Module 2: Protein Structure and Function
1/77
Earn XP
Description and Tags
Module 2 + Applied Lecture 2
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
78 Terms
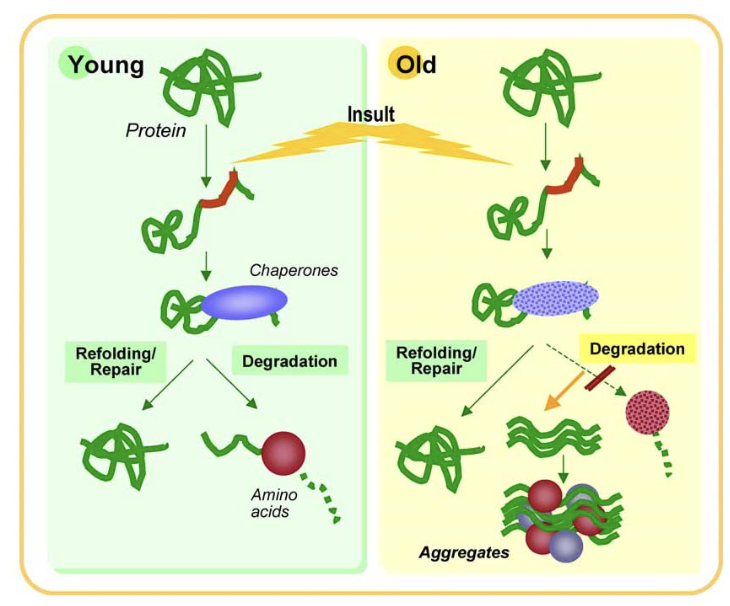
What happens when proteins fail to fold correctly in cells?
Most proteins fold correctly on their own using molecular interactions
If folding fails:
• Chaperones assist to prevent incorrect interactions
• If folding fails completely, aggregates may form (toxic to cells)
• Final solution: degrade misfolded proteins via the proteasome
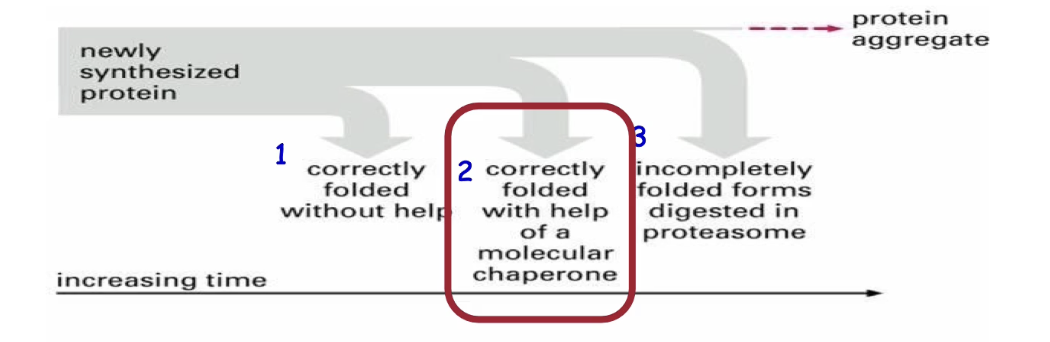
What are the two classes of chaperones and their general functions?
Classes:
Molecular chaperones (monomeric)
Chaperonin complexes (multimeric)
Functions:
Prevent inappropriate intramolecular and intermolecular interactions between amino acid residues
Help fold many proteins (not specific)
Ubiquitous—found in all organisms and compartments
How do molecular chaperones function, and what are some examples?
Bind hydrophobic residues on unfolded/nascent (newly formed) proteins to:
Prevent incorrect folding
Prevent premature folding
Prevent aggregation with other hydrophobic residues
Prevent inappropriate associations with other proteins
Do not direct folding—only prevent misfolding
Examples:
Hsp70 – cytosol and mitochondria
BiP – endoplasmic reticulum
DnaK – bacteria
Heat shock proteins (Hsp):
Expressed under stress (e.g., elevated temperature)
Help refold denatured proteins
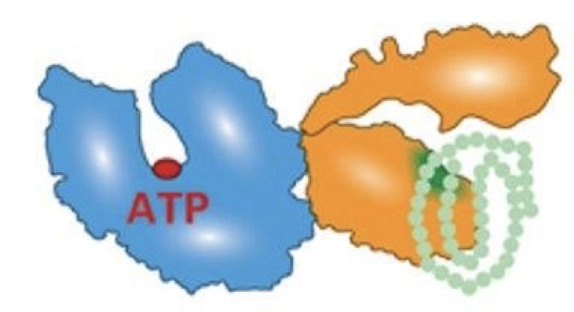
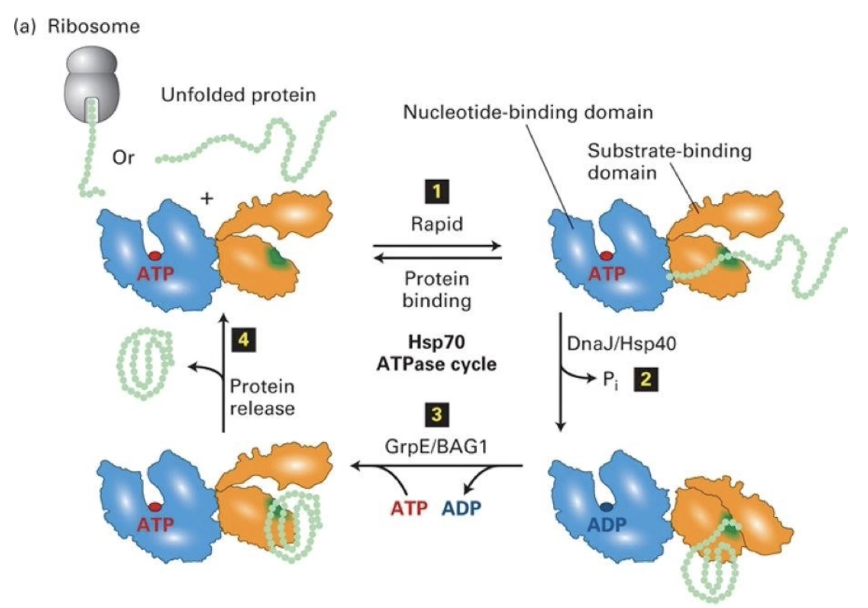
Describe how the Hsp70 molecular chaperone functions.
Composed of:
Nucleotide-binding domain (NBD): binds ATP
Substrate-binding domain (SBD): binds hydrophobic residues on unfolded proteins
Process:
Hsp70 binds unfolded protein
ATP hydrolysis to ADP (stimulated by DNAJ/Hsp40) causes conformational change
Protein folds
ADP released (assisted by GrpE/BAG1), ATP rebinds
GrpE/BAG1 is the nucleotide exchange factor
Folded protein released, cycle repeats
What are chaperonins and how are they structurally organized?
Large macromolecular complexes with internal folding chambers
Allow proteins to fold in isolation (prevent aggregation)
Examples:
TCiP (eukaryotic cytosol)
GroEL (bacteria and chloroplasts)
Hsp60 (mitochondria)
Structure:
2 large GroEL rings stacked back-to-back
Multiple proteins form the walls of the 2 GroEL subunits
GroES cap (lid) alternately seals top of each GroEL chamber
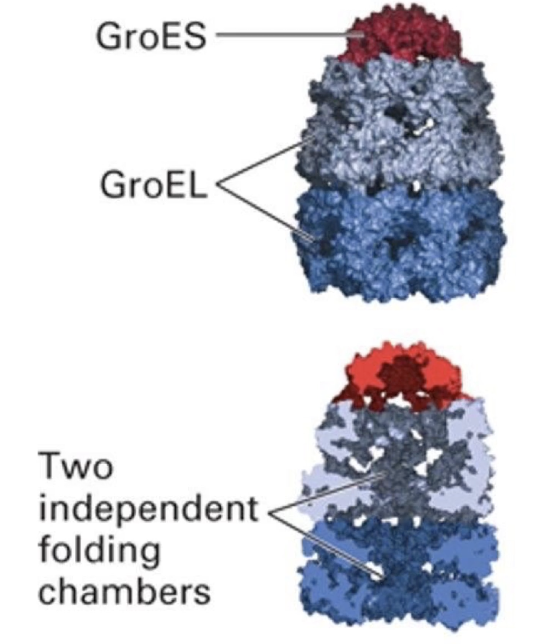
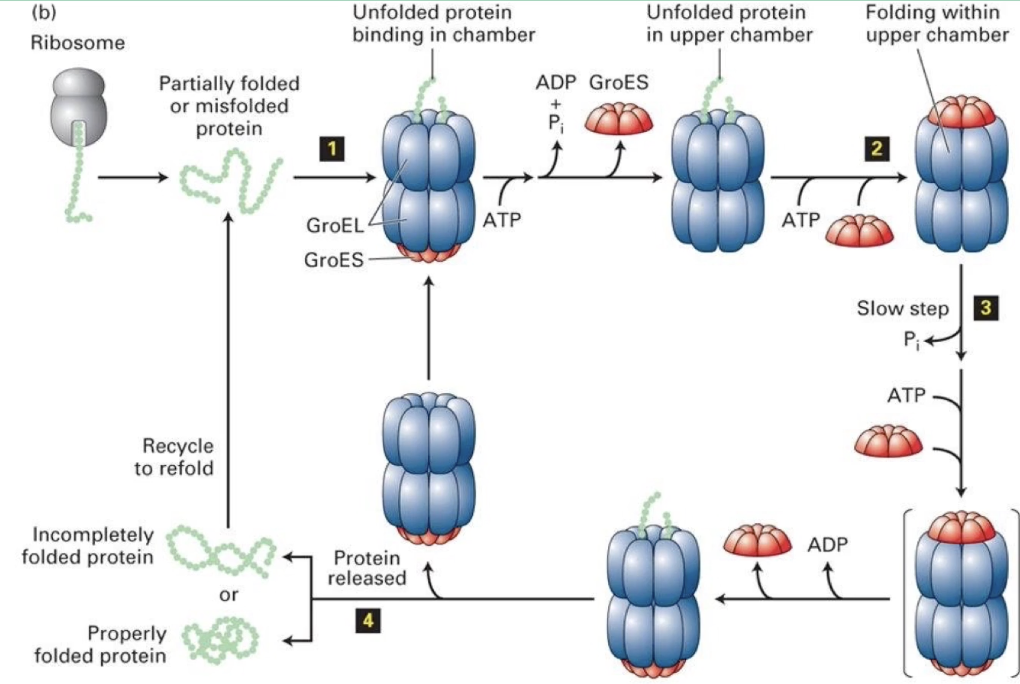
How does the bacterial GroEL/GroES chaperonin complex function?
Alternating chambers: Only one GroEL ring folds protein at a time
Folding process:
Spent chamber releases GroES + ADP
Opposite chamber binds new peptide + ATP
GroES cap seals chamber
Conformational change enlarges chamber for protein folding
After folding, ATP hydrolysis removes GroES cap
Protein diffuses out (folded or not)
Cycle repeats using the alternate chamber if needed
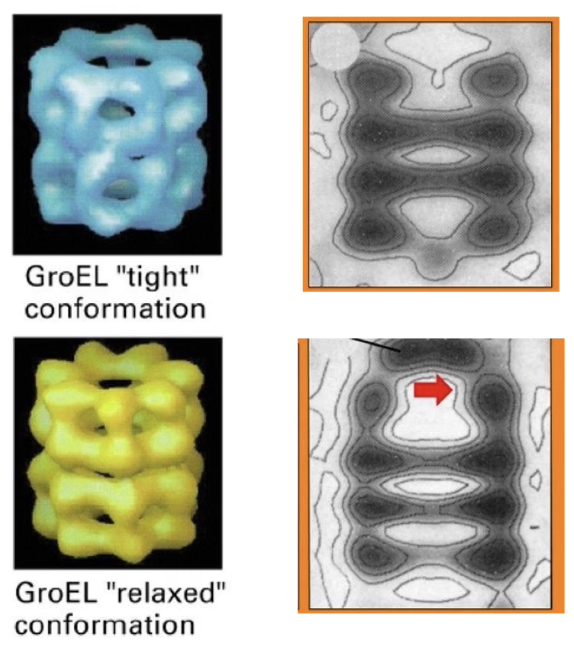
What are the two conformational states of GroEL and how do they differ?
Tight conformation: GroEL without GroES cap
Relaxed conformation: GroEL bound to GroES
Key differences:
Interior chamber is larger in relaxed state
Chamber opening narrows upon GroES binding
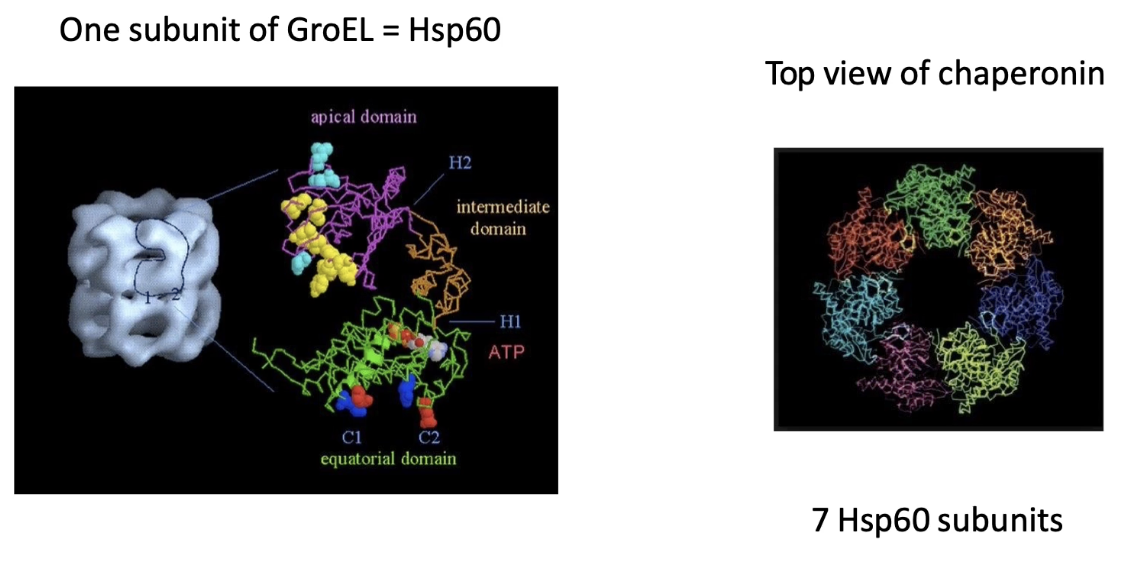
What is the structure of the GroEL chamber and its subunits?
