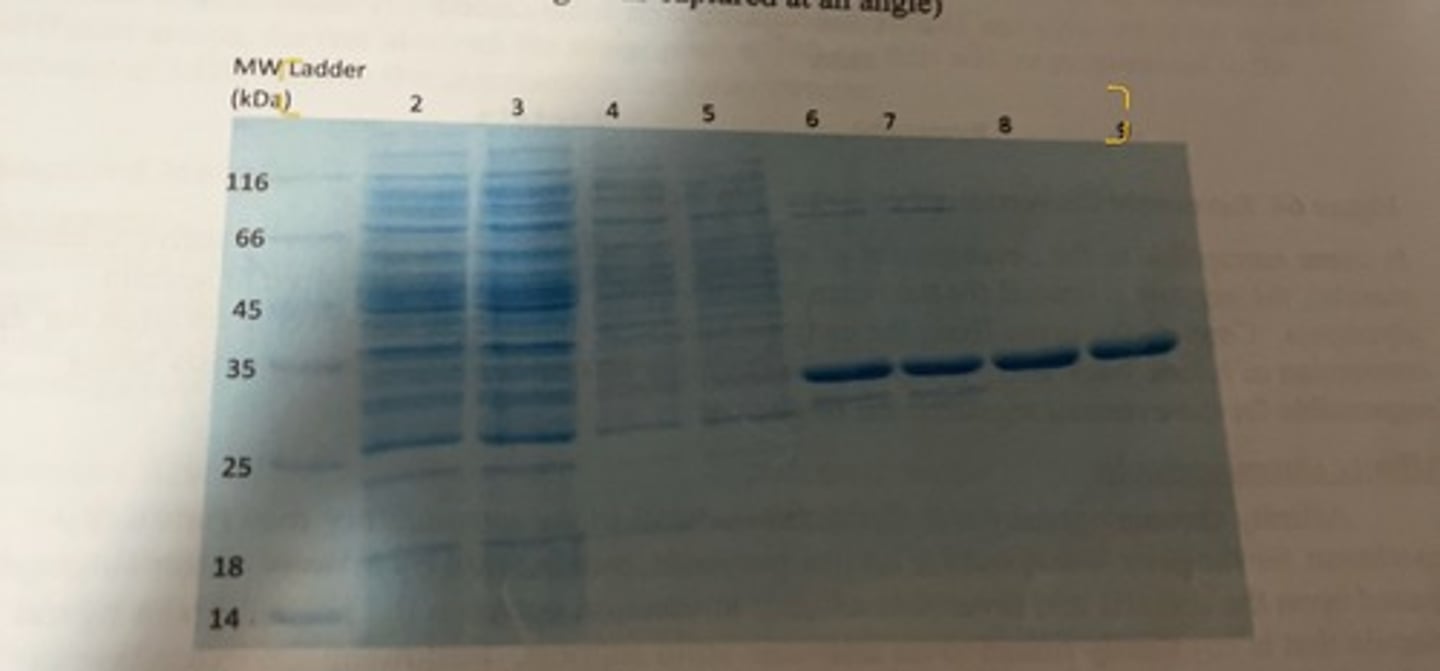
how can the purity of a protein sample be assessed
by the number of bands that are present.
a single band
suggests a pure protein
numerous bands
usually indicate that different types of proteins are present in the sample that was electrophoresed.
