Protein structure part II
1/40
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
41 Terms
single polypeptide chains that contain - residues are able to form distinct structural domains
domains are connected by relatively - stretches of the pp chain which act as -, allowing for domains to move about freely and independently of one another
domains often possess unique functions within the protein = -
having domains connected in a single protein permits much more - of its function
>100-200
unstructured, hinges
functional domain
efficient regulation
multidomain proteins arose when DNA sequences that encoded each domain became -, resulting in a new gene = -
joined, domain shuffling
modules are smaller, highly - structural/functional units that readily - into new proteins during evolution via -
conserved, integrate, gene or exon duplication and insertion
duplication of gene
subfunctionalization
neofunctionalization
degeneration/gene loss
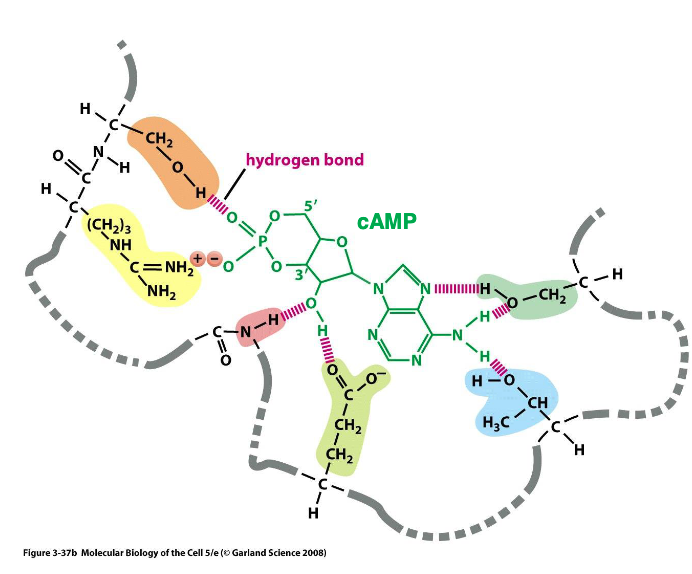
cAMP binds reversibly to the - domain, which is present in a diverse range of proteins performing vastly different biological function
cyclic AMP binding
-(three things) binding modules are present in a wide range of proteins, not just a particular protein family
cAMP, ATP, GTP
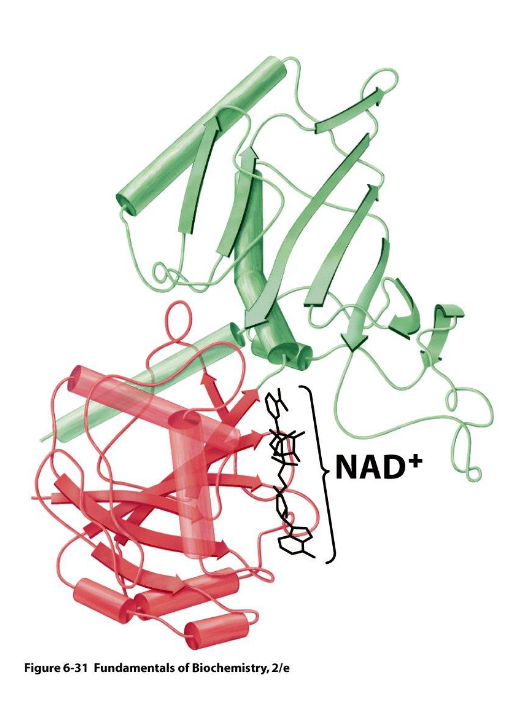
domains perform - functions in enzymes
ex. glyceraldehyde 3 phosphate dehydrogenase
1st domain binds -
2nd domain binds the - substrate and catalyzes its -, using NAD+ as an -
complementary
NAD+
GAP, oxidation, e- acceptor
domains and modules may be evolutionary -
domains and protein structures are conserved more than -
domains arose and were maintained over evolutionary time because they were able to
form -
tolerate amino acid - (three things) without losing stability or function
support essential biological - or form the basis for - new ones
sinks
AA sequence
stable protein folding patterns
deletions, substitutions and insertions
functions, advantageous
tertiary structure forms from interactions between lengths of amino acids from - of the polypeptide chain
the forces involved in folding a protein into its final tertiary structure are essentially the same as those discussed for secondary structure but in contrast to secondary structure, tertiary structure depends almost entirely on the interactions between -
the polypeptide chain folds, coils, and twists into the native conformation, which represents a stable (-) state for that particular sequence of amino acids
different regions
R groups
low delta G
why do we observe only a few thousand common folding patterns (domains)?