Chamber walls formed by Hsp60 proteins
Each GroEL ring = 7 Hsp60 subunits
Hsp60 domains:
Apical domain: rim of chamber
Equatorial domain: base/middle
Intermediate/hinge domain: between apical and equatorial
Each Hsp60 binds one ATP → 7 ATP needed per chamber
How does ATP binding affect the structure of Hsp60 subunits and GroEL function?
Tight conformation (no ATP):
Hsp60 subunits are bent at hinge
Relaxed conformation (with ATP + GroES):
Hsp60 subunits become elongated
Conformational change is coordinated across all 7 subunits
Drives functional change in the chaperonin complex
In eukaryotes (TRiC complex):
8 subunits undergo concerted movement like Hsp60
Coordinates the same chaperonin function through similar structural changes
What risks do unfolded proteins pose, and how are they removed from the cell?
Risks of unfolded/misfolded proteins:
Non-functional
Tend to aggregate, harming the cell
Tagged and removed by degradation via proteasome
Degraded proteins include:
Misfolded or denatured proteins
Excess or endocytosed proteins (proteins at high concentrations and proteins taken up into the cell)
Proteins regulated by cell cycle
Removal begins with ubiquitination
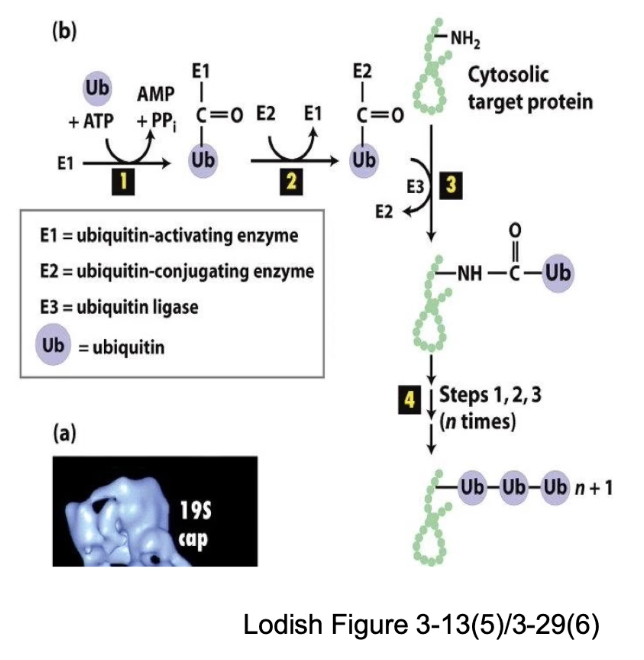
What are the steps of protein degradation via ubiquitination?
TAG: Ubiquitin covalent attachment to target protein
DEGRADE: Recognition by proteasome → protein cleaved into short peptides (7-8 residues)
Ubiquitin: small (76 residues), stable, folded protein reused after removal
Ensures selective degradation of unwanted proteins

What enzymes are involved in ubiquitination and what are their roles?
E1 (Ubiquitin-activating enzyme): Uses ATP to activate and bind free ubiquitin.
E2 (Ubiquitin-conjugating enzyme): Carries ubiquitin and helps transfer it.
E3 (Ubiquitin ligase): Recognizes specific target protein and attaches ubiquitin to lysine side chain.
E3 gives specificity to the degradation process.
Steps:
E1 binds ubiquitin using energy from ATP hydrolysis. Forms a high-energy E1~Ub complex.
Ubiquitin is transferred from E1 to E2 (ubiquitin-conjugating enzyme).
E3 ligase binds to a specific target protein. E3 interacts with E2 to transfer ubiquitin to the target protein.
Polyubiquitination: Multiple ubiquitins added → marks protein for degradation
How is the ubiquitination system amplified in the cell?
1 E1 → many E2s → hundreds of E3s
Each E3 targets specific proteins, ensuring broad yet specific degradation.
What is the structure and function of the proteasome?
Barrel-shaped complex with cap-like ends
Wall made of identical subunits (like chaperonins)
Core contains proteolytic enzymes
Function: cleaves proteins into peptides, unlike folding role of chaperonins
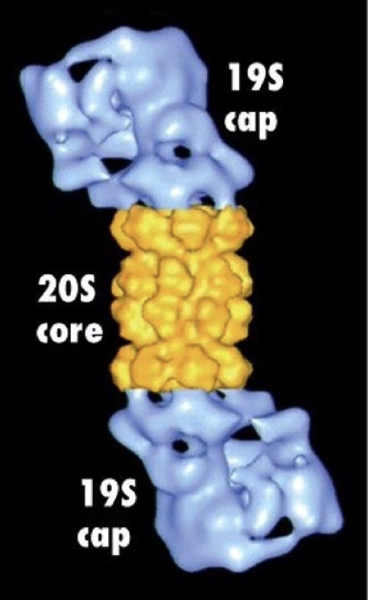
How does the proteasome degrade polyubiquitinated proteins?
Polyubiquitin tag is recognized by the proteasome cap
Target protein is unfolded as it enters the narrow opening of the cap
Ubiquitins are removed before entry → recycled
Protein enters core → cleaved into small peptides (2-24 aa)
Peptides degraded further by cytosolic proteases or lysosomes

What happens in spinocerebellar ataxia and how is it related to protein degradation?
Caused by mutated Ataxin-1 protein (misfolded)
Tagged with ubiquitin but cannot be unfolded
Gets stuck on the proteasome
Leads to:
Toxic protein aggregates
Blockage of proteasome function
Impaired degradation of other proteins
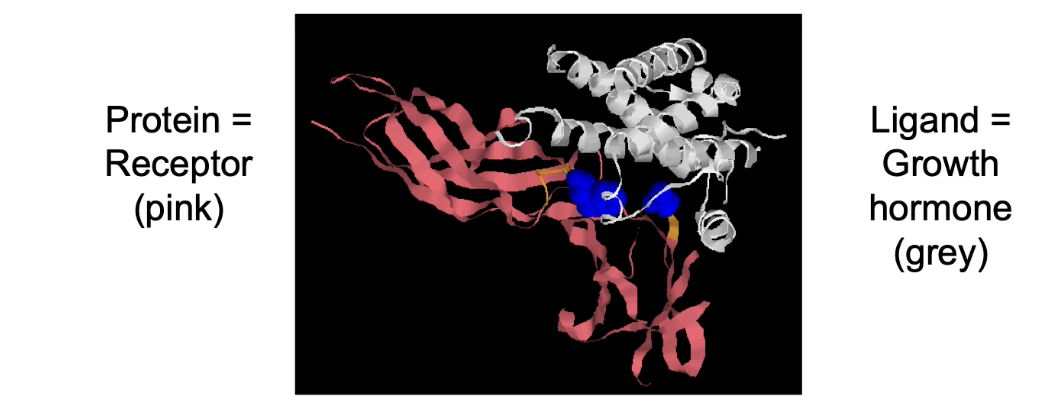
Why is proper protein structure important for protein function?
Protein structure determines its ability to bind specific molecules (ligands).
Without interaction with another molecule, a protein is essentially non-functional.
Examples of protein-ligand interactions:
Antibodies binding antigens (immune response)
Enzymes binding substrates (catalysis)
Transcription factors binding DNA (gene expression)
Cell-surface receptors binding signaling molecules (e.g., growth hormone receptors)
What two factors determine protein-ligand binding, and what do they mean?
Specificity:
Protein’s ability to bind a unique ligand or closely related ones.
Affinity:
Strength of the interaction between protein and ligand.
High affinity = molecules stay bound longer.
Low affinity = molecules dissociate quickly.
Both depend on molecular complementarity: how well the shapes and chemical properties of the interacting surfaces match.
What is molecular complementarity and why is it important?
Describes how well two molecular surfaces fit together.
Relies on non-covalent interactions (e.g., hydrogen bonds, Van der Waals, ionic bonds).
Good shape fit + compatible amino acid R-groups = strong, specific binding.
Otherwise, thermal motion rapidly breaks the molecules apart.
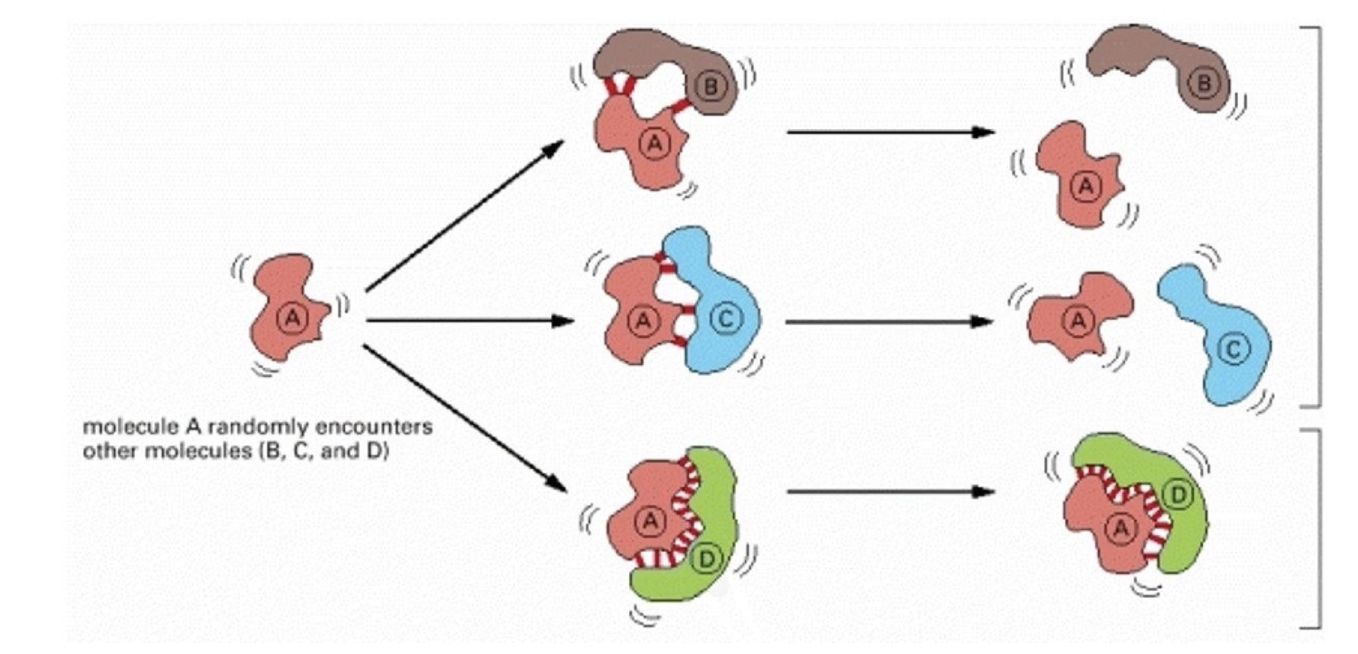
Example:
A stable complex forms when surfaces are complementary (Protein A + D).
Poor fit or same-charge interactions (e.g., two negative R-groups) prevent stable binding (Protein A + B)

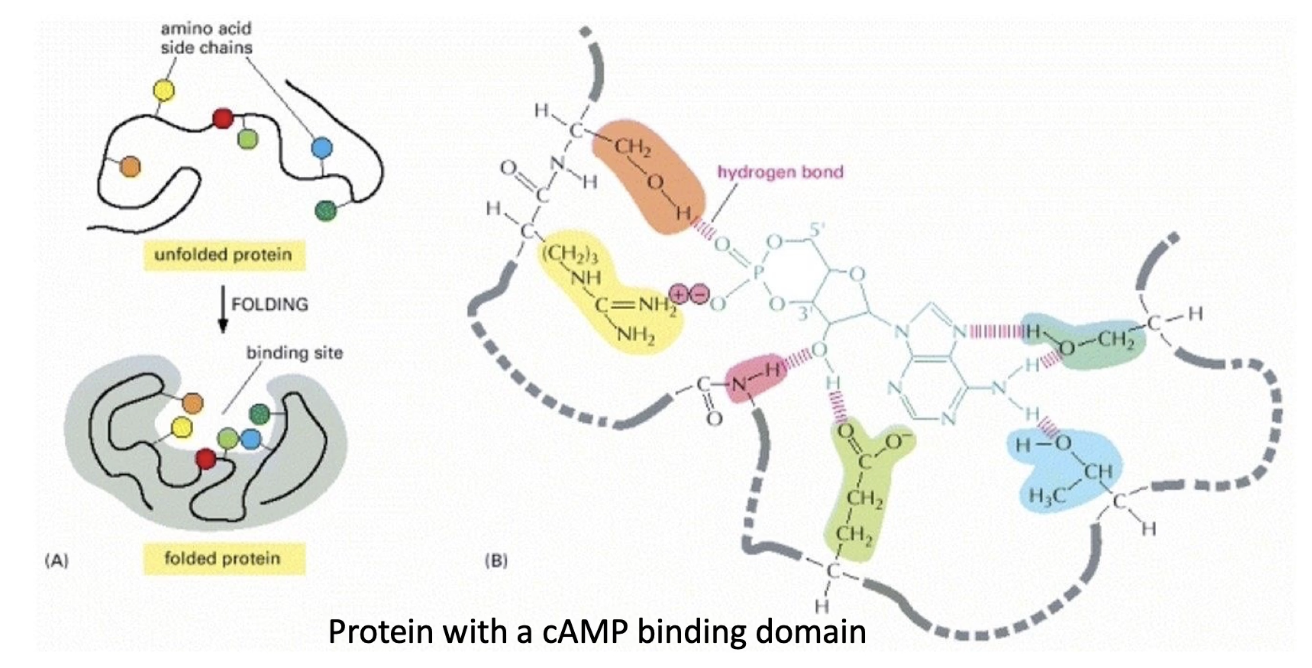
What is a ligand-binding pocket, and how is it formed?
A ligand-binding pocket is a specific 3D cavity in a protein where a ligand binds.
Formed when a protein folds, bringing together amino acid residues from different parts of the primary sequence.
Example:
cAMP-binding domain has a pocket formed by 6 key amino acids.
These residues directly interact with cAMP via hydrogen and ionic bonds.
The pocket’s shape ensures only cAMP fits well, not ATP, ADP, or cGMP.
Mutating even one residue can reduce affinity by altering shape or interaction ability.