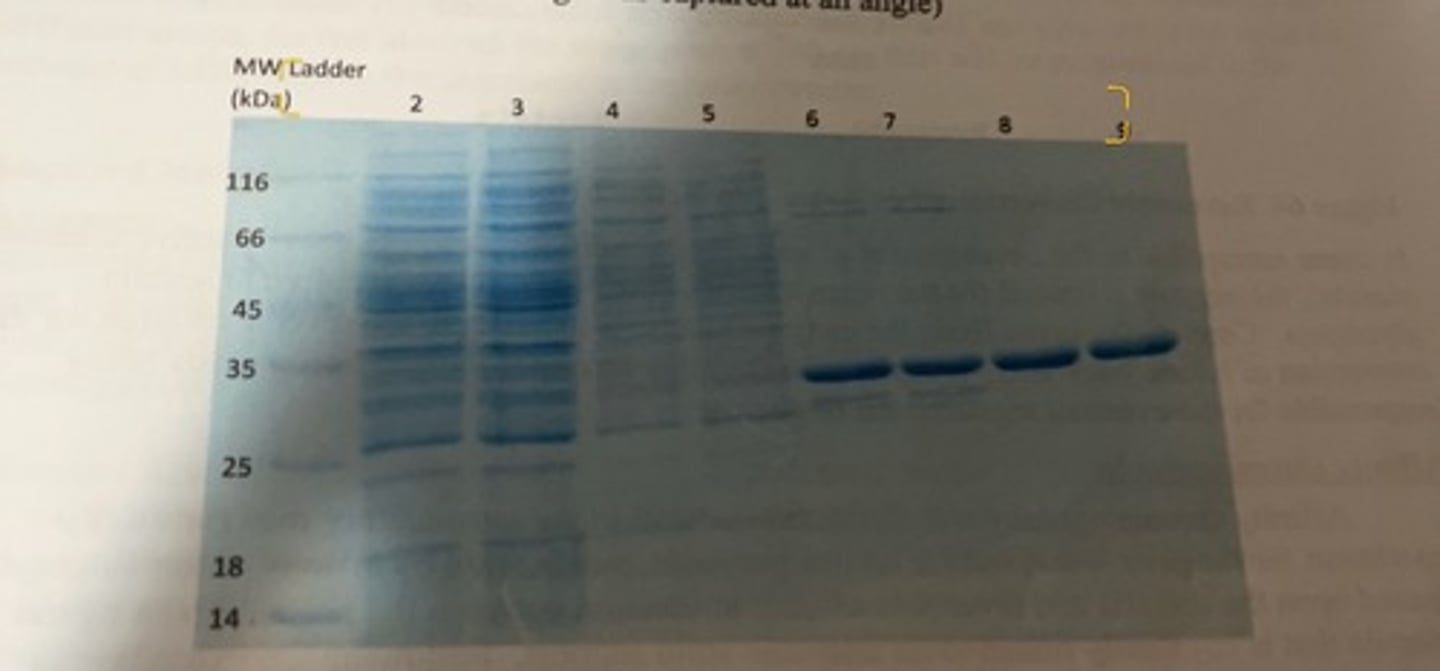
E. coli specific protein gel
-approximately 28 kDa
-this shows 2 different purification methods.
-lane 2 and 3 depict all of the various proteins that are present in the cell lysate and one can see that there are numerous bands of different sizes proteins that are present.
-land 6 and 7 are for the purified fraction following affinity chromatography. there are still 4 or 5 distinct bands visible including the protein we want (the thick dark band) which indicates that the purified fraction is not fully pure. the affinity chromatography purified fraction was then loaded on a IEX column and different fractions were collected as the concentration of NaCl was slowly increased.
-lanes 8 and 9 represent the purest fraction for the desired protein, and one can see that is it now highly pure as only a single band is visible.
in experiment 7 week 2, what was purified
lactate dehydrogenase (LDH) from chicken breast tissue
what technique was used
affinity chromatography
what is the goal
measure the amount of enzyme activity at various stages of the purification by spectrophotometric enzyme assay
-you will also perform several Bradford assays to enable you to calculate specific activity and the purification factor for the purified enzyme.
-you will also prepare your SDS PAGE gel that you will use for week 3 to analyze your purifications.