evolution and natural selection, if it aint broke, don’t fix it
gene duplication and mutations that created advantageous new functions from an already functional and stable protein were selected and maintaned over time
protein families are evidence of conservation, members perform similar functions and have homologous amino acid sequences and 3D conformations
Levinthal Paradox
protein folding cannot be a random process
the more + a hydropathy value, the more likely a side chain will be found in the - of a protein and vice versa
interior
non-polar residues like Leu, Ile, Met, Val, and Phe occur mostly in the - of proteins
polar residues like Asn, Gln, Ser, Thr, Tyr are usually on the protein -
charged residues like Asp, Glu, Arg, His, Lys are typically found on the -
interior
surface
exterior
a folded protein is - (less/more) energetically favourable
this is due to the - effect
hydrophobic core regions contain non-polar side chains, once protein is folded they do not need to be surrounded by -, those water molecules can go and be -
more
hydrophobic
water cages, chaotic
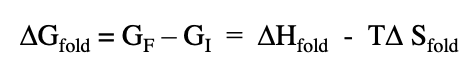
delta G fold refers to
stability of the final folded state vs stability of the initial unfolded state
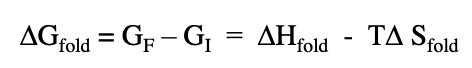
delta H fold refers to
difference between amount of energy to break all bonds in the unfolded state vs breaking all bonds in the folded state
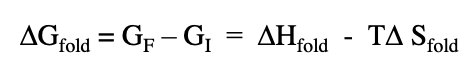
T delta S fold
difference in stability associated with molecular motion or freedom
folding is primarily - driven and is a competition between the - of the folded polypeptide vs the - of the bulk water
stabilization forces that to occur during polypeptide folding to produce a favourable enthalpy that tilts energy equation towards - and a -
entropically, reduced entropy, increased entropy
folding, net negative delta G fold
overall, delta G fold is slightly negative, but there is not much -holding a folded polypeptide together
this explains why many proteins can be easily - by increased temperature or detergents that disrupt hydrophobic interactions
net free energy
denatured
Forces that stabilize native protein structure
hydrophobic effect
VDW
H-bonding
electrostatic interactions
chemical cross-links
hydrophobic effect is a major - to initiate protein folding
driver
because the interiors of native proteins are closely packed, VDW are a major - force of - polypeptides
- proteins do not benefit from VDW because distances between molecules are too -
stabilizing, folded
unfolded, great
H bonds make only a - contribution to overall protein stability during - process
unfolded proteins can also form H bonds with -
H bonds play a key role in determining and refining - though
if protein folded in a way that prevented H bonding, the stabilization energy would be - and the structure would be -
minor, folding
water
folded structure
lost, unstable
association of two ionic residues of opposite charge is called an - or -
75% of charged residues are involved in ion pairs and are located mostly on the protein -
ion pair, salt bridge
surface
disulfide - bonds form within and between polypeptide chains as they fold into their native conformations and/or subunits associate
cytoplasm is a reducing environment, so intracellular proteins do not contain -
covalent
disulfide bonds
cationic metal ions can coordinate several - around them to form discrete domains or motifs
charged a.a. residues
protein denaturation
heat
pH
detergents
chaotropic agents
detergents associate with - residues and interfere with - interactions
nonpolar, hydrophobic
chaotropic agents are - or - that increase solubility of - residues in water
ions, small organic molecules, nonpolar
in vivo, chaperones are involved in folding of many proteins to -
ensure correct folding, prevent aggregation, correct misfolded proteins
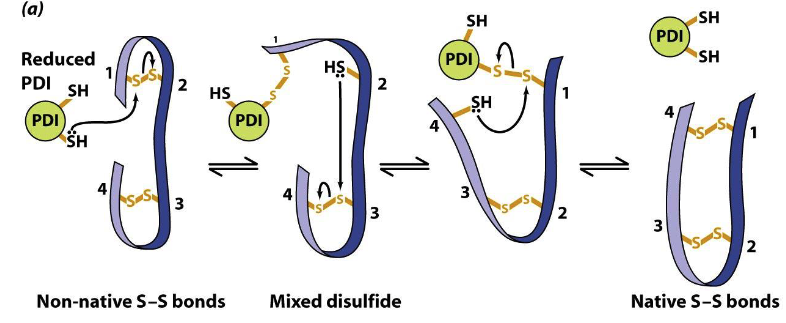
re-arrangement of non-native disulfide bonds via reduced PDI
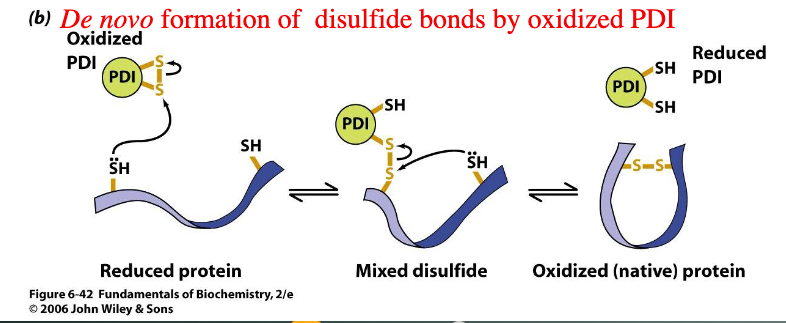
de novo formation of disulfide bonds by oxidized PDI
there are 18 human diseases caused by deposition and accumulation of insoluble protein aggregates, collectively called -
amyloids
when enough amyloid accumulates in the affected tissue, normal cellular functions are -, leading to cell - and eventual -
disrupted, death, organ failure
amyloid structures have striking similarities
the likelihood of at least 2 mutant polypeptides aggregating to nucleate the fibril is very low, which presumably explains the long latent period for spontaneous amyloid diseases
gross assemblies of beta sheet structure, which is virtually indestructible
in Alzheimers gamma secretase cleaves APP into AB fragments that can aggregate into - and eventually large fibrils, referred to as -
oligomers, plaques
accumulation of Tau, a - protein, causes - within neuronal cells, which also lead to neuronal cell -
Tau appears to be - by accumulation of AB oligomers
microtubule stabilizing, tangles, death
dysregulated
prions are - versions of WT proteins
misfolded