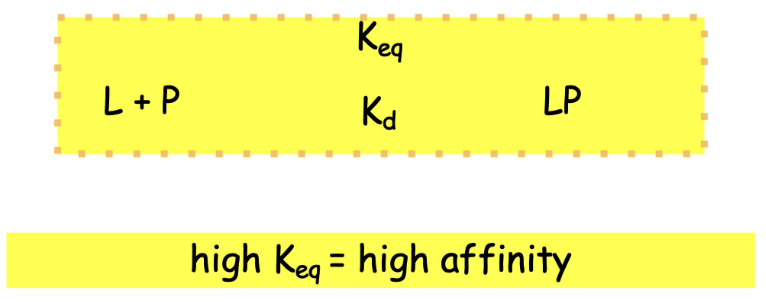
How is protein-ligand binding affinity measured, and what do Keq and Kd represent?
Binding affinity relates to the free energy of interaction.
Keq (equilibrium constant):
High Keq → strong affinity (binding favored, reaction tends right → LP complex).
Low Keq → weak affinity (reaction favors dissociation → L + P).
Kd (dissociation constant):
High Kd = weak binding (more dissociation).
Low Kd = strong binding (more stable complex).
Formula:
P + L ⇌ PL (Protein + Ligand ⇌ Protein-Ligand complex)
What is Keq and how is it calculated in protein-ligand binding?
Keq (equilibrium constant) indicates binding affinity.
Formula:
Keq = [LP] / [L][P]
A higher Keq = stronger binding = more complex formed.
What is Kd and how does it relate to binding affinity?
Kd (dissociation constant) = inverse of Keq.
Kd = [L][P] / [LP]
Lower Kd = higher binding affinity
▸ Less dissociation = more stable complex
▸ Commonly used to describe ligand-protein interactions
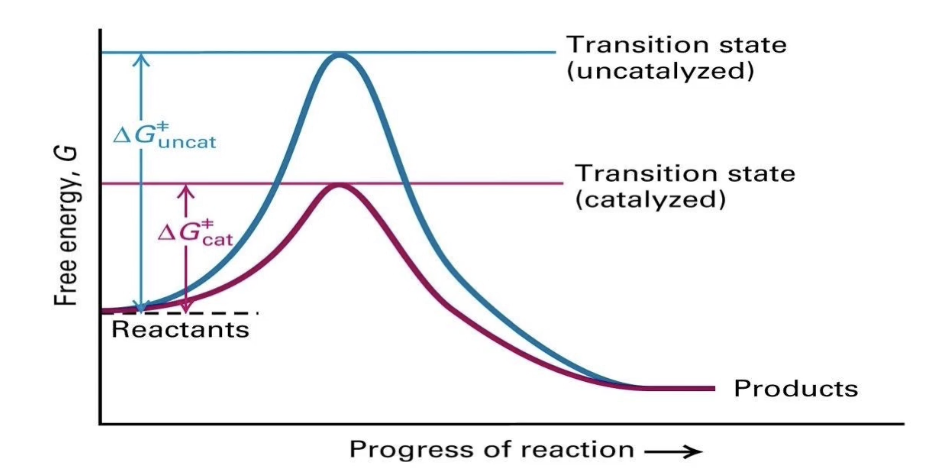
How do enzymes affect the free energy of a reaction?
Enzymes do not change the overall ΔG (free energy change).
They lower the activation energy (Ea) by stabilizing the transition state.
This increases the reaction rate without altering products or reactants.
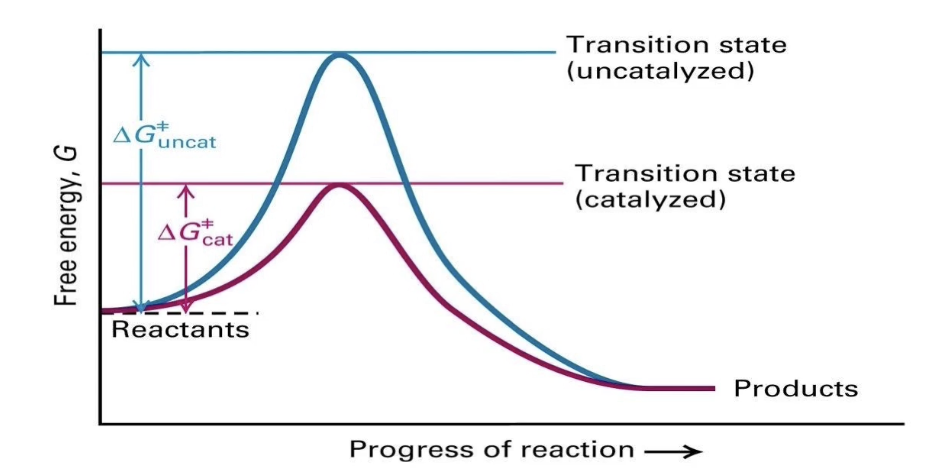
What does a reaction energy graph show about enzyme catalysis?
Uncatalyzed reaction (blue line):
▸ High energy transition state → slower reactionCatalyzed reaction (pink line):
▸ Lowered transition state energy → faster reactionEnzymes facilitate transition but don’t change final energy states
How do enzymes bring substrates together to increase reaction speed?
Enzymes have specific binding sites for substrates.
These sites exhibit molecular complementarity.
Enzymes align substrates in proper orientation → facilitates bonding.
Can increase reaction rate by 10⁶ to 10¹² times.
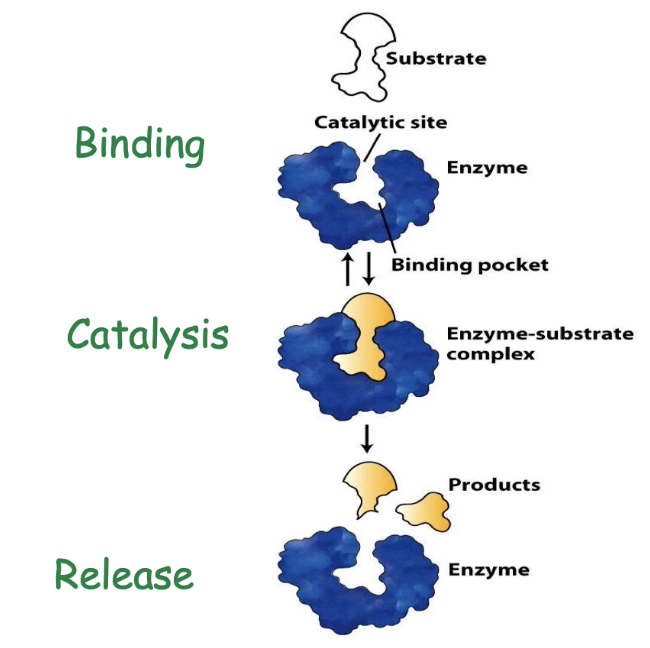
What features of enzyme-substrate interaction ensure efficiency?
Enzymes show:
▸ High specificity: Only bind correct substrate
▸ High affinity: Tight binding improves catalysisFunctional regions of the active site:
▸ Binding site/pocket - determines specificity
▸ Catalytic site - promotes reaction
How is enzyme activity measured, and what is Vmax?
Enzyme activity = rate of product formation
As substrate concentration increases, reaction rate increases.
At saturation (all active sites filled) → rate plateaus = Vmax
Vmax is fixed for a given amount of enzyme.
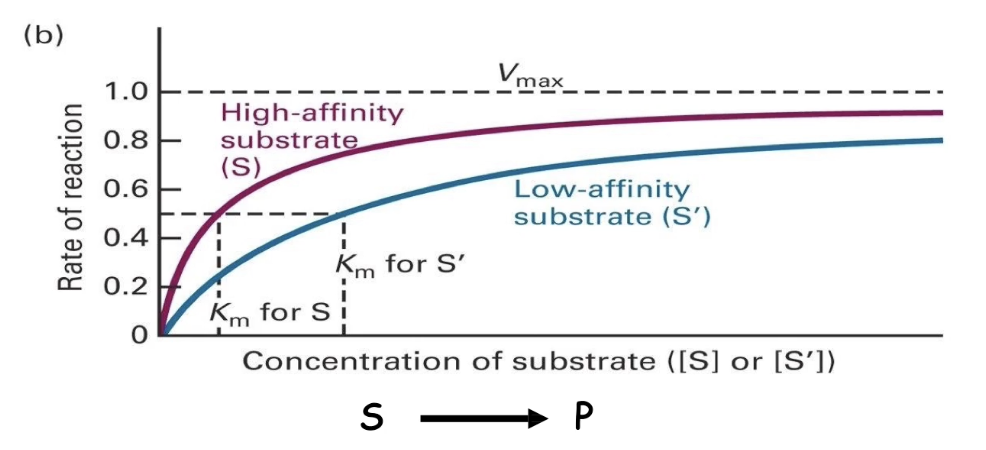
How does substrate affinity affect reaction rate and Vmax?
Both high- and low-affinity substrates reach the same Vmax (if enzyme concentration is constant).
Low-affinity substrates need higher concentrations to reach Vmax.
This is due to more frequent dissociation of enzyme-substrate complex.
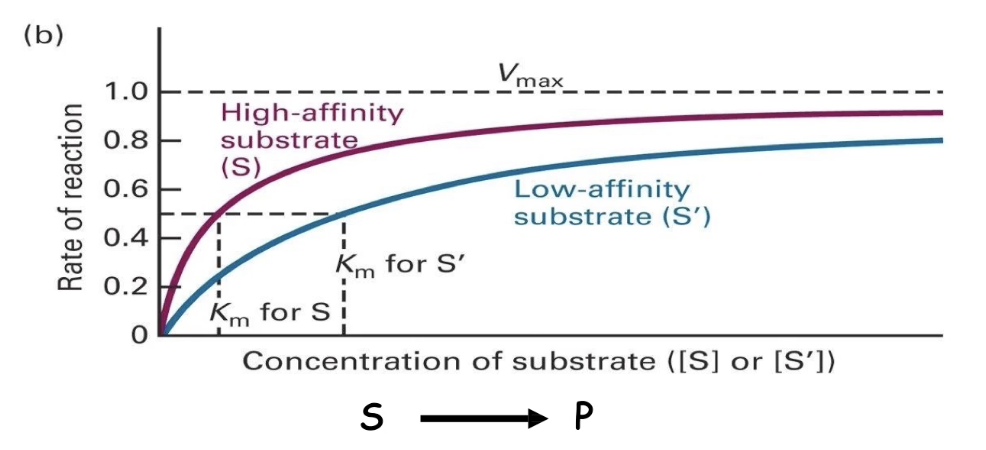
What is Km and how does it relate to enzyme affinity?
Km (Michaelis constant) = substrate concentration at ½ Vmax
Measures enzyme-substrate affinity:
▸ Low Km → high affinity (reaches ½ Vmax faster)
▸ High Km → low affinity (needs more substrate)
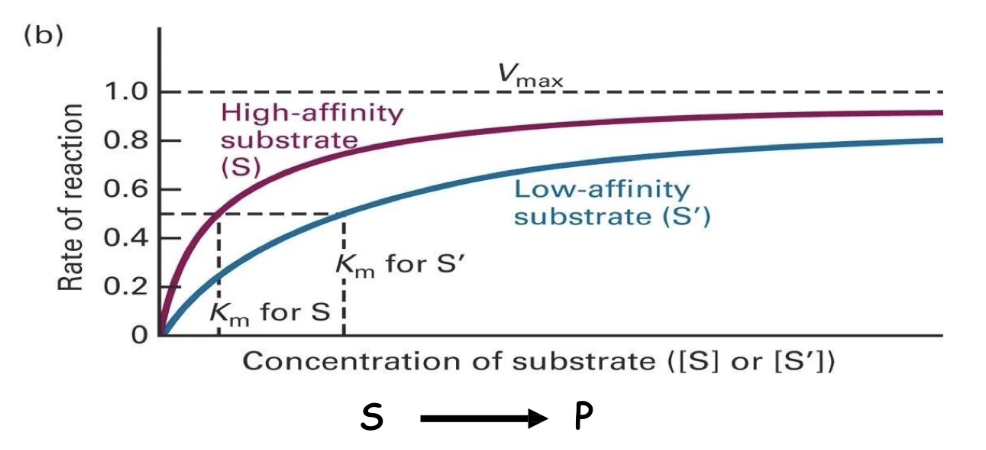
How do Km values reflect enzyme kinetics and substrate behavior?
Km reflects the concentration needed to make the enzyme work at half-speed.
Lower Km:
Tighter binding
Reaches Vmax faster
Higher Km:
Weaker binding
Slower approach to Vmax
What does sucrose synthase do, and how is its activity measured?
Catalyzes reversible reaction between sucrose ⇄ glucose + fructose
We focus on sucrose breakdown for this analysis
Activity is measured by product formation (glucose or fructose)
Increasing sucrose concentration increases reaction rate until Vmax is reached
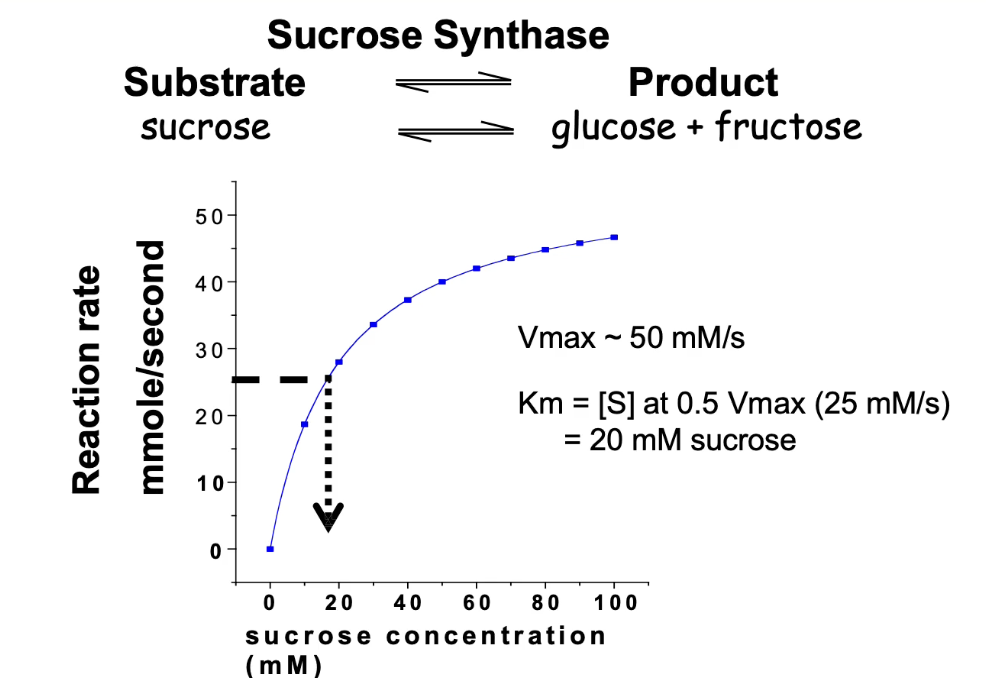
What is Vmax and how is it determined for sucrose synthase?
