Renal 1 Main Points
1/110
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
111 Terms
What are the non potassium sparing drug types? What conditions should you look out for?
Look out for hypokalemic patients!
1) Thiazide diuretics
Inhibits the Na/Cl cotransporter in the distal convoluted tubule (inhibits the reabsorption of Na and Cl into the cell, increasing the concentration of Na and Cl in the lumen)
Further down the line, there is a Na transporter into principal cells of the collecting duct, which increases the concentration of Na inside the cell, for which the the Na/K ATPase on the basolateral side works (Na reabsorbed, K secreted)
This increases the amount of K in the lumen for excretion; hence, thiazide drugs do not spare K, as they actually lead to increased excretion of it.
2) Loop diuretics
These drugs inhibit Na/2Cl/K transporter in the thick ascending limb of the loop of Henle. Preventing these components from being reabsorbed.
This leads to an increase in these electrolytes in the lumen for excretion, which undergo similar processes down the line as thiazide diuretics, leading to an increase in K excretion.
This process leads to a larger effect of K excretion because TAL is responsible for 20-40% of Na reabsorption, whereas DCT = 10-12%
Describe the parts of the nephron and their responsibility for sodium reabsorption.
1) Proximal convoluted tubule = 60% of sodium reabsorption
2) Thick ascending limb (of the loop of Henle) = 20-40% of sodium reabsorption
3) Distal convoluted tubule = 10-12% of sodium reabsorption
4) Collecting duct = 2-4% of sodium reabsorption
The % of responsibility decreases as the flow progresses
What are the types of drugs that reduce potassium loss during sodium diuresis? What are thier effects for general diuresis?
1) Aldosterone receptor antagonists
Blocks aldosterone from binding, which decreases the following:
Uptake of Na/K ATPase (basolateral side)
Uptake of Na transporters into the cell and the K transporter secretors (out of the apical side)
This decreases the amount of Na reabsorption and K secretion, which helps preserve bodily K concentrations
2) ENaC blocker
Directly inhibits the ENaC channels (sodium channels on the apical side of principal cells), which decreases the amount of Na in the cell, decreasing the amount of Na Na/K ATPase transporters from “working.”
This decreases the amount of K that is transported into the cell, needed to drive the electrical gradient driving K secretion out of the apical side
These drugs help create mild diuresis (their main effect is with inhibition of reabsorption of Na), but only 2-4% of Na reabsorption occurs here, so the main point of choosing these drugs is for their K-sparing effects
What are the major types of diuretics?
1) Carbonic anhydrase inhibitors
2) Loop diuretics
3) Thiazide diuretics
4) K-sparing diuretics (aldosterone receptor antagonists + ENaC blockers)
5) Osmotic diuretics
6) ADH antagonists
1) What is the MOA of carbonic anhydrase inhibitors?
2) What are the effects of carbonic anhydrase inbitors on: site of action, Na, K, HCO3, and pH of urine?
1)
MOA: Inhibits CA, which typically helps reabsorb filtered HCO3 back into the body. Inhibition increases excretion of HCO3, Na, and water, leading to diuresis.
MOA of this: CA typically converts carbonic anhydride (HHCO3) into water and CO2, which can freely pass through the cell membrane, where CA can convert back into carbonic anhydride and then HCO3 and H
HCO3/Na symporter on the basolateral side of the cell
Na/H antiporter on the apical side of the cell -> H used to create HHCO3
CA inhibitors inhibit CA as well as the ability of the symporter and antiporter to function!
This leads to large amounts of HCO3 being excreted -> metabolic acidosis
Because blood HCO3 decreases over time (it is self-limiting), less HCO3 is filtered, so there is less substrate for CA inhibitors to act on.
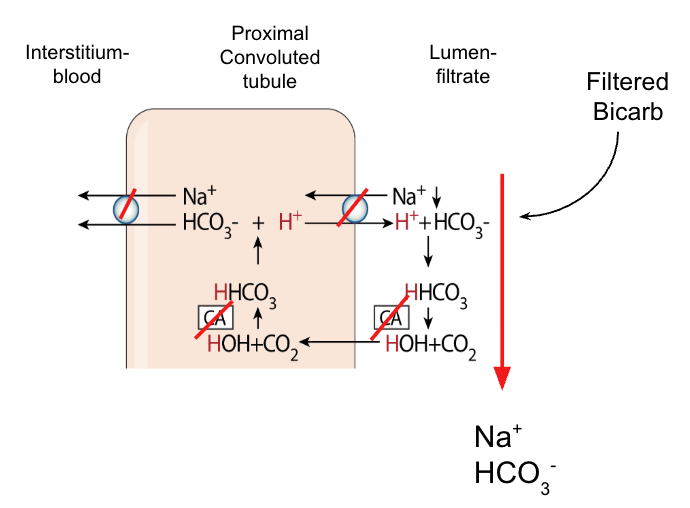
2)
Site of action: Proximal convoluted tubule (PCT)
Na effect: ↑ Na⁺ excretion
K effect: ↑ K⁺ excretion
HCO3 effect: ↑↑↑ HCO3 excretion
pH of urine: ↑↑
1) What is the MOA of loop diuretics?
2) What are the effects of loop diuretics on: site of action, Na, K, Ca, and pH of urine?
1)
MOA: Inhibit the Na/K/2Cl transporter in the TAL, which decreases intracellular amounts of these ions, which also prevents the following from happening:
K from leaking back out into the lumen, which would typically cause a more positive charge in the lumen, which would cause Ca and Mg to transport paracellularly to the blood -> so there is an increase of Ca and Mg excretion with the use of loop diuretics
Intracellular Na -> prevents Na/K ATPase on the basolateral side from working as much decrease of bodily Na (K, and Cl as well through K/Cl symporter)
Increase of Na in the lumen space increases the amount of Na that can be transported into the ENaC transporter in the principal cell of the collecting duct which can increase the amount of K excreted in lumen (by a lot → since there is a lot of Na in the lumen 20-40% responsibility)
In alpha-intercalated cells down the line the increase of K can increase work of K/H antiporter pushing H out of the cell decreasing pH of the urine.
