Applications of pathogen genomics
1/33
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
34 Terms
What is the role of genome sequencing in studying pathogenic microorganisms?
Genome sequencing identifies all the genes and mutations present in a microorganism.
This allows scientists to detect genes related to virulence, transmission, and antimicrobial resistance, helping track how pathogens evolve and spread.
How does whole genome sequencing (WGS) help understand drug resistance?
WGS reveals mutations in genes that confer resistance to antibiotics. For example, mutations in the gyrA gene can cause fluoroquinolone resistance, while beta-lactamase genes break down beta-lactam antibiotics.
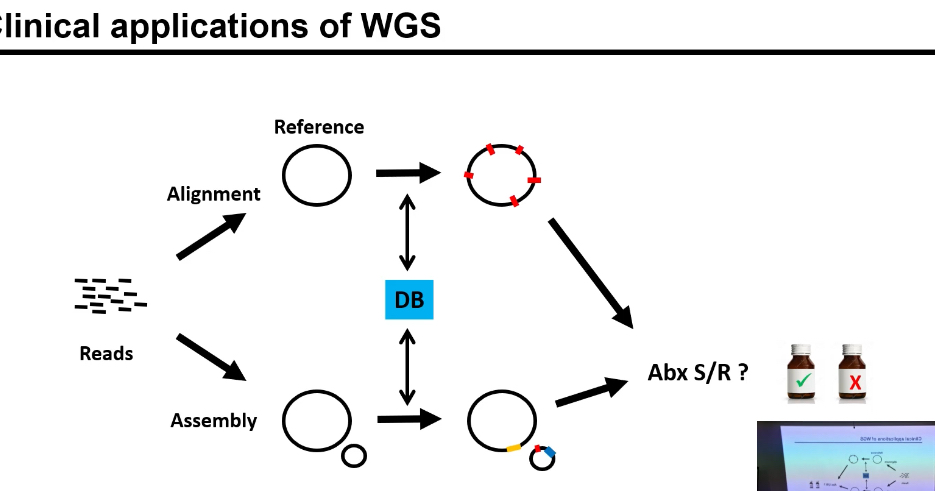
Whole-genome sequencing (WGS) is performed on a bacterial isolate.
Sequencing produces many short DNA reads
Two parallel analysis routes
1⃣ Alignment-based approach
• Reads are aligned to a reference genome.
• Differences between sample and reference are identified:
• SNPs,
• insertions/deletions,
• resistance mutations.
• These variants are compared against a resistance database (DB).
• Known resistance markers are detected.
• This is used to predict antibiotic susceptibility or resistance (Abx S/R?).
2⃣ Assembly-based (de novo) approach
• After WGS, the short DNA reads are not aligned to any reference genome.
• Instead, reads with overlapping sequences are stitched together to reconstruct the genome as accurately as possible.
• This produces contigs (long continuous stretches of DNA, but usually not a single complete chromosome).
• The contigs are then annotated to identify genes and genetic elements.
• Identified genes are compared against a known antibiotic resistance gene database (DB).
• This approach is especially good at detecting:
• Entire resistance genes,
• Plasmid-borne resistance,
• Mobile genetic elements (transposons, integrons).
• Based on which resistance genes are present, antibiotic susceptibility or resistance (Abx S/R?) is predicted.
What are the three main mechanisms that lead to antimicrobial resistance?
1) Modification of drug targets (e.g., gyrA mutation for fluoroquinolone resistance),
2) Enzymatic degradation of drugs (e.g., beta-lactamases),
3) Altered drug influx/efflux (efflux pumps expel drugs or porins reduce uptake).
Why is identifying genetic markers for resistance important?
Genetic markers help predict whether a pathogen is resistant or sensitive to a drug, enabling faster and more precise antibiotic selection without relying solely on slow phenotypic tests.
What is resistance genotyping?
Resistance genotyping uses genome sequencing to detect known resistance genes or mutations and predict whether a pathogen will respond to a specific antibiotic.
Step 1: Obtain the bacterial sample
• Sample comes from:
• blood,
• urine,
• sputum,
• wound swab, etc.
• Contains many bacterial cells of the same strain.
Step 2: Extract bacterial DNA
• Lyse the bacterial cells.
• Purify genomic DNA (and plasmid DNA if present).
• This DNA contains:
• chromosomal genes,
• plasmid-encoded resistance genes.
Step 3: Choose resistance targets
• Decide what resistance you’re testing for.
• Targets can be:
• known resistance genes (e.g. β-lactamase genes),
• known SNPs in drug target genes.
• These targets are known markers of resistance.
Step 4: Detect resistance genes / mutations
PCR-based genotyping (most common clinically)
• Design primers specific to resistance genes or mutations.
• Run PCR.
• Result:
• band present → gene present,
• band absent → gene absent.
qPCR
• Same idea as PCR but quantitative.
• Faster and more sensitive.
• Often used for rapid diagnostics.