Vmax = maximum velocity of enzyme-catalyzed reaction
At Vmax, all enzyme active sites are saturated
For sucrose synthase:
Vmax ≈ 50 mmol/sec
Half of Vmax = 25 mmol/sec
How do you calculate Km from a Michaelis-Menten graph?
Km = substrate concentration at ½ Vmax
For sucrose synthase:
½ Vmax = 25 mmol/sec
Corresponding [substrate] = 20 mmol
Interpreting Km:
Low Km = high affinity
High Km = low affinity
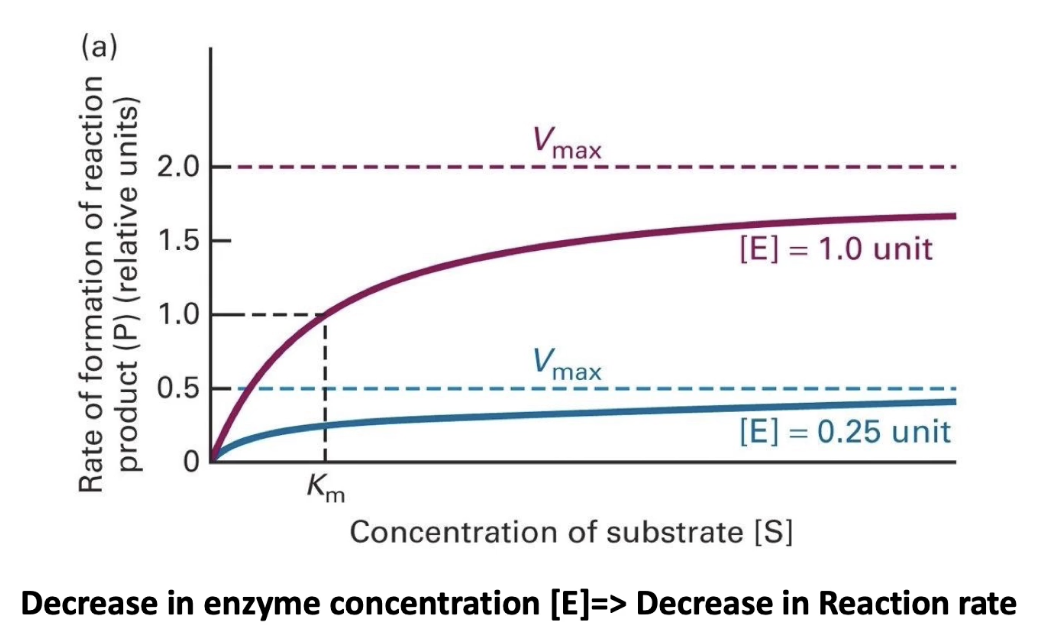
What happens to Vmax and Km when enzyme concentration changes?
Vmax changes with enzyme concentration:
More enzyme = higher Vmax (more active sites)
Less enzyme = lower Vmax
Km stays the same:
Km reflects enzyme-substrate affinity
Does not depend on enzyme concentration
Same enzyme = same Km
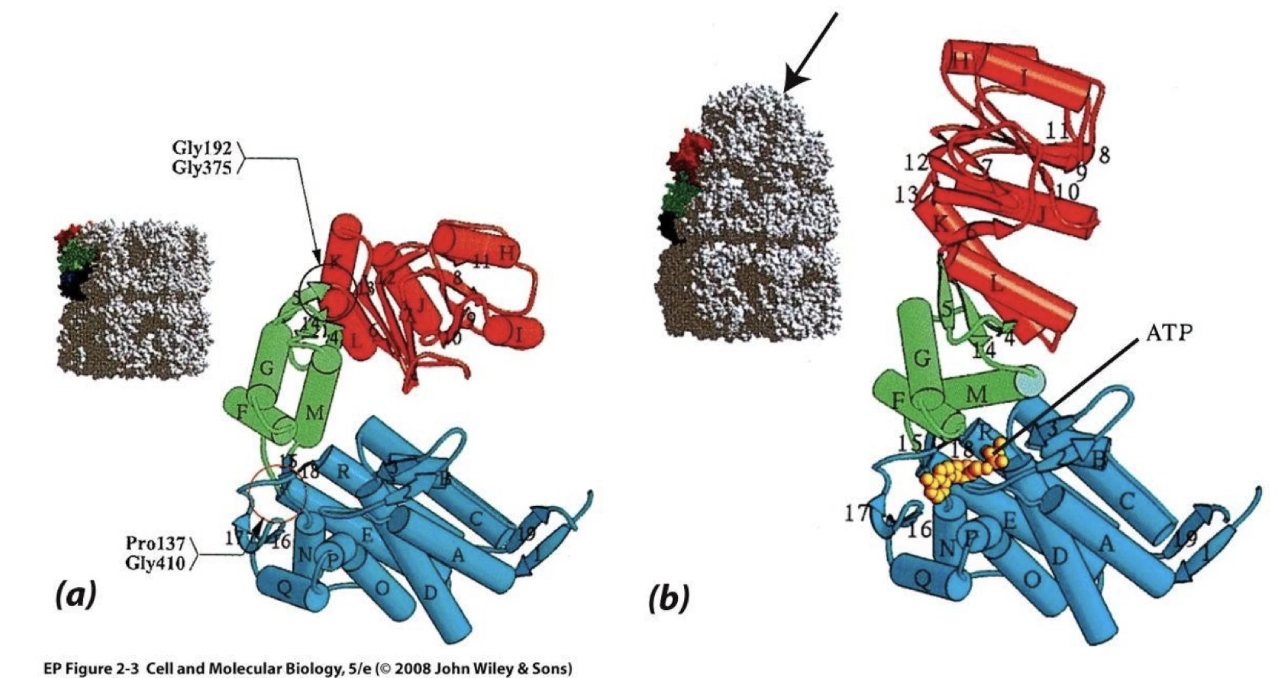
What is the structure and function of Protein Kinase A (PKA)?
PKA = enzyme that phosphorylates target proteins
Binds two substrates:
ATP (phosphate donor)
Target peptide/protein
Structure:
Small domain (glycine lid, top)
Large domain (bottom)
Hinge connects domains
Together form the kinase core (active site)
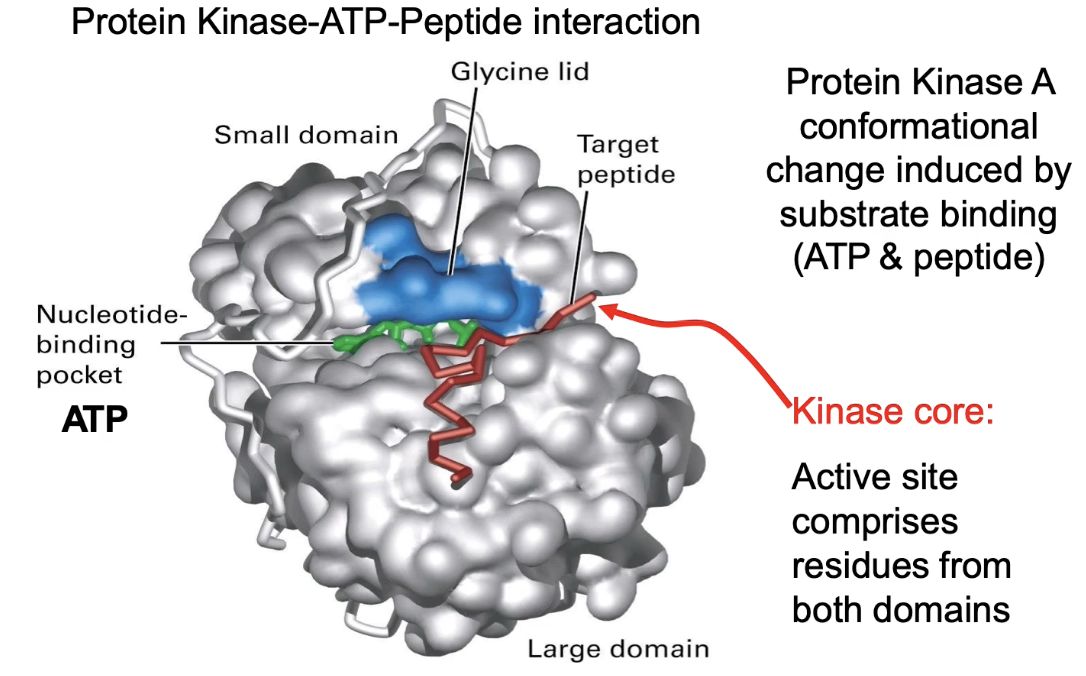
How does PKA achieve substrate specificity and molecular complementarity?
ATP-binding site:
High specificity for ATP
Low affinity for similar nucleotides (ADP, GTP, cAMP)
Target peptide recognition:
Recognizes sequence: Arg-Arg-X-Ser-Y
X = any amino acid
Y = hydrophobic amino acid
Glutamic acid residues in large domain mediate binding?? was never mentioned
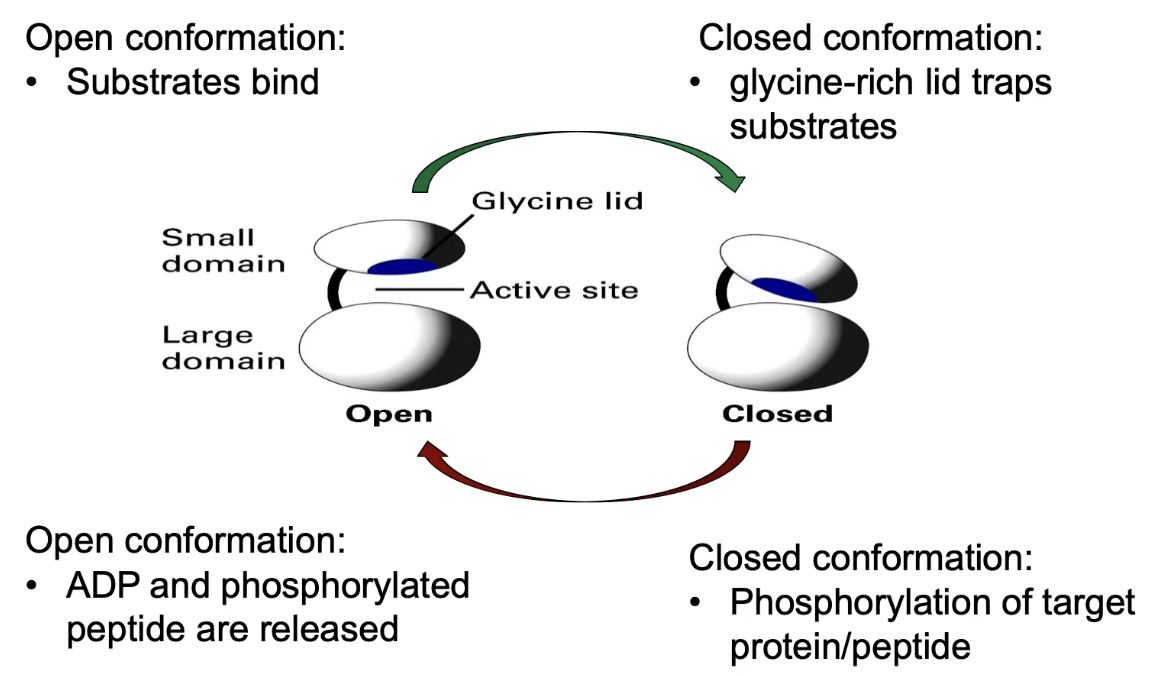
What conformational changes occur in PKA upon substrate binding?
ATP + target peptide bind to open conformation of PKA
Binding causes hinge to shift → domains close
Glycine lid traps substrates in active site
Allows phosphate transfer from ATP → peptide
After reaction:
Products = ADP + phosphorylated peptide
Both have lower affinity → exit when enzyme reopens
Why is regulating protein activity in the cell important, and how is it achieved?
Thousands of proteins are present in a cell, but not all are active at once
Regulating activity ensures proteins are only active when needed
Protein activity is often regulated by changes in protein shape
5 major mechanisms for regulating protein activity:
Allosteric regulation
Signal-induced regulation of protein levels
Covalent modification
Proteolytic cleavage of precursor forms
Formation of enzyme complexes
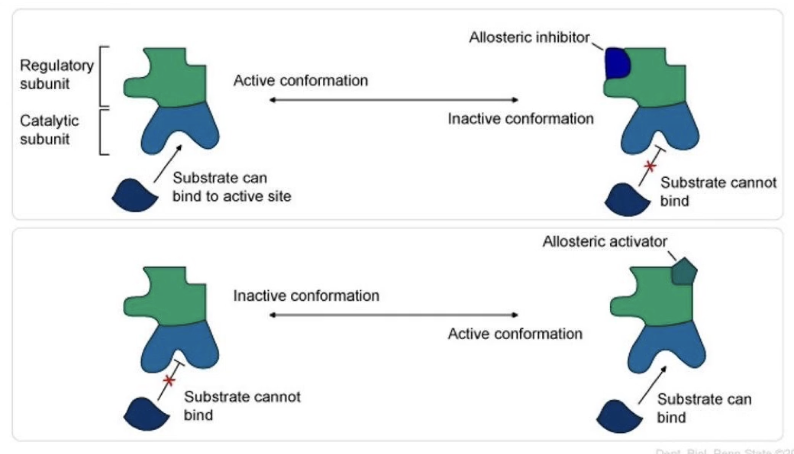
What is allosteric regulation, and how does it work?
Allosteric regulation = regulation of protein function by binding of a molecule at a site other than the active site
Binding causes a conformational (shape) change
Molecules involved are called allosteric modulators
Positive modulators (activators): enhance protein activity
Negative modulators (inhibitors): reduce protein activity
How is PKA regulated through allosteric activation by cAMP?
Inactive PKA: tetramer (2 regulatory [R] subunits + 2 catalytic [C] subunits)
R subunit has a pseudosubstrate domain that blocks the catalytic site → enzyme inactive
cAMP (allosteric activator) binds to R subunits at nucleotide binding sites
Causes a shape change → pseudosubstrate detaches from catalytic site
Catalytic subunits (monomers) are released and become active
High [cAMP]: active PKA
Low [cAMP]: inactive PKA
No protein cleavage occurs—just changes in folding
![<ul><li><p class=""><strong>Inactive PKA:</strong> tetramer (2 regulatory [R] subunits + 2 catalytic [C] subunits)</p></li><li><p class=""><strong>R subunit has a pseudosubstrate domain</strong> that blocks the catalytic site → enzyme inactive</p></li><li><p class=""><strong>cAMP </strong>(allosteric activator) <strong>binds to R subunits</strong> at nucleotide binding sites</p><ul><li><p class="">Causes a shape change → pseudosubstrate detaches from catalytic site</p></li><li><p class="">Catalytic subunits (monomers) are released and become active</p></li></ul></li><li><p class=""><strong>High [cAMP]:</strong> active PKA</p></li><li><p class=""><strong>Low [cAMP]:</strong> inactive PKA</p></li><li><p class="">No protein cleavage occurs—just changes in folding</p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/a4c92edf-934f-4122-9c0a-ae12bc865e1b.png)
What structural changes happen in PKA upon cAMP binding and release?