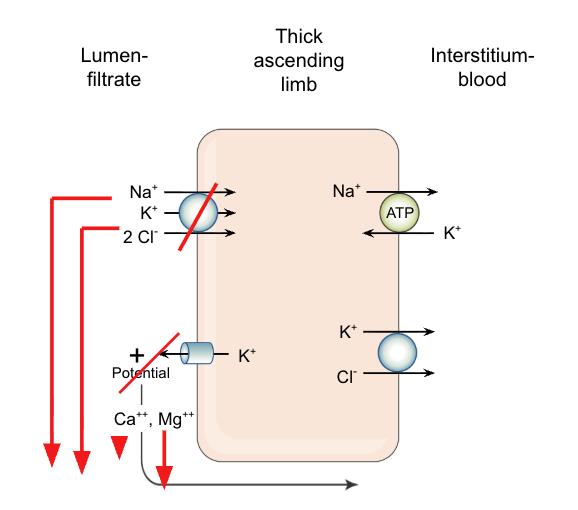
2)
Site of action: Thick ascending limb (TAL)
Na effect: ↑↑↑↑ Na⁺ excretion (20-40% Na reabsorption responsibility)
K effect: ↑ K⁺ excretion
Ca effect: ↑↑ Ca²⁺ excretion
pH of urine: ↓
1) What is the MOA of thiazide diuretics?
2) What are the effects of thiazide diuretics on: site of action, Na, K, Ca, and pH of urine?
1)
MOA: inhibition of Na/Cl symporter into the cell (apical side in the DCT)
This decreases the amount of intracellular Na
This calls for an increase of Na pull into the cell from the basolateral side with the cell down this gradient through the Na/Ca antiporter (Ca is favored within the body)
This pull of Ca into the body increases the Ca reabsorption from the lumen
The pull of Ca into the body decreases the amount of Ca in the kidney lumen meaning a decreased chance of developing kidney stones (Ca products) by essnetially pushing Ca back into the body
Increase of Na in the lumen space increases the amount of Na that can be transported into the ENaC transporter in the principal cell of the collecting duct which can increase the amount of K excreted in lumen
In alpha-intercalated cells down the line the increase of K can increase work of K/H antiporter pushing H out of the cell decreasing pH of the urine.
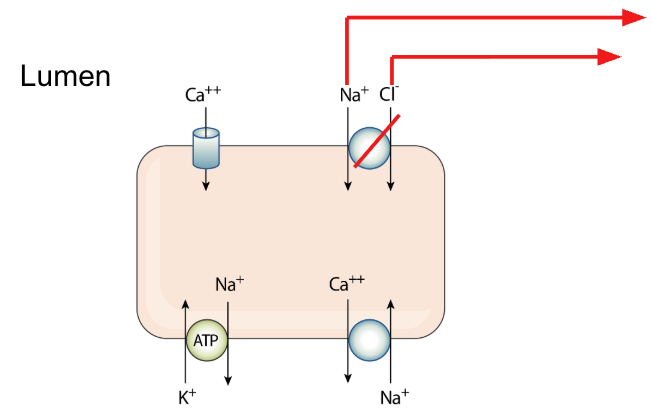
2)
Site of action: Distal convoluted tubule (DCT)
Na effect: ↑↑ Na⁺ excretion (10-12% Na reabsorption responsibility)
K effect: ↑ K⁺ excretion
Ca effect: ↓ Ca²⁺ excretion
pH of urine: ↓
What are the effects of potassium-sparing diuretics on: site of action, Na, K, and pH of urine?
Aldosterone receptor antagonists + ENaC blockers
Site of action: Collecting duct (principal cells)
Na effect: ↑ Na⁺ excretion
K effect: ↓ K⁺ excretion (spares K⁺)
pH of urine: ↑
1) What is the MOA of osmotic diuretics?
2) What are the effects of osmotic diuretics on: site of action, Na, and K?
1) MOA:
Initially “dilutes” blood by pulling water from intracellular compartment
Then dramatically increases urine output (if GFR is adequate). Because tubular urine moves so fast, reabsorption of solute is decreased (from PCT and descending limb).
Finally “concentrates” blood as excess water is excreted.
2)
Site of action: PCT and descending limb
Na effect: ↑ Na⁺ excretion (secondary)
K effect: ↑ K⁺ excretion
What are ENaC blockers typically used for/with?
Reducing K-wasting by other diuretics → typically used with thiazide diuretics
Reduce Li+-induced diabetes insipidus (treatment)
What is Li+- induced diabetes insipidus? What type of disease is this?
This is a type of: Nephrogenic diabetes insipidus
Lithium can enter ENaC channels in the principal cells in the collecting ducts which can:
Interferes with ADH signaling (even after binding) → resistance
Reduces aquaporin-2 expression
Prevents water reabsorption
This decreases the amount of water the body can reabsorb leading to the inability of the kidney to concentrate the urine → excessive, dilute urine output + extreme thirst
What is the MOA of ADH antagonists/agonists?
Agonists (MOA of ADH)
Help increase free water and urea reabsorption see below (opposite) → helps increase aquaporin water channels on apical side
Urine is left concentrated (very yellow)
Antagonists (diuretic effect)
Block vasopressin (V2) receptors in the collecting duct (lower part in the medulla)
Prevent insertion of aquaporin water channels and urea channels on the apical side of the principal cell
Aquaporin and Urea transporters already exist on the basolateral side → need ADH to upregulate on apical side
Water cannot be reabsorbed → leads to increase diuresis
What is hyponatremia? How is the condition of hyponatremia different when comparing loop and thiazide diuretics?