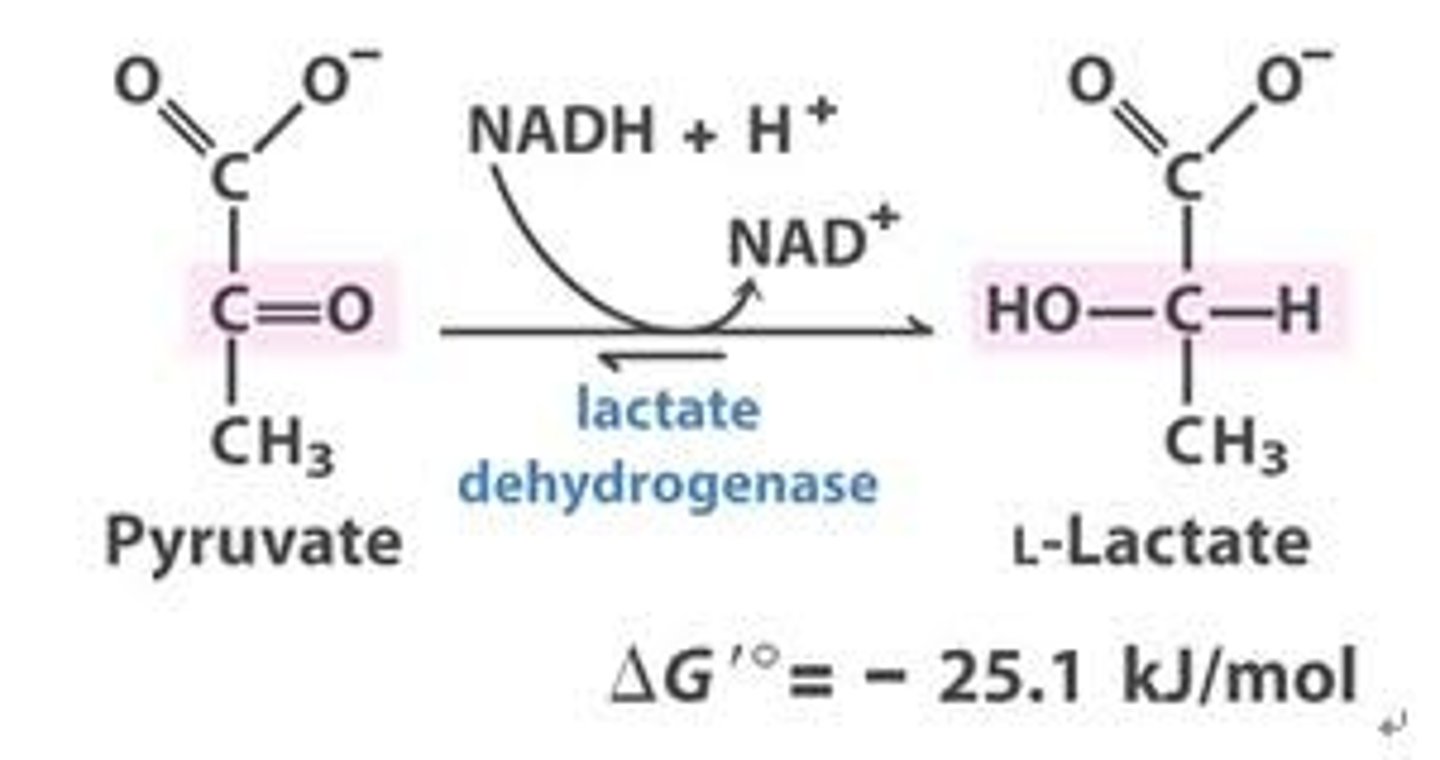
lactate dehydrogenase reaction
catalyzes the reversible reduction of pyruvate into lactate and is abundant in many tissues including the heart, liver and skeletal muscle.
tissues susceptible to the development of anaerobic conditions
-heavily active skeletal muscle
when is lactate dehydrogenase crucial
in tissue susceptible to the development of anaerobic conditions, the enzyme is crucial for the regeneration of the NAD+ that is required to sustain glycolysis.