With cAMP bound:
Pseudosubstrate retracts
Cannot bind catalytic site → PKA is active
When cAMP is released:
Pseudosubstrate extends
Blocks catalytic site → PKA is inactive
Allows cyclical activation/inactivation of PKA
Change is due to protein folding, not cleavage
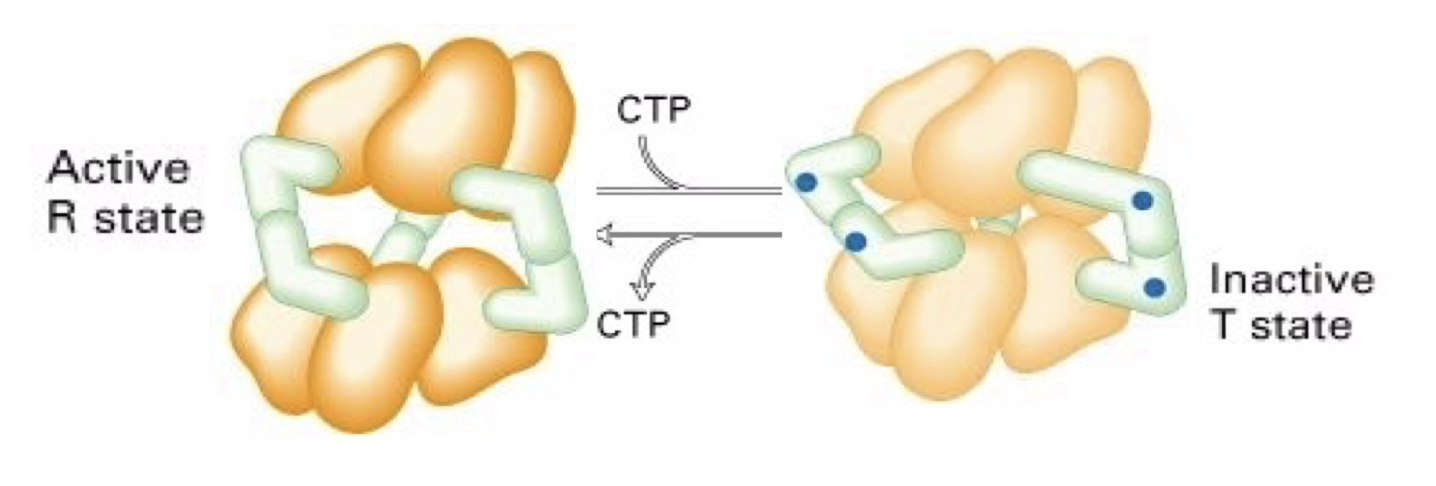
How is the enzyme ATCase regulated allosterically by CTP?
ATCase structure: 6 catalytic (yellow) + 6 regulatory (green) subunits
CTP = allosteric inhibitor
Binds to regulatory subunits → conformational change
Enzyme twists into inactive/tense (T) state → hides substrate binding sites
Low CTP: enzyme shifts to active/relaxed (R) state
CTP concentration controls ATCase activity
What is the role of allosteric regulation in feedback inhibition in biochemical pathways?
Prevents unnecessary synthesis of end-products
Example: In a 5-step pathway A → E:
End product E binds to enzyme 1 → inhibits its activity
Stops the pathway if E is abundant → saves energy/resources
Real example:
CTP synthesis pathway
ATCase catalyzes step 2
CTP (end product) is a negative modulator and binds ATCase → inhibits it
Prevents overproduction of CTP
CTP helps make DNA/RNA
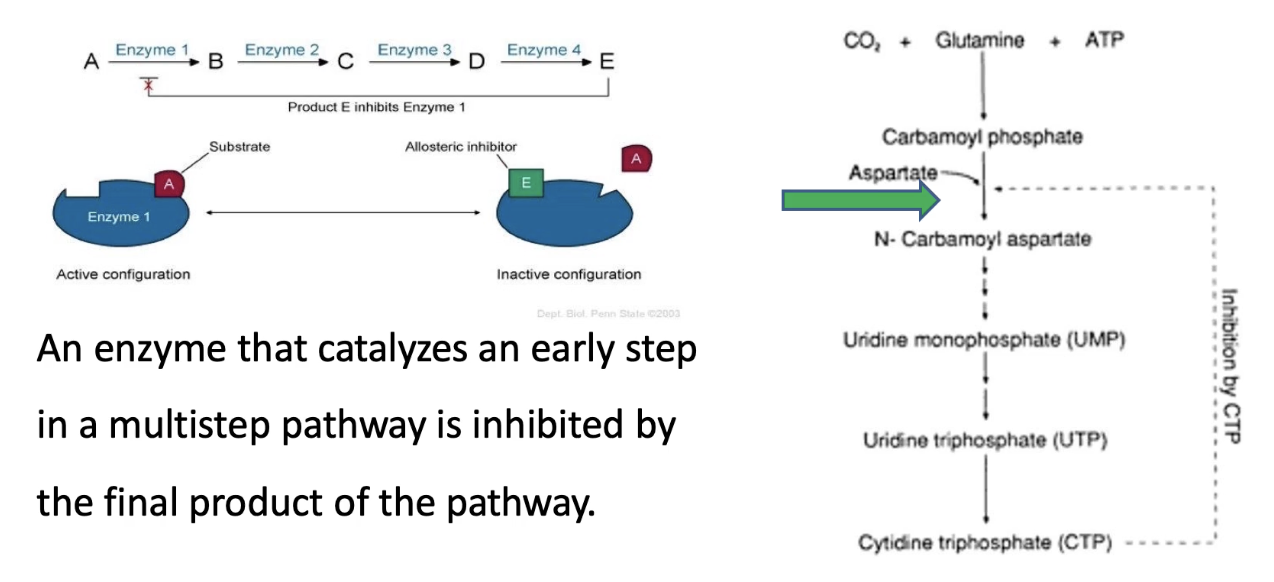
What are allosteric modulators, and how do they affect ATCase?
Allosteric modulators bind to sites other than the active site and change enzyme activity.
ATCase (Aspartate Transcarbamoylase):
CTP = allosteric inhibitor
ATP = allosteric activator
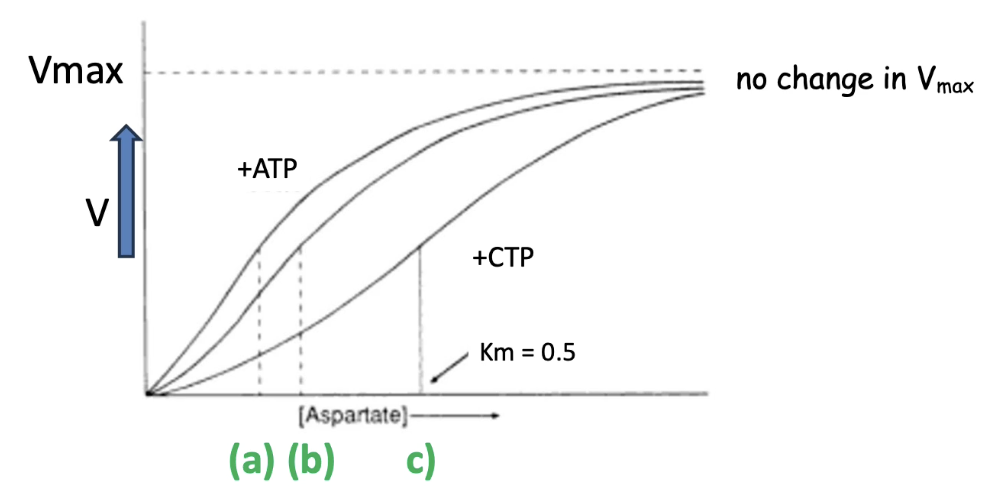
How do CTP and ATP affect the enzyme kinetics of ATCase?
Graph interpretation:
X-axis: Substrate concentration (aspartate)
Y-axis: Reaction rate
Unmodified ATCase:
Middle curve; Km = baseline affinity for aspartate
ATCase + ATP:
Top curve
Lower Km = higher affinity for aspartate
ATCase + CTP:
Bottom curve
Higher Km = lower affinity for aspartate
Vmax stays the same for all curves:
Number of catalytic sites doesn't change
Only availability of active sites is altered by allosteric modulators
What is cooperative allostery and how does it affect multimeric enzymes?
Definition: Binding of a ligand to one subunit affects binding affinity of other subunits
Occurs in multimeric enzymes, not monomeric ones
Initial binding triggers conformational changes in all subunits
Leads to increased overall affinity for the ligand (↓Km)
Result: Sigmoidal ("S"-shaped) enzyme kinetics curve
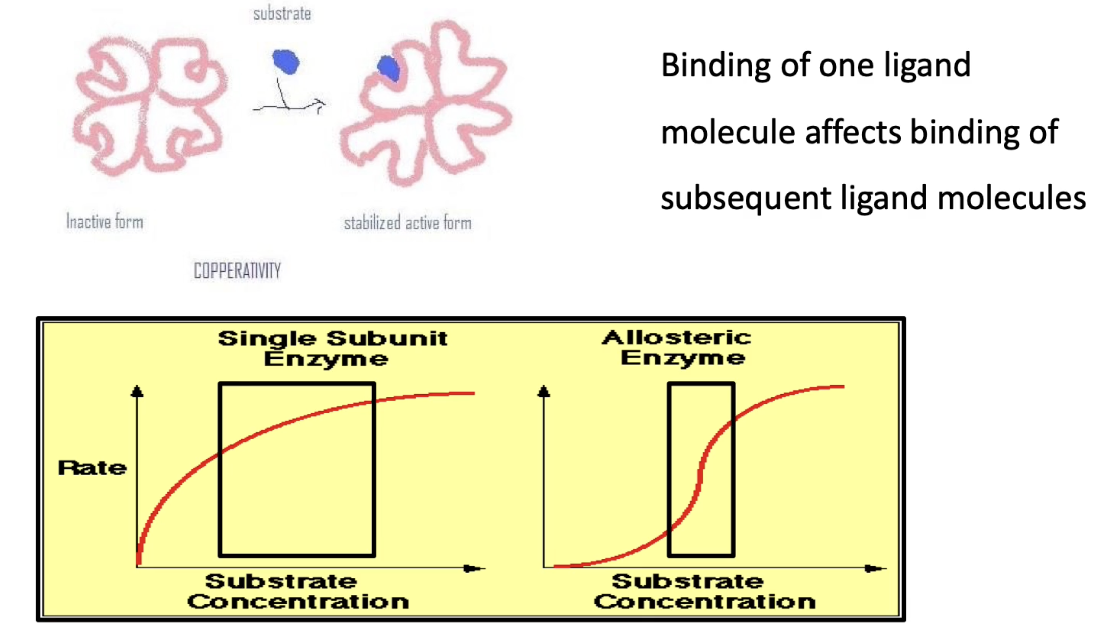
How does the enzyme kinetics curve differ for monomeric vs. multimeric enzymes?
Monomeric enzyme (blue curve):
Traditional hyperbolic Michaelis-Menten curve
Requires large increase in ligand concentration to go from 10% → 100% activity
Multimeric enzyme with cooperativity (red curve):
Sigmoidal curve due to cooperative allostery
Requires small increase in ligand concentration for same increase in activity
Allows for more sensitive and rapid response in cells
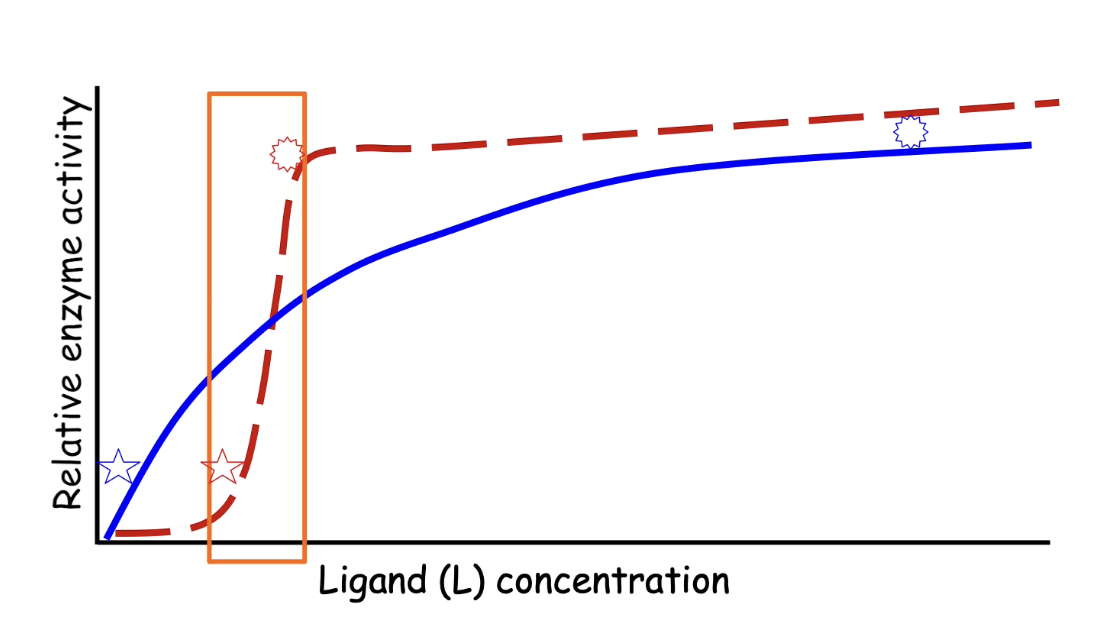
How does hemoglobin demonstrate cooperative allostery?