Hyponatremia: Low concentration of sodium in the blood, often caused by an excess of water in the body, which dilutes the sodium
NORMAL PHYSIO:
1) Thick ascending limb pumps out NaCl using transporters like Na/K/2Cl but does not transport water → this makes the medulla full of Na and Cl (Na is the driver of extracellular fluid osmolality, and it’s attendant anions) and the lumen dilute
Lumen: Hypotonic
Medullary space: Hypertonic
2) This gradient allows ADH to pull water out later in the collecting duct
Thiazides
Causes more hyponatremia
Since the MOA occurs in the distal convoluted tubule, the initial step can occur: there is reabsorption of Na/K/and Cl in the TAL
The drug increases the excretion of Na as well from the body due to blocking the Na/K transporters in the DCT (but this is in the cortex, so there is no effect in the medullary environment where ADH functions)
When the lumen fluid makes it back into the medullary area in the collecting duct, ADH works as per usual, reabsorbing water, leading to:
Increased Na excretion from the body (Na is not reabsorbed properly)
Increased H2O reabsorption into the body
Loop
Works in the medulla → disrupts the medullary osmotic gradient so that there is an increase of electrolytes in the lumen by the time the fluid reaches the ADH working site
Because there is a decrease in Na and Cl in the medullary space, ADH cannot pull much water out of the collecting duct
You lose both Na and water, preventing dilution of plasma sodium → making hyponatermia less likely
What is nephrogenic diabetes insipidus? How is it treated? What is the MOA for this?
Nephrogenic diabetes insipidus: a kidney disorder where the kidneys cannot concentrate urine, resulting in excessive, dilute urine output (polyuria) and extreme thirst (polydipsia)
ENaC blockers for Litium induced diabetes insipidus
Thiazide diuretics for non-litium
Although thiazides are diuretics, in NDI they paradoxically decrease urine output.
Thiazides inhibit Na⁺/Cl⁻ reabsorption in the distal convoluted tubule
Mild natriuresis and volume depletion
The body compensates by increasing Na⁺ and water reabsorption in the proximal tubule
Less fluid reaches the collecting duct/excreted
Even though ADH functionality is inhibited in this condition there is less filtrate for it decreasing overall urine output.
This is a typical result of thiazide diuretics
What therapy should be used for kidney stones? What is the MOA?
Thiazide diuretics
See Thiazide diuretic flashcard here is a photo for warm up (if you get this card first lol)
What drug would you recommend for a patient with severe hypercalcemia with advanced carcinoma?
Loop diuretics (after IV hydration of pateint)
Mechanistically, there is an increased excretion and decreased reabsorption through paracellular spaces of Ca and Mg due to the lack of K leakage from the intracellular space (due to Na/2Cl/K transporter inhibition)
This is the only diuretic that increases the Ca excretion
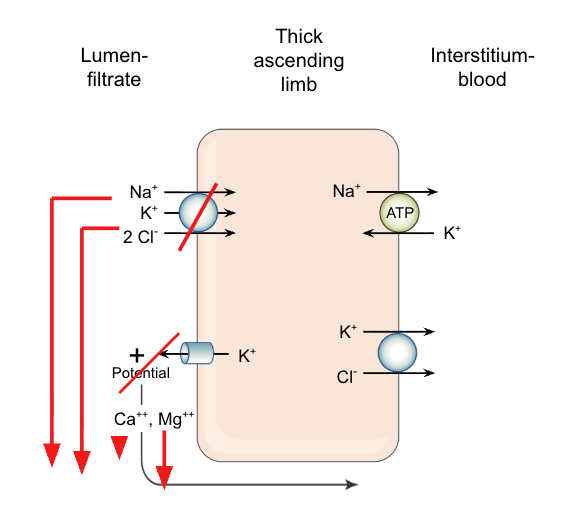
What is a sulfonamide moiety? Which diuretics have it? What are alternatives
It is a chemical group (–SO₂NH–) → sulfonamide drug allergy sensitivity potential found in:
1) Loop diuretics (all except ethacrynic acid)
2) Thiazide diuretics
3) Carbonic anhydrase antagonist
Alternatives:
Ethacrynic acid
If they want a loop diuretic
Potassium-sparing diuretics (aldosterone receptor antagonist + ENaC blockers)
For K-sparing needs
Explain the parts of the different parts of a nephron and which are responsible for water reabsorption and which are not.
1) Proxminal convoluted tubule → yes water reabsorption
2) Descending limb (of loop of Henle) → yes water reabsorption
3) Thin ascending limb → NONE
4) Thick ascending limb → NONE
5) Distal convoluted tubule → NONE
6) Collection duct → conditional → ADH-dependant
What hormones control water and sodium in the kidney?
Sodium → Aldosterone
Water → ADH
Where in the kidney are calcium, potassium, and phosphate reabsorbed and secreted (if applicable)?
1) Calcium
Reabsorbed: PT, TAL (both paracellular), DCT (intracellular)
2) Potassium
Reabsorbed: PT, TAL
Secreted: Collecting duct (principal cell) → increased by aldosterone
3) Phosphate
Reabsorbed: PT (through Na cotransportation)
What is the role of each nephron segment?
1) Glomerulus → filtration
2) Proxminal convoluted tubule → Bulk reabsorption
Filtrate stays isotonic
3) Descending limb (of loop of Henle → creates gradient)
Reabsorbes water only → concentrates the filtrate
4) Thin ascending limb
5) Thick ascending limb
Reabsorbes Na, K, Cl, → and Ca, and Mg byproxy → dilutes filtrate and concentrates medullary environment
Impermeable to water
Tubular fluid becomes dilute
6) Distal convoluted tubule “fine tuning”
Reabsorbes Na + Cl
Impermeable to water
7) Collection duct “final regulation”
Aldosterone adjusts for sodium reabsorption → indirectly affects water by
ADH adjusts for water reabsorption
How does the proximal tubule conduct reabsorption and secretion of Na (and co.), nutrients, proteins, peptides, and organic molecules? In other words what are the main functions of the proximal tubule?