reverse reaction
in the liver, the enzyme functions to catalyze the reserve reaction for the conversion of lactate back into pyruvate
-the other enzymes for gluconeogenesis are responsible for the eventual regeneration of glucose.
purification by affinity chromatography
-based upon the specific and reversible affinity interaction between the protein and a ligand molecule that is covalently linked to an insoluble solid support.
how does affinity chromatography work
following the extraction of proteins from the cells of the tissue into solution, a small volume of the crude lysate is applied to the top of the column and allowed to flow down into the column. once the sample has entered the column, a wash buffer is continuously added to the top of the column to promote the flow of protein solution within the column and to prevent the column from becoming dry. proteins that have an affinity for the ligand affixed to the solid support will bind to the column, whereas proteins that have no affinity for the affixed ligand will simply flow through the column unimpeded. ideally, after the addition of a volume of wash buffer equal to several times the volume of the column itself, only the protein that had an affinity for the affixed ligand will remain in the column as the rest of the non-binding proteins and other cellular material will have been washed away. the bound enzyme can be removed from the column by the addition of an elution buffer.
elution buffer
will be a high concentration of a ligand to which the protein will bind.
what happens with the addition of elution buffer
-the protein will bind to the ligand in the elution buffer (which is flowing down through the column), more frequently than it binds to the ligand affixed to the solid support, with the result that protein is eluted from the column.
what ligand-solid support is used in the experiment
Cibacron blue F3GA
-it is marketed as Blue Sepharose TM 6 Fast Flow
-has a functional group that resembles NADH
-the bound LDH will be eluted with 2 mM NADH
assay
one must have some type of assay to measure the amount of protein of interest that is present at any step of the purification procedure.
-since the protein you are attempting to purify for this experiment is an enzyme, you will be able to determine the amount of the enzyme that is present by performing a spectrophotometric assay.
what is this assay based off of
-the assay for your purification is based upon the fact that the coenzyme for LDH (NAD/NADH) absorbs light differently depending on its chemical state.
the reduced for, (NADH)
absorbs light at 340 nm
the oxidized state (NAD)
does not absorb light at 340 nm
how can the progress be followed
-for each mole of pyruvate converted to lactate by the enzyme, 1 mole of NADH will be oxidized to NAD+
-the progress can be followed by measuring the decrease in absorbance at 340 nm.
-if the concentration of NADH and pyruvate is the same for different assays, the rate at which the absorbance at 340 nm falls will be proportional to the amount of LDH enzyme that is present in the assay cuvette.
what column do we use
equilibriated cibacron blue F3GA affinity column
acrylamide
a potential neurotoxin
safety precautions for acrylamide
-wear gloves during gel preparation
-after the gel has polymerized, the falcon tube with the polymerized gel in it may be discarded into the trash
how do you monitor the polymerization of your gel
by inspecting the remaining resolving gel polymerization mixture in the falcon tube that you did not use.
how was the chicken breast prepared
the chicken breast lysate was prepared by grinding chicken breast with cold lysis buffer and then centrifuging it at 10,000 x g to remove tissue and cellular debris. the supernatant from the centrifugation step is the lysate
where do we expect to find the most lactate dehydrogenase in test tubes
the fractions that contain flow-through immediately following addition of the elution buffer.
what does TEMED do
promotes the polymerization of the acrylamide
-you will have limited time to get it right once you have added the polymerizing stacking gel mixture between the plates.
how is the polymerized gel stored
-stored in the polymerization bag with the comb still in the gel.
-remove the polymerization bag containing the gel from the casting block and close the open end of the polymerization bag using the ziploc seal.
how do you ensure the gel does not dry out
fold the top of the bag and seal it with masking tape.
-wrap a damp paper towel around the sealed polymerization bag.
-use another piece of tape to label the wrapped gel with your name.
why do we have to dilute the chicken lysate
for the enzyme and protein assays because there is so much material in the undiluted lysate that we would not be able to obtain accurate results.