Hemoglobin = tetramer: 2 α-subunits + 2 β-subunits
Has 2 conformations:
T-state: Low oxygen affinity (inactive)
R-state: High oxygen affinity (active)
Oxygen acts as an allosteric activator
Binding of one O₂ → switches all subunits to high-affinity R-state
Promotes rapid and complete O₂ binding

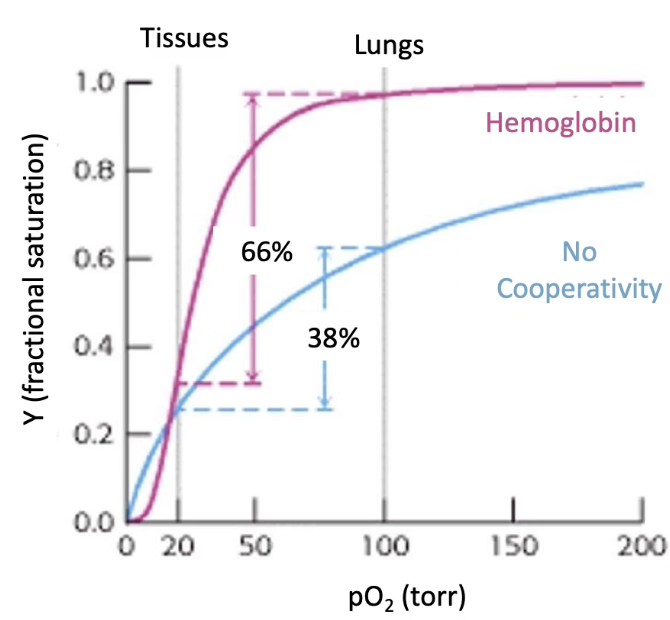
Why is cooperative allostery beneficial for hemoglobin?
Hemoglobin must:
Pick up O₂ in lungs (high pO₂ ~100 torr)
Release O₂ in tissues (low pO₂ ~20 torr)
Graph comparison:
Y-axis: Fractional O₂ saturation
X-axis: Partial pressure of oxygen (pO₂)
Myoglobin (monomeric):
Hyperbolic curve; ~38% saturation difference between lungs & tissues
Hemoglobin (cooperative):
Sigmoidal curve; ~66% saturation difference
Efficient O₂ loading and unloading
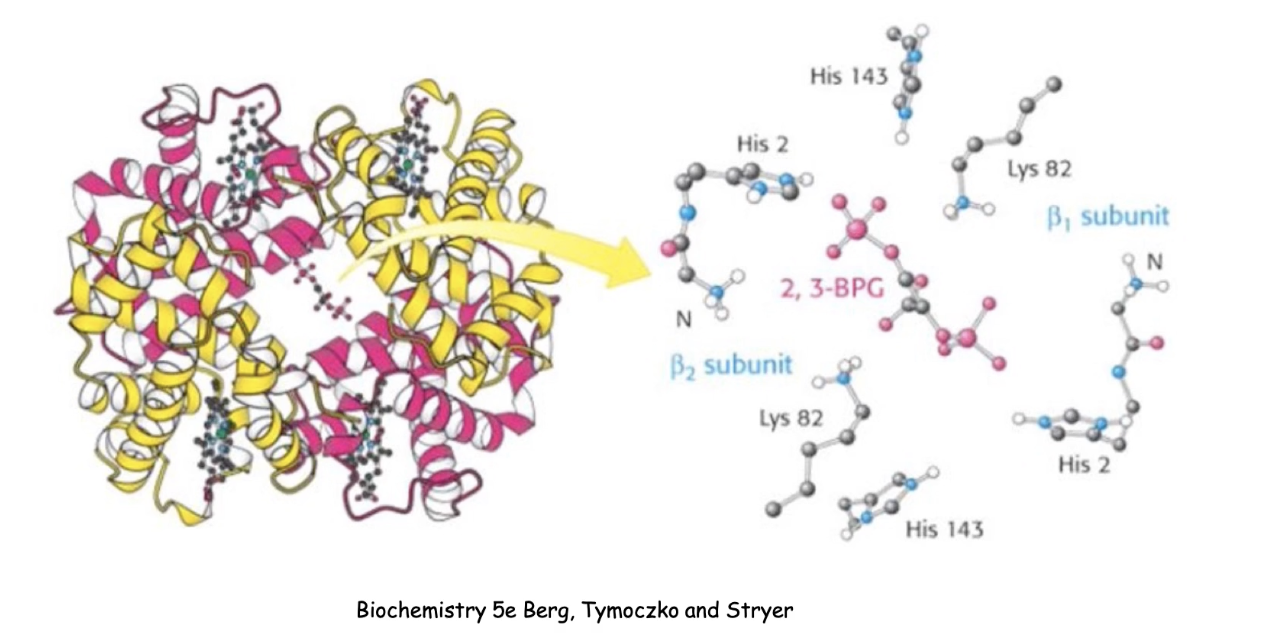
What is the role of 2,3-BPG in regulating hemoglobin function?
2,3-BPG = allosteric inhibitor of hemoglobin
Found in tissues (areas of low O₂)
Function:
Binds to an effector site in the middle of the hemoglobin tetramer. Forms noncovalent interactions with specific amino acid residues.
Decreases O₂ affinity
Promotes O₂ release in tissues
Enhances oxygen delivery efficiency
What is the difference in oxygen affinity between fetal and adult hemoglobin? Why?
Fetal hemoglobin has a higher affinity for O₂ than adult hemoglobin.
Necessary because fetus obtains O₂ from maternal blood (not lungs).
Fetal hemoglobin binds less 2,3-BPG → maintains higher O₂ affinity.
Why does fetal hemoglobin have a lower affinity for 2,3-BPG?
To prevent 2,3-BPG from decreasing its oxygen affinity.
Allows fetus to effectively draw oxygen from maternal circulation.
What is covalent modification and what are some examples?
Regulation by attaching chemical groups covalently to proteins
Common types:
Phosphorylation – adds phosphate group
Acetylation – adds acetyl group
Methylation – adds methyl group
Carboxylation – adds carboxyl group
Phosphoregulation
A reversible covalent modification
A phosphate group is added to amino acids with hydroxyl groups:
Serine, threonine, tyrosine
Kinase enzymes use ATP to add phosphate (inactivation or activation)
Phosphatases remove phosphate to reverse effect
Changes protein shape or chemical properties, altering activity
Describe the phosphoregulation of CDK (Cyclin-Dependent Kinase).
CDK has a substrate-binding pocket blocked in inactive form
Phosphorylation causes a conformational change:
Adds negative charges
Forms new ionic interactions with positively charged regions
Moves red domain → opens substrate-binding site
Phosphorylation results in addition of 2 negative charges
Result: CDK becomes active and can phosphorylate target proteins

Why is phosphorylation a dominant form of protein regulation in cells?
Seen in all organisms and in many protein types
Approx. 3% of yeast proteins are kinases/phosphatases and 2.5% of Arabidopsis proteins are kinases
Suggests phosphorylation is essential for regulating diverse functions
What is proteolytic cleavage and how does it regulate protein activity?
Irreversible mechanism to make lots of protein in inactive conformation
Cleaves protein at specific sites to activate it
Allows storage of inactive precursors (e.g., enzymes, hormones)
Common in digestive enzymes, clotting factors, caspases, collagen
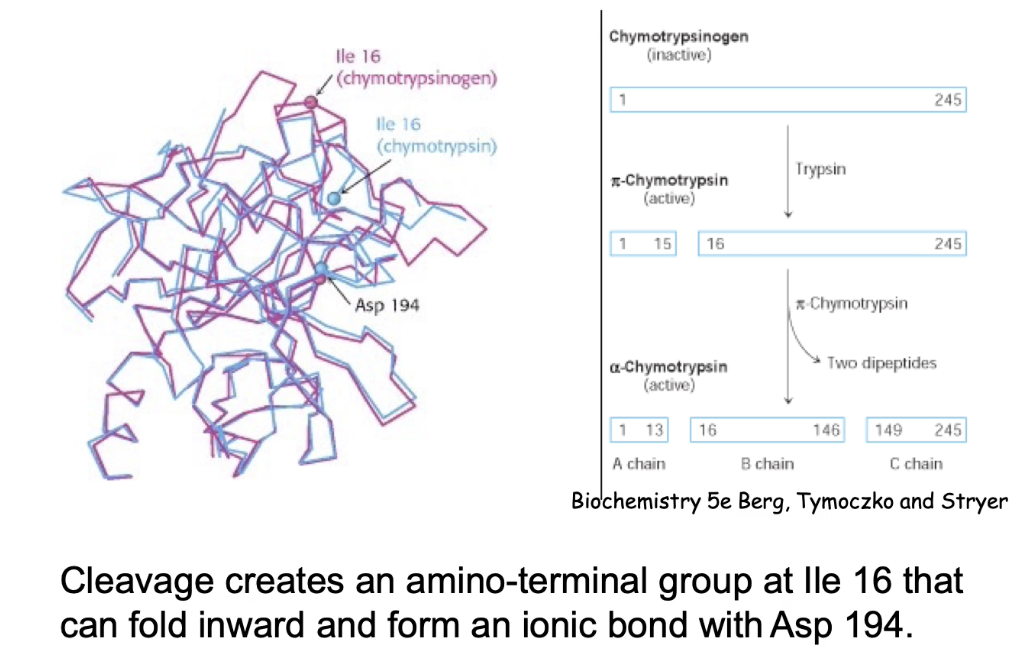
How is digestive enzyme chymotrypsin activated by proteolytic cleavage?
Starts as inactive chymotrypsinogen - can be safely transported through cells
Activated by two cleavage events:
Trypsin cleaves between AA 15–16.
Another enzyme cuts two more sites, releasing two dipeptides → forms A, B, and C chains
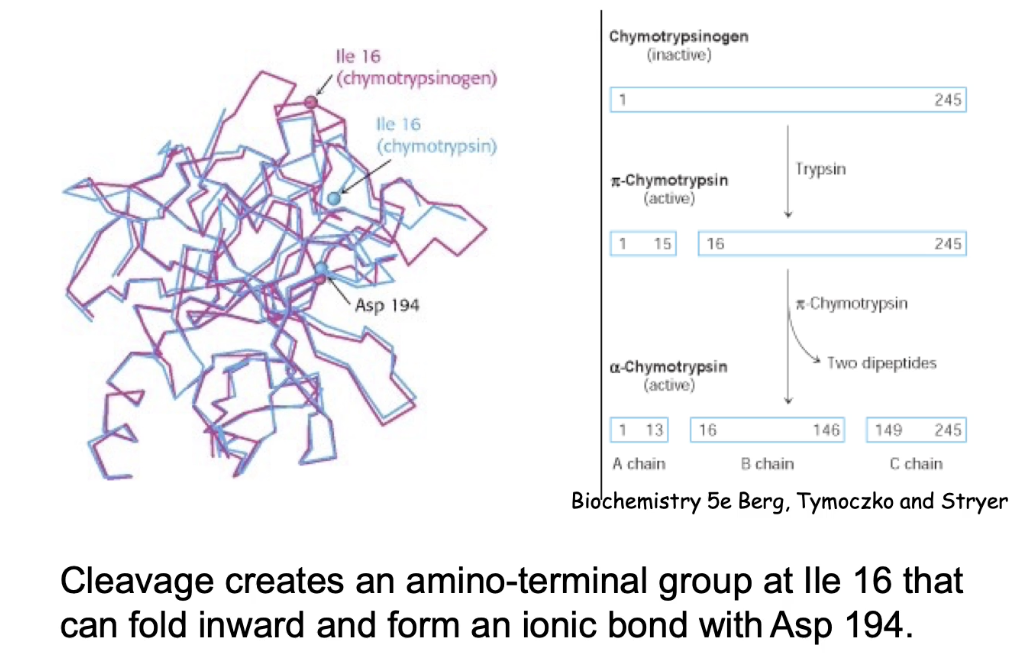
What structural changes occur during chymotrypsin activation?
Cleavage allows folding of isoleucine-16 inward.
Ionic bond forms between Ile-16 (amino group) and Asn-194.
Results in 3 polypeptide chains (A, B, C).
Conformational change creates substrate-binding domain → enzyme becomes active.
What key feature makes proteolytic cleavage irreversible?
Broken peptide bonds cannot be reformed.
The protein cannot return to its original inactive state.
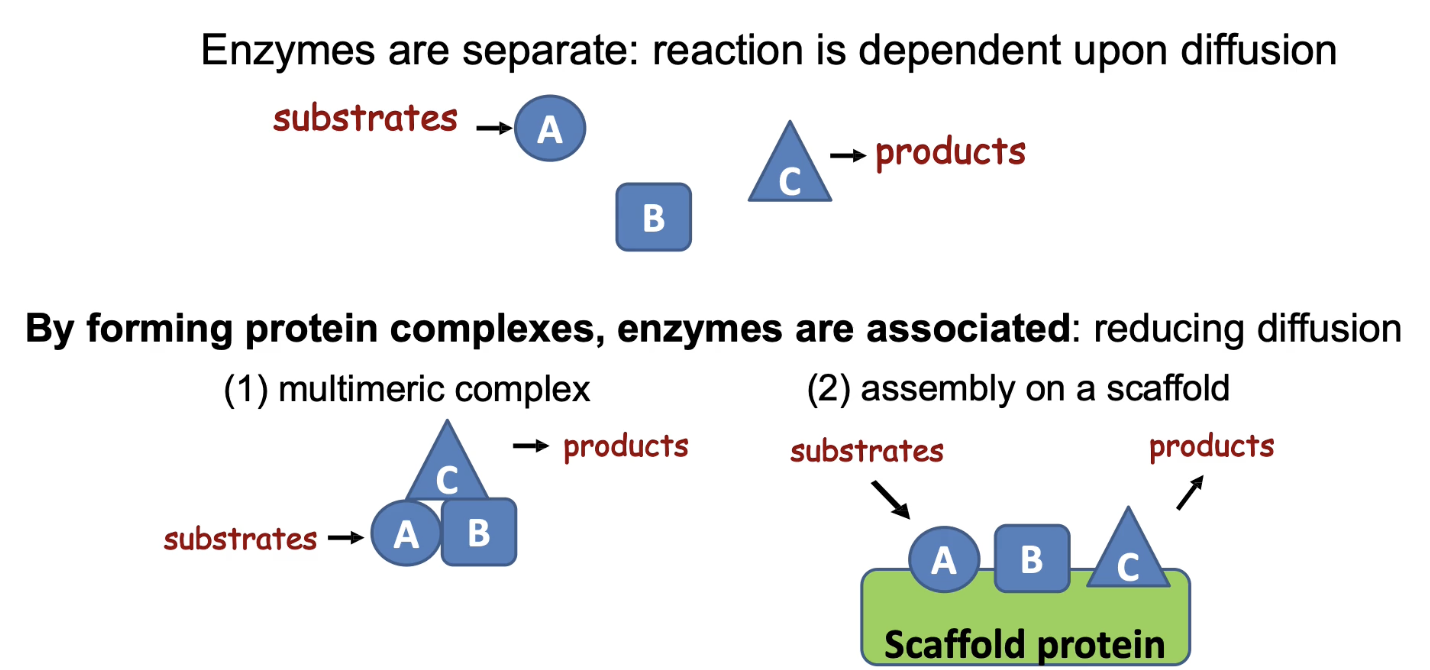
How do protein/enzyme complexes regulate activity and increase pathway efficiency?