Sequencing-based genotyping
• Sequence:
• specific resistance genes, or
• whole genome (WGS).
• Compare sequence to resistance databases.
• Detect:
• known genes,
• point mutations,
• new variants.
Step 5: Analyse the genotype
• Identify:
• which resistance genes are present,
• which alleles or mutations they carry.
• This produces a resistance genotype.
Example:
• bla gene present → β-lactam resistance
• gyrA mutation → fluoroquinolone resistance
Step 6: Interpret resistance phenotype
• Translate genotype → predicted resistance.
• Some genes:
• guarantee resistance,
• others depend on expression/context.
• Results are often combined with:
• phenotypic susceptibility testing.
What challenges exist when predicting resistance using genomics?
Challenges include multiple resistance mechanisms, numerous contributing genes, different lineages, and expression-based mechanisms (like efflux pumps) that may require transcriptomic data.
How do efflux pumps contribute to antibiotic resistance?
Efflux pumps actively remove antibiotics from the bacterial cell, reducing the drug’s intracellular concentration.
They can make bacteria resistant to multiple drugs simultaneously.
What databases are used in resistance genotyping?
CARD (Comprehensive Antibiotic Resistance Database), ResFinder (for acquired resistance genes), and ReSeqTB (for TB strain resistance data) are major curated bioinformatics resources.
How is WGS used in clinical applications for tuberculosis?
TB isolates are sequenced to predict drug resistance profiles, identify lineages, and track transmission. UKHSA now routinely sequences all TB isolates in England to inform clinical decisions.
What was the NIX-TB trial and how was genomics applied?
The NIX-TB trial sequenced 355 Mycobacterium tuberculosis isolates to study resistance to Pretomanid. Mutations or deletions in the ddn gene were associated with Pretomanid resistance.
What is GWAS and how is it used in pathogen genomics?
Genome-Wide Association Studies (GWAS) identify statistical links between genetic variants (like SNPs) and phenotypes such as antibiotic resistance. It helps discover new resistance genes(markers)
How does a GWAS plot show significant associations?
The x-axis shows genetic variants and the y-axis shows their -log10(p-value). Peaks above a significance threshold indicate variants strongly associated with the phenotype, like resistance.
What are the four main approaches used in GWAS for pathogens?
1. SNP-based GWAS
• Tests single nucleotide polymorphisms one position at a time.
• Reference-based: SNPs are defined relative to a reference genome.
• Most classic human GWAS use this.
• Limitation: misses large insertions, deletions, or novel sequences.
2. Gene-based GWAS
• Variants are grouped by gene instead of tested individually.
• Tests whether variation within a gene is associated with the phenotype.
• Requires annotation (you must know where genes are).
• Useful for rare variants with small individual effects.
3. k-mer-based GWAS
• Uses short DNA sequences of length k (e.g. 31 bp).
• No reference genome required.
• Detects:
• SNPs,
• indels,
• structural variation,
• presence/absence variation.
• Especially useful in bacteria and plants.
4. Unitig-based GWAS
• Unitigs are longer, non-redundant sequences built from overlapping k-mers.
• Represent variable regions across genomes.
• Best for pangenome analysis (core + accessory genome).
• Assess variation across the pangenome, not just a single reference.
Concrete example:
Sequence: ATGCTAGC
k = 4 → ATGC, TGCT, GCTA, CTAG, TAGC
These k-mers overlap heavily and are redundant.
A unitig represents this region once instead of many times.
You do not start with a known reference genome.
You start with:
a bacterial isolate, and
its sequencing reads from WGS.
From those reads, you:
extract k-mers, or
assemble unitigs.
These sequences represent the genome of that specific bacterial cell/isolate.
You then test whether the presence, absence, or frequency of those sequences is associated with a phenotype (e.g. resistance).
These approaches examine genetic variation across genomes or pangenomes to find associations with traits.
What are the advantages and limitations of using GWAS in infectious disease research?
Advantages:
It can identify both known and novel resistance markers, providing insight into multiple drug classes and guiding future diagnostic or therapeutic development.
Limitations:
It requires large isolate datasets, and expression-based mechanisms (e.g., efflux pumps) are difficult to capture. Population structure can also confound associations.
How does genomic surveillance work in public health?
WGS data from clinical isolates are compared to detect genetic similarities, construct phylogenetic trees, and identify clusters indicating outbreaks or transmission events.
How are phylogenetic trees interpreted in outbreak tracking?
Closely related isolates cluster together, suggesting recent transmission or a common source. Distant branches imply unrelated infections or different origins.