1) Reabsorption of Na + nutrients
INITIAL: lumen is quite positive → leads to Na being reabsorbed paracellularly
3Na/2K ATPase on the basolateral side of the proximal cells drives reabsorption by creating an electrochemical gradient
This increases the amount of Na going into the body and decreases the amount in the cell → gradient of Na from lumen into the cell
Through Na/Nutrient transporters (ie. SGLT1 and SGLT2 transporters)
Glucose exits through the basolateral side through GLUT transporters
Through Na/H channels on apical side as well
Pull of Na into the cell causes the lumen to become more negatively charged creating a gradient pull for Cl- into the body paracellularly
Water will follow = osmosis
Most of K, Ca, and Mg is absorbed through solvent drag (with water)
2) Reabsorption of proteins and peptides
Proteins bind to receptors
Endocytosis of proteins which are then broken down into amino acids -> into the blood
3) Secretion of organic ions → DRUG APPLICATION
Basolateral side -> organic cation transporters (OCT) and organic anion transporters (OAT)
Apical side -> Antiporters (of similar charges) -> Cl- and H+
What are the definitions for the following terms?: effective circulating volume, hypo-, hyper- and isotonic solution, osmolality and osmolarity.
Effective circulating volume
The amount of blood the heart is actually pumping = CO
Hypotonic solution
Water moves into cells
Lower solute concentration than inside the cell
Hypertonic solution
Water moves out of cells
Higher solute concentration than inside the cell
Isotonic solution
No net water movement
Same solute concentration as inside the cell
Water moves toward spaces with higher solute concentration
Osmolality
Number of solute particles per liter of solution (Osm/L)
Volume
Osmolarity
Number of solute particles per kilogram of solution (Osm/Kg)
Mass
Increase of plasma osmolality, a decrease of BP and blood volume increases what?
Thirst
What is the MOA of ADH in vasculature?
It binds to V1 receptors causing vasoconstriction (helps with Low BP and volume that can be measured from baroreceptors leading to the increase release of ADH in the first place)
NOTE: angiotensin 2 increases vascular tone as well (predominant)
Describe the glomerular blood supply from the artery.
Renal artery → Smaller arteries → Afferent arteriole → glomerular capillaries (still relatively high pressure due to further journey blood has to take still) → efferent arteriole → second capillary network → venules → renal vein → inferior vena cava
Describe the glomerular filtration membrane structure and their functions. How does hypertension affect the capillaries?
1) Fenestrated capillary endothelium (has pores)
Allows for water + small solutes through
Blocks RBCs, platelets
2) Glomerular basement membrane
Thick, negatively charged -> repels large and negatively charged proteins (like albumin)
3) Podocytes/Podocyte slits
Provides structural support to glomerular capillaries
Exposed to higher pressures than the systemic capillaries
Higher pressures = more damage to these capillaries from hypertension than other capillaries
Slit membranes repel large, negatively-charged proteins (like albumin) -> prevents them from being freely filtered
Damage to podocytes lead to what? What condition is this characteristic of? What about both endothelial cells and podocytes?
1) More proteins in the urine → Diabetes
2) Blood cells entering renal tubules → blood in urine → some infections + autoimmune diseases
What is GFR and what is it’s function in kidney function?
Glomerular filtration rate (GFR) → volume of plasma filtered by all glomeruli in the kidney per mL/min
Helps to measure kidney health
Reflected in plasma creatinine concentration
What is plasma creatinine and what is it used for?
1) Plasma creatinine is a waste product from muscle metabolism -> produced at a fairly constant rate
Freely filtered at the glomerulus (but not reasborbed, secreted, degraded, or synthesized in the lumen of the kidney)
Some is secreted (not important)
2) Used as a marker for GFR (kidney health)
Can calculate using equation:
Creatinine clearance (Cc)(i.e. GFR in this case) = [estimate of GFR using urine (UC) x V(of urine)] / Plasma creatinine (PC)
Scr and GFR are inversely proportionate to one another so if GFR increases there is less Scr and vice versa
Glomerular filtration depends on what two things?
Net filtration pressure and filtration surface
What are the two regulatory factors for GFR?
Macula Densa
Secretes
ATP: vasoconstrictor
Adenosine: vasodilator
Measures the kidney’s GFR through NaCl concentrations in the distal convoluted tubule
If NaCl is higher -> release of ATP
Leads to afferent arteriole constriction
If NaCl is lower, -> release of adenosine
Dilation of the afferent arteriole
Granular cells
1) Secrete renin through stimulation of baroreceptors in the following:
Lower BP
Lower stretch
Lower NaCl (sensed by the macula densa)
Higher sympathetic activity (b1)
Renin -> activation of RAAS
Angiotensin II -> efferent constriction to help increase/maintain GFR → release of aldosterone
Also constricts the afferent arteriole, but the release of protsaglandin in this area makes this vasoconstriction less than in the efferent.
2) SANS activation
Releases NE
Vasoconstriction → Reduces blood flow to the kidney (alpha1 receptors)
Less blood to vital organs → Decreases GFR
NE also constrict mesangial cells (beta receptors) in the bowman’s capsule
Lowers SA for glomerular filtration → Decrease GFR
Stimulates RAAS (Activates beta1 receptors) on granular cells
Release renin → aldosterone release
How do SANS and RAAS systems affect RBF?
SANS
Constricts renal arteries, afferent, and efferent arterioles → moves predominant blood flow to the brain/heart
Decrease RBF
Decrease GFR
RAAS
Triggered by low BP and volume
Constricts efferent arterioles > afferent (protective mechanism produced locally by the kidney endothelial in response to RAAS/SANS stimulation)
Helps to maintain GFR
Does not help in hypertensive states → just increases the pressure in the glomerular capillaries = increased damage
How is the RAAS system regulated and what does it produce that affects the kidney?