Brings enzymes in a pathway close together
Reduces diffusion time between steps
Achieved by:
(1) Protein-protein interaction domains = multimeric complex
(2) Scaffold proteins that bind multiple enzymes
Enhances reaction rate and metabolic efficiency
Applied Lecture
Chaperons and Chaperonins
What does protein folding theoretically involve, and how does this compare to what happens in a real cell?
In theory (ideal conditions):
Folding follows specific rules
Proteins fold into their lowest stable energy state
In reality (inside the cell):
Folding is affected by internal and external stressors
These stressors can promote unfolding or misfolding
What are the key findings and implications of Anfinsen’s experiment on protein folding?
Demonstrated in vitro refolding of denatured ribonuclease
Led to Anfinsen’s Dogma:
Each amino acid sequence encodes a unique, defining 3D conformation
Protein folding is:
Spontaneous
Reversible
Unique
Folding driven by thermodynamic molecular forces

How likely is correct protein refolding in vivo, and what cellular conditions impact it?
Correct refolding is more probable:
At low protein concentrations (less interaction between proteins)
At low temperatures (weaker hydrophobic interactions)
BUT:
Cells are crowded and in a warm environment, increasing chance of aggregation
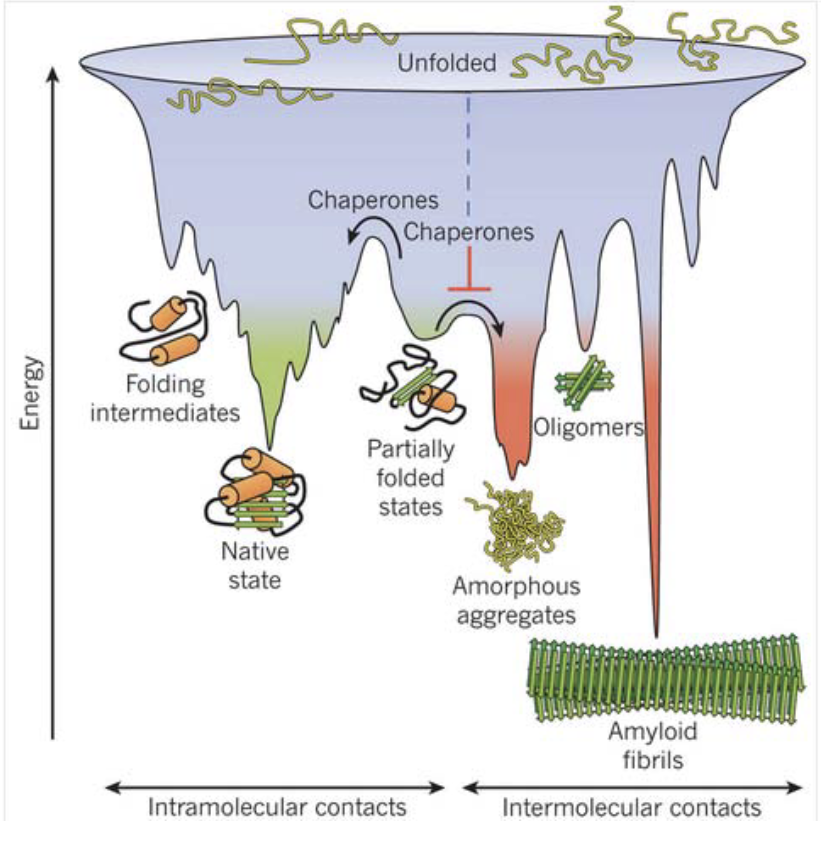
What is the competition between folding and aggregation, and what role do chaperones play?
All proteins aim to fold into low energy, stable states - want to stay in native states
But in vivo, they may misfold or aggregate
Chaperones:
Act at the center of this folding vs aggregation battle
Help prevent aggregation
Ensure efficient and correct folding
What are chaperones, and why are they important?
Found in all organisms, from bacteria to humans
Highly conserved evolutionarily
Prevent aggregation of nascent (new) proteins
General Mechanism:
Prevent inappropriate interactions between residues
Increase efficiency and accuracy of protein folding
What are the two main types of chaperones and their mechanisms?
Molecular chaperones:
Bind short segments of unfolded proteins
Stabilize them and prevent aggregation
Prevent premature folding or incorrect interactions
Chaperonins:
Form folding chambers
Sequester unfolded proteins to provide isolated, optimal folding conditions
What do molecular chaperones bind to, and how do they function?
Bind to hydrophobic R groups on nascent polypeptides, uses ATP
Prevent:
Association with other proteins
Premature folding
Aggregation of hydrophobic residues
Examples of Molecular Chaperones (Heat-Shock Proteins - HSPs):
HSP70 – cytosol and mitochondria
BiP (Grp78) – endoplasmic reticulum
DnaK – bacteria
What are heat-shock proteins (HSPs), and how are they regulated?
Chaperones that respond to stress
Upregulated during:
Heat shock
Other stresses (oxidative stress - low/no oxygen conditions, infection, heavy metals - toxins) and diseases of protein folding
Downregulated once favorable conditions return
Stressors increase probability of HSF binding to HSE to produce HSPs
Functions:
Stabilize unfolded proteins during stress
Prevent aggregation
Assist in refolding during recovery

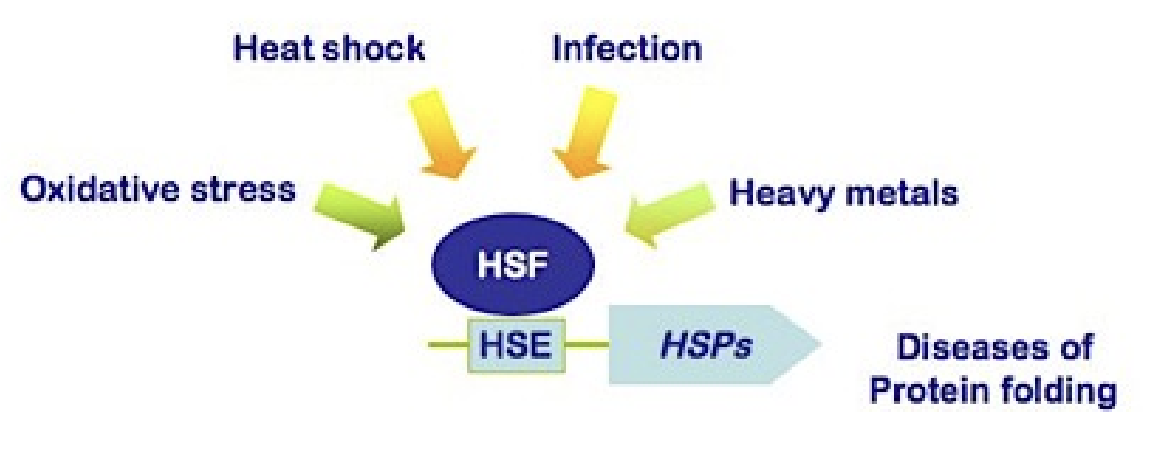
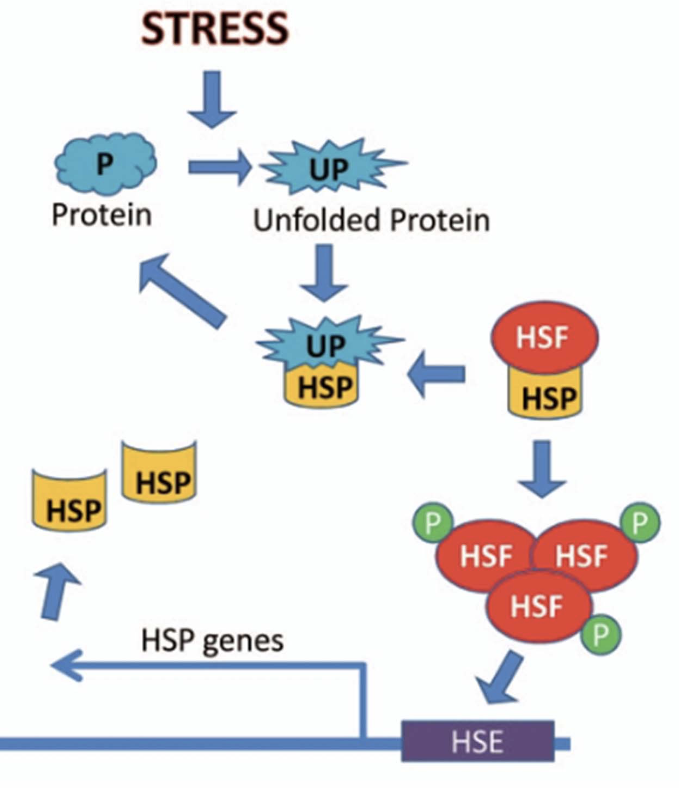
How is the heat shock response activated at the molecular level?
When proteins unfold:
HSPs dissociate from Heat Shock Factors (HSFs)
Freed HSFs trimerize (phosphorylated) and become active
Activated HSFs enter nucleus → increase transcription of HSP genes
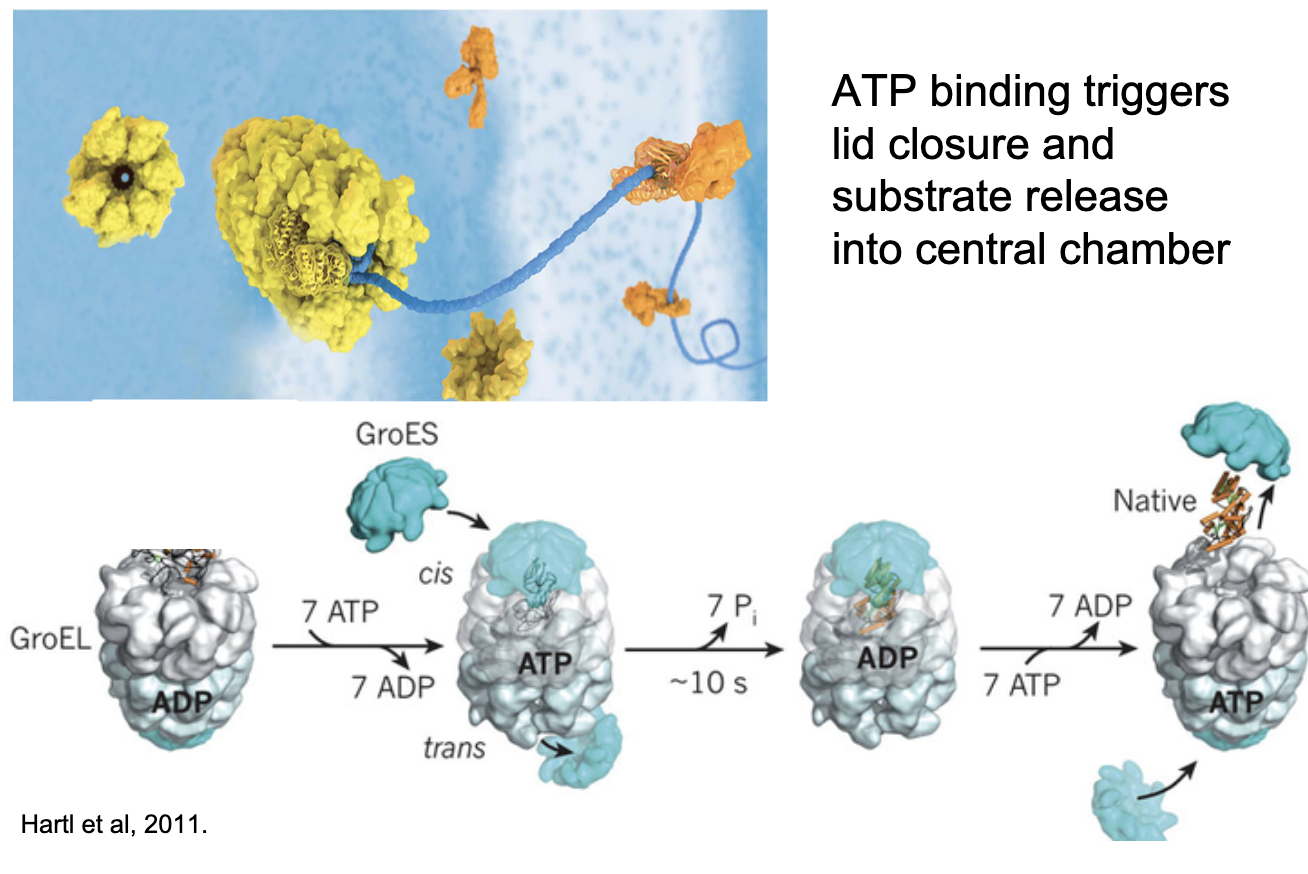
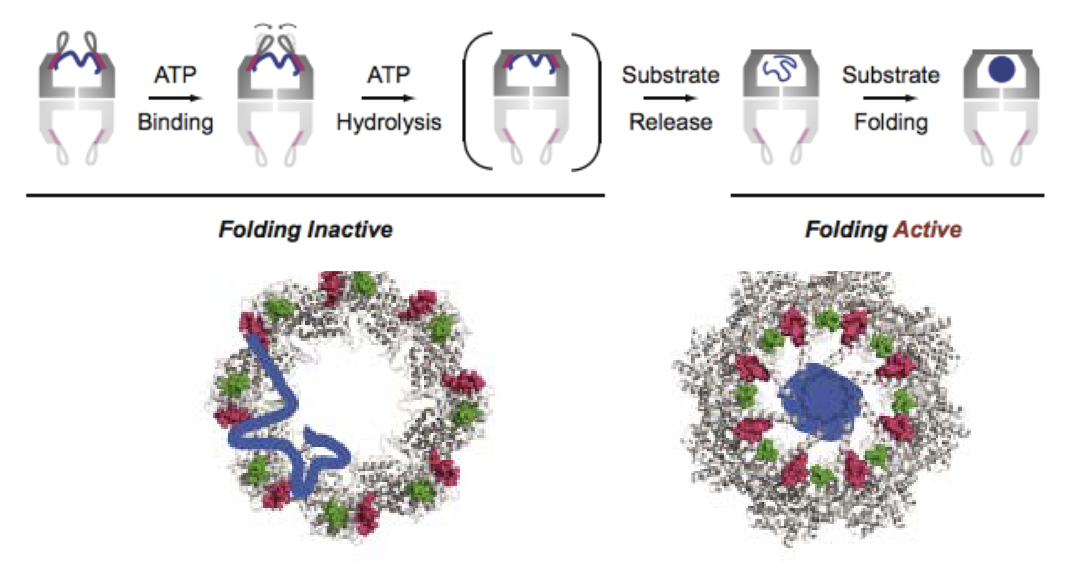
What is the general mechanism of chaperonins and how do they facilitate protein folding?