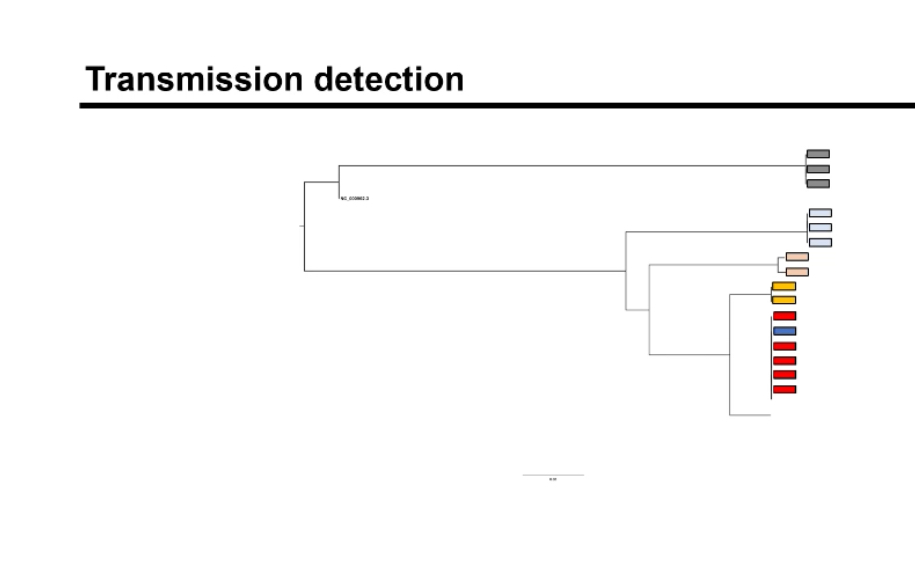
What was the 2011 E. coli outbreak in Germany and how did genomics help?
A virulent Shiga toxin-producing E. coli strain caused >40 deaths. Rapid genome sequencing and open-source data sharing identified the strain source and linked cases worldwide.
How did genome sequencing contribute to managing COVID-19?
Global sequencing enabled real-time tracking of SARS-CoV-2 variants, guiding public health interventions and informing vaccine design. Over 16 million genomes were sequenced by 2023.
What role does sequencing play in national surveillance systems?
It helps monitor transmission patterns, detect emerging variants, and inform interventions at local, national, and global levels.
What is the NeoHIEC study and what did it investigate and what were the findings?
The Neonatal-Healthcare associated Infection Epidemiology Cohort collected swabs from infants to study bacterial colonization and resistance transmission in neonatal units.
It found that Klebsiella and E. coli shared identical resistance elements (like CTX-M-15 on Tn3 transposons), suggesting horizontal gene transfer and cross-species exchange.
What is the significance of the CTX-M-15 beta-lactamase gene?
CTX-M-15 is an extended-spectrum beta-lactamase that breaks down cephalosporins. Its presence in multiple species highlights the spread of resistance through mobile genetic elements.
How do mobile genetic elements spread antibiotic resistance?
Elements like transposons and plasmids transfer resistance genes between species or strains, facilitating rapid dissemination of resistance within microbial communities.
How does WGS assist in TB clinical trial monitoring?
Sequencing pre- and post-treatment isolates helps distinguish relapse (same strain persists) from reinfection (new strain acquired), informing whether treatment failure occurred.

What are mixed infections and how do they affect genomic analysis?
Mixed infections contain more than one strain. They can complicate interpretation of resistance or relapse by masking minority variants, requiring computational separation of strain data.
What is k-means clustering and how is it used in genomic trials?
K-means clustering groups genomic data points to distinguish major and minor strains within a sample, helping identify mixed infections or reinfections in TB studies.
How does genomic data inform the diagnosis of infectious diseases?
It identifies pathogen species, strain types, and resistance genes directly from sequence data, allowing faster diagnosis compared to traditional culture-based tests.
How does genomic data improve treatment and management?
Clinicians can tailor antibiotics based on resistance genotypes, avoid ineffective drugs, and use epidemiological data to control transmission sources.
What is the microbiome and how is it related to pathogen genomics?
The microbiome is the community of microorganisms in a specific niche. Metagenomics sequences all microbial DNA, revealing both culturable and unculturable species and their genes.
How can microbiome analysis inform human health?
Studies link microbiome composition to diseases like asthma or Clostridium difficile infection. Fecal microbiota transplants can restore healthy microbial balance to treat infections.
What is the difference between microbiome and metagenome?
The microbiome refers to the organisms present, while the metagenome refers to the total genetic material recovered from that environment.
How do bioinformatics tools support genomic analysis?
Tools align sequence reads to reference genomes, assemble contigs, and compare to resistance databases to predict antibiotic sensitivity or resistance.
What are the steps in a sequencing and analysis workflow?
DNA extraction, sequencing (e.g. Illumina), read alignment, variant calling, resistance gene identification, phylogenetic analysis, and data interpretation.
How is genomic data visualized to identify outbreaks?
Sequence data are converted into phylogenetic trees or similarity matrices showing how closely related isolates are, which helps identify chains of transmission.
Why is time-to-result an important challenge in clinical genomics?
While WGS offers high accuracy, sequencing and analysis can be time-consuming, which may delay urgent treatment decisions compared to rapid diagnostic tests.