Activation: Release of Renin (from granular cells in afferent arteriole)
Decrease BP
Decrease afferent arteriole stretch
Mechanoreceptors in granular cells
Decreased NaCl delivery to the macula densa
Increased SANS activity
Renin converts angiotensinogen into angiotensin 1 -> ACE converts it into angiotensin 2
Angiotensin 2
Stimulates aldosterone release -> increases Na reabsorption and increases K excretion in the principal cells in the collecting duct
Stimulates increased thirst
Vasoconstriction of the efferent > afferent arteriole
Explain how aldosterone regulates total body sodium and volume but does not affect the plasma osmolality while ADH regulates body water, but its primary purpose is regulation of plasma osmolality.
Aldosterone
Aldosterone increases Na⁺ reabsorption and K⁺ secretion by upregulating ENaC and Na⁺/K⁺ ATPase in the collecting duct.
MOA: Na/K ATPase creates initial gradient → increase Na into the cell (increase Na intracellularly → pushes K out)
Water follows Na⁺, so both Na⁺ and water are retained together → MUST REMEMBER THO THAT WATER REABSORPTION CAN ONLY OCCUR ID ADH IS THERE → water will follow if qquaporin channels are present
Cl- ions follow the Na+ to balance charge
Total body volume increases but plasma osmolality remains unchanged because Na⁺ concentration stays relatively constant.
ADH
ADH binds to V2 receptors and inserts aquaporin channels in the collecting duct, increasing reabsorption of free water.
This water movement is driven by the hypertonic medullary gradient (Na/2Cl/K channel in thick ascending limb helped to create).
ADH reabsorbs water without Na⁺, it dilutes plasma Na⁺ concentration and therefore decreases plasma osmolality.
How does ADH is released in response to non-osmotic stimuli (what is this?)? What does this result in?
NON-osmotic stimuli (low MAP/ECV, pulmonary disease, malignancies, surgery)
Ex: Drop in MAP leads to stimulation of ADH secretion as a defense mechanism (regardless of osmotic status)
To ensure perfusion of vital organs (i.e. brain) by
Vasopressor effect
Water retention in kidney (volume preservation without regard for osmolaltiy)
Increased thirst
This leads to increased reabsorption of free water content into the body = hyponatremia + increased urine concentration.
How do aldosterone and ADH effect urine concentrations?
Aldosterone
Decreases Na concentration in the urine + water follows
Decreases overall volume in the lumen (neglegible)
ADH
Increases urine concentration (takes free water out essentially)
Increase osmolality of urine.
How can you identify a patient with active ADH production vs aldosterone vs both?
Aldosterone
Urine sodium content (low = aldosterone active)
Hypokalemia → increased K excretion
ADH
Increased thirst
Highly concentrated (low volume) urine
Hyponatremia
Both (HF/cirrhosis)
Increased thirst
Hyponatremia (ADH)
Low urine Na (aldosterone)
Low urine volume
Why it is critical to control water intake in a patient with low MAP-caused hyponatremia (such as ADHF)?
Low MAP leads to increased free water reabsorption through ADH release → if patient drinks more water on top of this can worsen dilutional hyponatremia
Want to prevent cerebral edema (deadly).
Draw out the mechanisms that regulate arterial blood pressure that act fast and slow (which do what?).
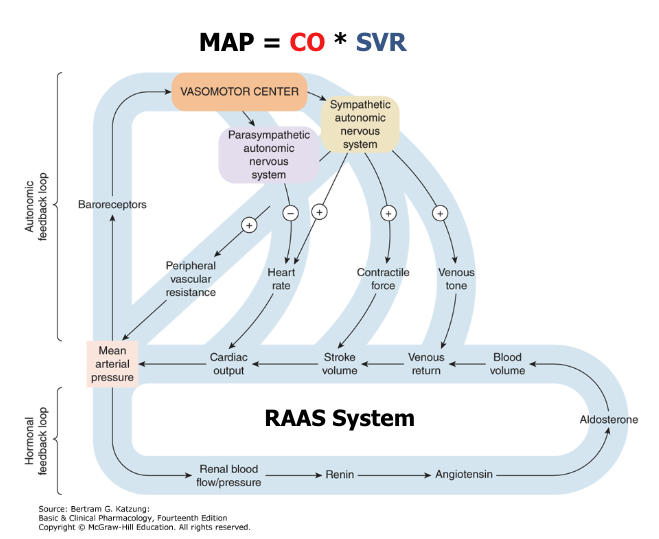
Autonomic system = quick acting
Kidney (RAAS system) = slow
Strategies for combating hypertension affects which three factors?
1) Decreasing SV
2) Decreasing SVR
3) Decreased body fluid volume
For essential hypertension to occur what must happen?
1) Increase in one or more of the following:
Increased SV (and CO) → typically seen in younger pateints (acute)
Increased SVR → most common for primary/essential hypertension (conceptually)
Increased Body fluid volume → diet impact + increased thirst (from fluid intake and reabsorption → fluid retention leads to increased fluid volume in the arterial system provided heart is not failing)
AND
2) Resetting/recalibration of baroreceptors (increased activity of one of the three above has to be prolonged to the point where the body adapts to the new, higher blood pressure)
What is the typical progression of essential hypertension?
Begins with prehypertension in persons aged 10-30 years (by increased cardiac output)
Advances to early hypertension in persons aged 20-40 years (in which increased peripheral resistance is prominent)
Progresses to established hypertension in persons aged 30-50 years
Advances to complicated hypertension in persons aged 40-60 years
What are known risk factors for primary/essential HTN?
Obesity, lack of exercises, genetics, stress, testosterone predominant sex hormone
What factors would help you identify if a patient has primary HTN vs. secondary HTN?