Chaperonins are barrel-shaped protein complexes that aid protein folding.
ATP binding triggers lid closure and substrate release into the central chamber.
Folding occurs inside the chamber, isolated from the cellular environment.
They provide a controlled chemical environment for folding.
Two possible models:
Mechanical Force Model: Chaperonin holds and folds protein.
Passive Release Model: Protein is released inside the chamber, folds due to internal conditions.
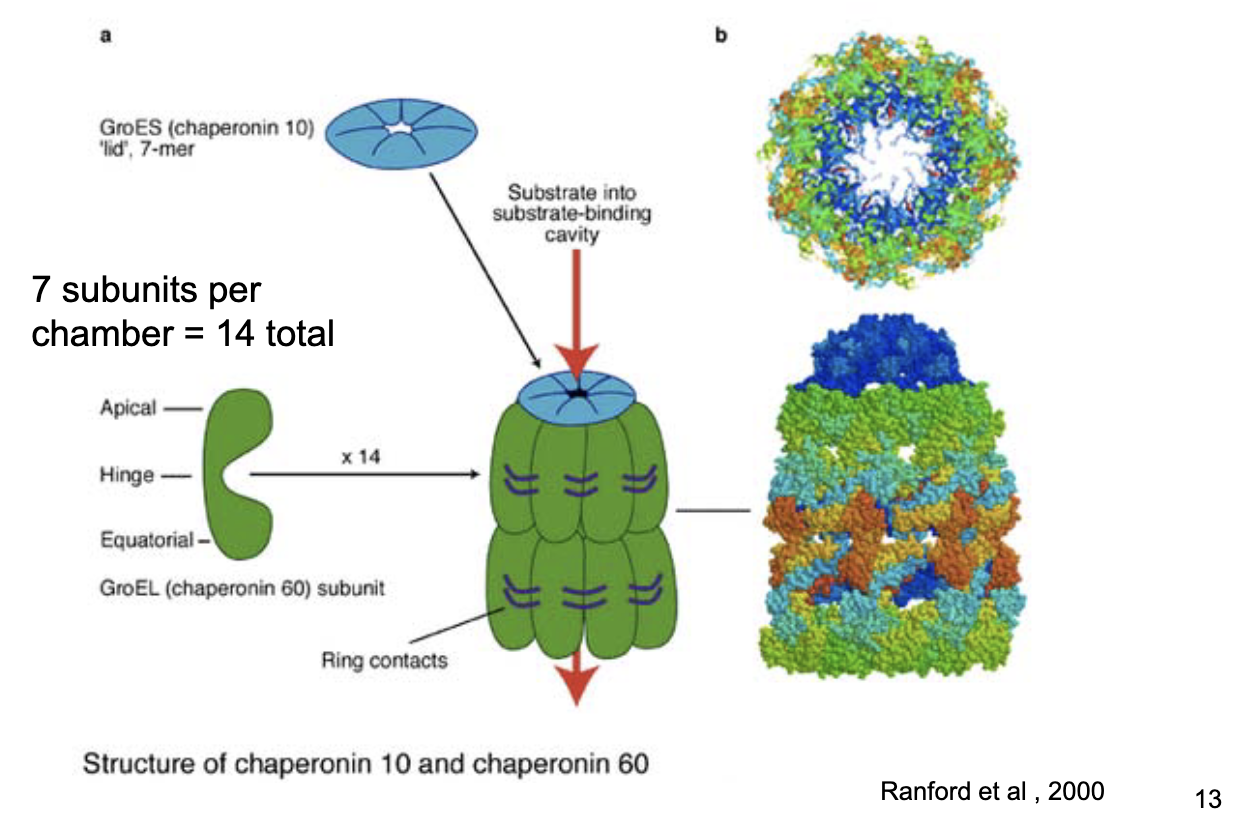
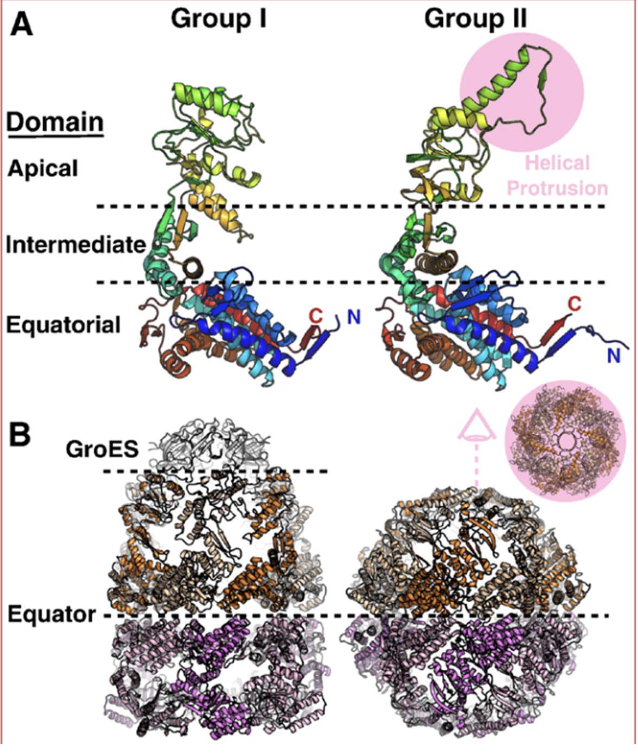
Bacterial chaperonin (GroEL-GroES) structure
2 rings × 7 subunits = 14 total.
Detachable lid (GroES).
Chaperonin 10 (GroES cap) and chaperonin 60 (GroEL chamber).
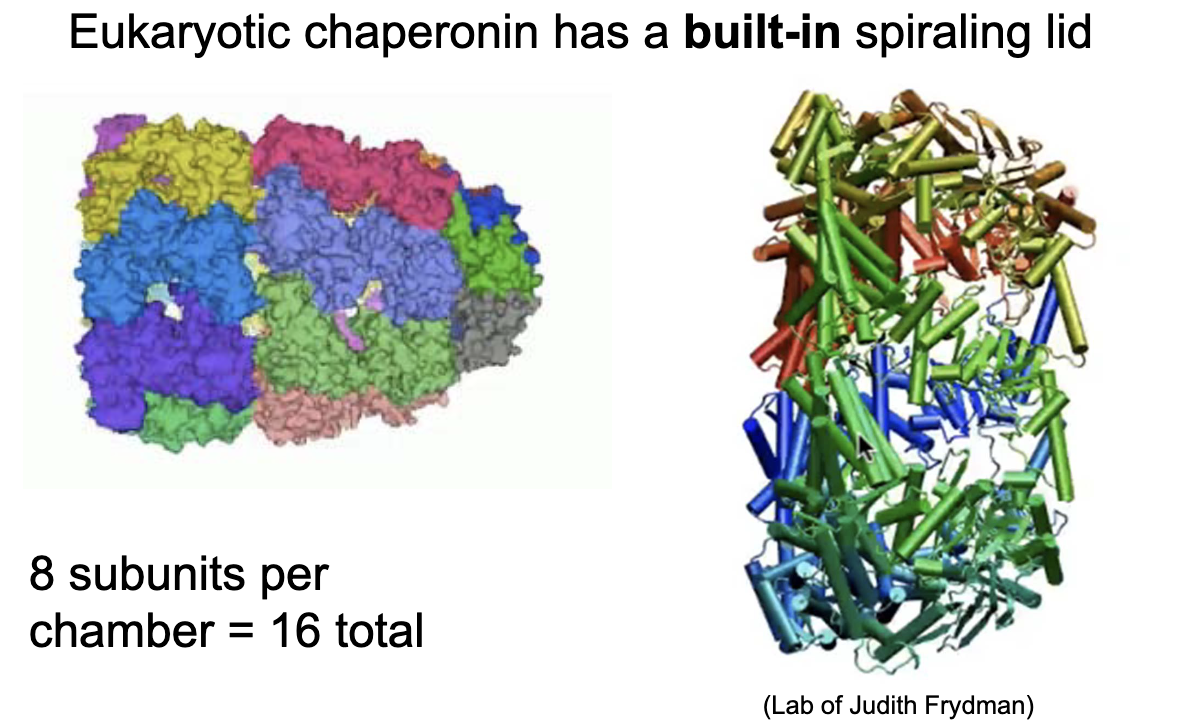
Mammalian chaperonin (Group II, e.g., TRiC/CCT) structure
8–9 subunits per ring; homomeric or heteromeric.
2 rings = 16–18 subunits total.
Built-in spiraling lid; no detachable cap (twists close).
ATP hydrolysis triggers conformational change and lid closure.
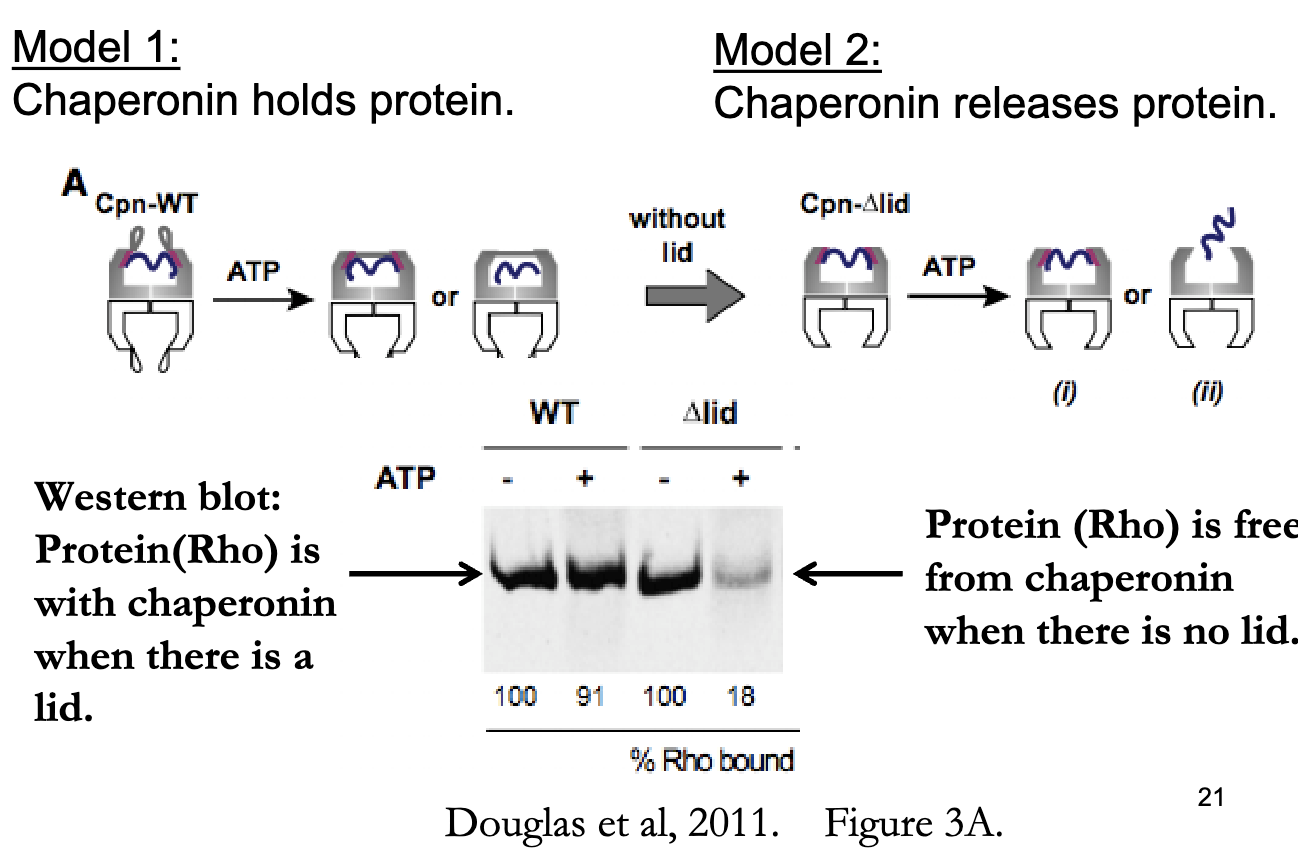
What experimental evidence supports the passive release model of chaperonin-assisted folding? (Hypothesis 1)
Hypothesis 1:
If folding requires release into the chamber, then removing the lid should allow the protein to escape → supports passive release model
Experiment:
Purified extracts of rhodanese + chaperonins
Incubated ± ATP for 10 minutes
Analyzed using native gels
Conditions Tested:
With lid
Without lid
Results:
With lid: Protein (Rho) is trapped in chaperonin
Without lid: Protein (Rho) is free = supports release model
Conclusions:
No lid = protein escapes
ATP is required for Rho protein to dissociate from chaperonin
Thus, Folding likely occurs after release, not through physical manipulation → supports passive chemical environment model
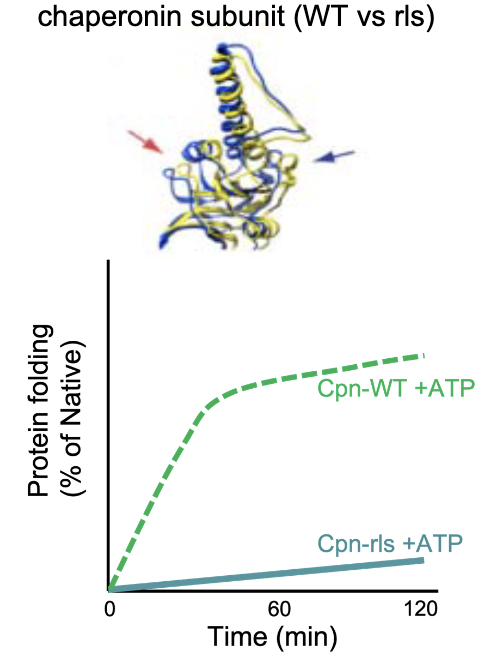
What experimental evidence supports the passive release model of chaperonin-assisted folding? (Hypothesis 2)
Hypothesis 2: If release is blocked, folding fails.
Used mutant Cpn-rls (modified Loop 11 prevents substrate release).
Lid closes but substrate isn’t released from edges of chaperonin.
No folding occurred = folding requires release into chamber.
Conclusion: Chaperonins “facilitate” protein folding
Why are chaperonins important beyond initial protein folding?
Prevent protein misfolding and aggregation.
Maintain protein quality control as cells age.
Misfolded proteins accumulate with age and can lead to protein aggregation diseases.
Chaperonins help counteract cellular stress and preserve protein integrity over time.