Primary:
Gradual onset (often age 20–50)
No clear secondary cause (normal labs/workup)
Often associated with:
Family history
Obesity
High salt intake
Sedentary lifestyle
Secondary:
Resistant to ≥3 drugs (including diuretic)
Sudden onset or worsening (acute) → in patient that previously has well controlled BP
Young age (before puberty, esp. with no family history or hypertension exists)
Hypokalemia (suggests hyperaldosteronism → caused by plaque formation which signals a decrease of BP by decreasing blood volume → macula densa with sense and cause granular cells to release renin)
Can be medication induced
What is secondary hypertension? What conditions can it be caused by?
Caused by identifiable underlying condition (not idiopathic like primary) + potentially treatable
More acute caused than primary
Caused by conditions like:
Renal artery stenosis:
Decreases RBF and GFR resulting in kidney “thinking” that MAP is low which increases RAAS and leads to increased fluid retention as well as systemic vasoconstriction in response to aldosterone and angiotensin II (respectively).
Hyperaldosteronism:
Increased salt and water retention by kidney.
Cushing’s syndrome:
Increased production of angiotensinogen and increased sodium and water retention in kidney.
Pheochromocytoma:
Tumor of adrenal medulla producing too much adrenal cathecholamines – epinephrine, norepinephrine, dopamine – leading to powerful activation of adrenergic receptors.
What medications can cause secondary hypertension?
Oral contraceptives
The estrogen content in them leads to sodium retention and activation of RAAS.
NSAIDs
Interfere with prostaglandin-mediated afferent arteriole vasodilation in the kidney, which leads to a decreased GFR and RAAS activation that then induces hypertension.
Prostaglandins also help with vasoconstriction in other parts of the body → decreased effect
Calcineurin inhibitors
Immunosuppressant medications frequently used in autoimmune conditions and to prevent organ rejection after transplant. These medications cause hypertension via vasoconstrictive effects on the renal vasculature and sodium retention.
Steroids
Via sodium and water retention.
Stimulants
This includes psychiatric medications, such as amphetamines, and decongestants (sympathomimetics!).
Novel cancer therapies
Many new cancer therapies, including vascular endothelial growth factor (VEGF) inhibitors have been ground-breaking in their impact on treatment of malignancies, however, these drugs lead to a disruption of normal vascular homeostasis as they inhibit production of the vasodilator nitric oxide and lead to increased vascular resistance.
How does hypertenion effect large and small vessels throughout the body?
Large:
Causes tears in the tunica intima (endothelium)
Leads to bleeding in the tunica media (Muscle layer) along the length of the vessel → dissection
Tunica media provides structural integredy → destruction of it decreases this = bleed (acutely deadly)
Promotes atherosclerosis → damage allows LDL cholesterol to deposit in sub-endothelial space
Small:
Causes hyaline transformation (hyaline atherosclerosis) of the smooth muscle layer in small arteries and arterioles
Leads to deposition of proteins that replace smooth muscle → remodeling
Decrease delivery of blood through arterioles → ischemic damage + rupture
NOTE: HTN causes atherosclerosis not the other way around → only happens when there is A LOT of atherosclerosis in the aorta → but does not happen generally because atherosclerosis does not occur in every blood vessel to cause HTN
What is hypertension caused damage to the retinal vascular system called? What defines it?
Hypertensive retinopathy
Signs develop late in the disease
Funduscopic examination shows:
Arteriolar constriction
Arteriovenous nicking
Vascular wall changes
Flame-shaped hemorrhages
Cotton-wool spots
Yellow hard exudates
Optic disk edema
Treatment:
Controlling blood pressure and, when vision loss occurs, treating the retina.
How does HTN affect the cardiovascular system?
It increases afterload (heart has to pump against higher resistance)
Heart compensates → Left ventricular concentric hypertrophy
Sarcomeres added in parallel
Wall thickens (fibrosis → ventricular remodeling)
Increases heart tissue O2 demand but same numebr of cells and capillaries exist so no way to compensate
Consequences:
Problems with relaxation → diastolic dysfunction (HFpEF)
↑ O₂ demand (but no increase in capillaries) → ischemia
Long-term:
Overload of atria (high pressure needed to fill stiff ventricle → HFpEF) + damage to heart vasculature = AFIB
Systolic dysfunction (HFrEF) from prolonged myocyte death + fibrosis
Also:
Vascular damage → ↓ coronary perfusion
How does HTN affect the kidneys?
Hyaline transformation of afferent arteriole → decreases perfusion to glomerular sclerosis:
1) Glomerular ischemia
Increased RAAS (angiotensin II)
Sensed from the macula densa → released and works on all nephrons (damaged and healthy)
Vasoconstriction effect to efferent arteriole
Profibrotic → speeds up damage to kidney and heart
Increased glomerular pressure
Increased fibrosis
2) Loss of myogenic control of glomerular BP
Glomerulus not protected from systemic BP oscillations/increase
Why is the release of angiotensin II problematic when considering HTN?
Background:
Angiotensin II can work everywhere (not specific to only damaged nephrons from where macula densa sensed decrease blood flow from hyaline atherosclerosis)
Angiotensin II causes vasoconstriction to the efferent arteriole (some to the afferent but more predominant in the efferent on the account of prostaglandin production by angiotensin II as a compensatory mechanism for the afferent).
Prostaglandin helps to keep minimal levels of RBF in face of hypovolemia
When working on a healthy nephron vasoconstriction on the efferent arteriole increases filtration pressure and GFR which only worsens the HTN and damage that it causes to the vasculature in the kindey
Glomerular membrane damage from increased filtration pressure means that more proteins are filtered through (more than the PCT can handle)
Leads to more proteins excreted through urine → proteinuria
Angiotensin II “helps” in the process of fibrose of injured glomeruli → remodeling → glomeruloscleosis = drop in GFR
This process continues in uncontrolled hypertension until kidney function is completely lost!
Summary: Hyaline atheroscleosis of the afferent arterioles resuilts in decreased filtration in the damaged nephrones and hyperfiltration int he areas with less/no damage.
In what case is angiotensin II needed (other than homeostasis)? What drugs can you not use at this time in ode to this?
Early development (fetus) → cannot use ACEi/ARB to pregnant individuals
Why are ARBs/ACEi’s igors fav drugs to use for HTN?
Kidney specific:
Inhibits and lowers BP
Protects the nephrons → by blocking ARBs and ACEis
How does HTN affect the brain?
The creation of hyaline atherosclerosis in the brain small arteries/arterioles induces micro bleeds → mini strokes
Afferent arteroles with hyaline atherosclerosis lose what ability?
To regulate renal blood flow (RBF) or glomerular filtration rate (GFR) because they have lost their smooth muscle.
Although the lumen is narrowed if pressure in systemic circulation increases → this increase will go to the glomerulus → results in hyperfiltration and filtration membrame damage
identify therapeutic approaches that can directly address these mechanisms for primary + secondary hypertension
What are first line agents for hypertension?
1) Thiazide diuretics
2) CCBs (DHP and non-DHP)
3) ACEi/ARBs
What does it mean if we see glucose in the urine?
The carrier proteins in the membrane on the proximal tubule are saturated (too much glucose) → diabetes indicator
What does PCT secretion depend on? What is secreted here?
Dependent on ATP. The following are secreted:
Drugs
H+
Creatinine
What is one way you can think about osmolality increasing or decreasing in a solution?
Sort of like a ratio:
Osmolality = solute/water → if water leaves the osmolality increases → if water comes into the solution the osmolality decreases
If the solution is getting more dilute the solution decreases in osmolality → water wants to travel to this area less (already balancing out)
If the solution is getting more concentrated the solution increases in osmolality → water wants to travel here more to even out the balance
What is the during of ENaC blockers compared to aldosterone receptor blockers?
They have a shorter duration of action
How is ADH regulated?
Osmoreceptors (MAIN)
Detect plasma osmolality (extracellular) above 280 Osm/Kg
Even 1% increase can trigger the release of increased ADH
290mosm/kg is where max antidiuresis of ADH occurs
As extracellular osmolality increases (water loss) → water leaves osmoreceptors (and other cells) → osmoreceptor cell “shrinkage” = signal for ADH release
Autoregulation
Baroreceptors
Decrease in MAP/ECV (low BP and volume) is detected by baroreceptors -> RAAS activation -> ADH release
ADH acts as a potent vasoconstrictor
What is the GDMT for HF?
1) ACEi/ARB/ARNI
2) BB
3) MRAs (minerocorticoid receptor antagonists → aldosterone inhibitor)
4) SGLT2 inhibitors
How does urea affect water reaborption?
Urea secretion into the interstitial space increases the osmolarity in the medulla to increase water reabsorption driven by ADH
What is aldosterone release controlled by?
1) Myogenic → baroreceptors
Central - stimulate SANS and renal
2) Tubuloglomerular feedback → Macula densa
Sensing drop in GFR → RAAS system activation
How does the body regulate GFR in HF? How can this lead to hyponatremia?
1) Reduced cardiac output reduces MAP → baroreceptors → signaling both SANS and RAAS.
This constricts afferent/ efferent arterioles and mesangial cells to redirect blood away from thekidneys and drastically lower GFR.
Prostaglandins preserve some flow through vasodilation.
2) Autoregulation attempts to oppose this, but without blood flow it has little effect.
Myogenic regulation will vasoconstricts when there is high pressure, but there is low blood flow with SANS and RAAS.
Tubuloglomerular feedback will attempt to constrict efferent arterioles in response toreduced NaCl due to reduced flow to bring up GFR.
3) This can lead to hyponatremia, as baroreceptors will sense large drop in Mean Arterial Pressure (and effective circulating volume) and trigger ADH release to defend MAP and preserve vital organs.
Large drop in MAP also increases thirst resulting in more drinking water and ADH preventing water loss in the urine leading to:
ECF dilution and reducing Na+ concentration
not “concentrations” - ECF has one concentration of sodium
In patients with HF what should be monitored?
NSAID use: protaglandin influence
During HF SANS is activated + RAAS = vasoconstriction in both afferent and efferent arterioles in the kidney which can cut blood flow off to the kidneys → acute kidney failure
Higher renal resistance = increased HTN/pressure
Water and salt intake
Increased water = volume overload
Increased salt intake = trigger osmorecepotrs → upregulates secretion of ADH = increased water retention + increased ECV = volume overload
Volume overload can worsen HF by increasing preload
Kindey damage → through low perfusion
Why do you pee when you drink alcohol?
Alcohol suppresses ADH secretion into the blood → prevents kidneys from reabsorbing water = diuresis
Increases solute/water ratio in the blood
What does the ALLHAT study show?
Thiazide-type diuretics are superior in preventing 1 or more major forms of CVD and are less expensive.
First-step antihypertensive therapy.
What is the LIFE study?
Selective AT1-receptor blockade with losartan is POTENTIALLY more effective than the BB atenolol in reversing LV hypertrophy and cardiovascular morbidity and mortality beyond blood pressure lowering
What are alpha and beta blockers used for in HTN?
They are NOT first line antihypertensive agents in the outpatient setting but can be added if patient has compelling indications
They are used in management of hypertensive EMERGENCIES where BP is managed acutely using IV drugs in ICU.
(Systolic BP >180 mM Hg & organ damage and/or Diastolic> 120 mm Hg & organ damage)
What are second line agents for HTN?
Loop Diuretics
Potassium-Sparing Diuretics
Aldosterone Antagonist Diuretics
Alpha-2 Central Agonists
Direct Renin Inhibitors
Alpha-1 Antagonists
Beta Antagonists
Direct Vasodilators
How ware hypertension emergencies defined?
A point where the patient’s BP is so high, their organs acutely start to shut down (different from severe HTN). Can look like:
High arterial pressure
Fibrotic necrosis in arteries
CNS effects
Headache, vision loss, coma
Renal disfunction (oliguria)
How is hypertension classified?
Normal:
<120 mmHg and <80 mmHg
Elevated:
120-129 mmHg and <80 mmHg
Hypertension
Stage 1:
130-139 mmHg or 80-89 mmHg
Stage 2:
≥140 mmHg or ≥90 mmHg
Which beta blockers are selective, which are not?
A-M except C and L are B1 selective
Which beta block is an ester with a fast onset and short half life?
Esmolol
What is the effect of cocaine on MAP (MOA)?
Excessive stimulation of the heart (increase SANS, HR, PVR = increase mean arterial pressure)
Cocaine blocks the reuptake of:
NE
Epi
Dopamine
This leads to increased SANS activation and activation of:
A1 receptors → vasoconstriction
B1 → increase HR, contractility
B2 → vasodilation
Why should you avoid beta-blockers in cocaine overdose?
In extreme SANS activation the only saving grace is that there is some vasodilation effect from B2 receptors. This is the only factor that allows for perfusion in the body → SO if you use a beta-blocker you are left with:
Pure alpha constriction (including coronary vessels → can trigger MI in patient with high HR)
What medications discussed have withdrawal symptoms?
Clonidine → downregulation of A2 → abrupt spot = increased NE effect in body
Selective B1 blockers (like metoprolol) → Long term use body will upregulate the amount of B1 receptors making the system more sensitive to catecholamines → rebound sympaetic surge
When would you lower BP using alpha/beta blockers? When would you not?
Use: In the acute setting + optimization for other health concerns like previous MI
Don’t: In settings where there might be unopposed alpha-activation potentially leading to coronary vasospasm + MI
Patient with HTN started on hydralazine returns with palpitations, tachycardia, chest pain, and edema. What is the mechanism causing these symptoms, and how do you fix it?
MOA: Vasodilation → decrease of BP leads to compensatory mechanisms like…
SANS activation
Incerase HR and contractility → tachycardia + chest pain (from increased O2 demand)
RAAS activation
From low renal perfusion
Na/water retention → edema
Fix with:
Adding beta blocker to prevent reflex tachycardia
Add diuretic → prevent fluid retention
OR reduce/avoid hydralazine (consider amplodipine which is first line)
What are some ways to deal with hypokalemia for pateints?
Add in K supplement or switch to K-sparing drugs (aldosterone receptor antagonists or ENaC blockers)
If the patient has history of MI and has HTN now how would this change treatment plan?
Give medications to help acute MI that can also help HTN → prioritize things like ACE + BB instead of optimizing thiazide treatment.
What channels interact with Ca? Which is “stronger”
Proximal convoluted tubule → solvent drag
Na/K/2CL channel → STRONGER
Na/K → mild hypercalcemia
How does HTN damage the heart?
1) Increased afterload leads to left ventricular hypertrophy
By addition of sarcomeres in parallel and an increase in myocardial oxygen demand.
However, the oxygen supply does not increase as this is not followed by commensurate increase in the capillary network. This results in remodeling of a stiffer heart unable to relax properly as evident in HFpEF and afib
Ventricular remodeling + ischemia = HF
Atrial remodeling + fibrosis = afib
2) Atherosclerotic damage to coronary arteries and their large branches
HTN damages the endothelium and increases LDL-C deposition in the wall of these blood vessels, and this decreases oxygen supply to the myocardium.
3) Hyaline atherosclerosis
Of the small coronary arteries and arterioles also leading to decreased oxygen supply to the myocardium
When a patient has the additional on top of HTN which should be first line drug approaches for their care?: HF, CKD, afib, low HR
HF
GDMT → prioritize ACEi/ARB/ARNI + B-blocker + MRA
Diuretics for symptoms
CKD
ACEi ro ARB first (renal protection)
Afib
BB or non-DHP CCB (rate control)
Low HR
AVOID BB
What is the ACC/AHA guidelines for HTN treatment?
Pretty much everyone has a BP goal of <130/80 → EXCEPT OLDER PATIENTS due to quality of life → HTN medications can lead to hypotension = falling → hospitalization
Stage 1
ASCVD risk < 7.5% → life style modifications
ASCVD risk ≥ 7.5% → Lifestyle modifications + 1 medication
Stage 2
Lifestyle modifications + 2 medications
DBP ≥ 90
Lifestyle modifications + 2 medications
SBP ≥ 130
Lifestyle modifications + 2 medications
What are the first steps of optimization + addition for HTN (if alone)?
Optimize first line treatments + add on other first line and optimize those before moving to 2nd or 3rd line therapies
What is the most common side effect in patients taking DHP CCBs? What is the MOA?
Edema. CCBs dilate arterioles without concomitant venodilation → increased venous pressure = capillary leak and increased interstitial fluid
How does HTN lead to afib?
Chronic HTN → LV hypertrophy → HFpEF which leads to decrease LV filling and increase of LV filling pressures
This pressure builds back up into the left atrium → dilation + volume overload = atrial myocyte injury + fibrosis
Fibrosis disrupts conduction → reentry circuits = AFIB
What lab should you look for when giving thiazides?
Sodium (decreases)
What lab should you look for when giving ARBS?
Potassium (increases) + Scr (increase)
What should you look for when giving CCBs?
Edema (increases)
What lab should you look for when giving MRA?
Potassium (increase)
What lab should you look for when giving loop?
CrCl > 30mL/min + sodium (decreases) → also would need a reason to use (like edema)
What should the monitoring process look like after giving a patient an ARB/ACEi?
Check labs at ~1 week
If:
K⁺ < 5.5
Scr ↑ <20%
→ Continue same dose
Do not increase dose until ≥2 weeks
Reassess labs before changing
How does a cluster randomized controlled trial (RCT) differ from a traditional RCT?
Traditional RCT: individual patients are randomized
Cluster RCT: entire groups (e.g., pharmacies, clinics, providers) are randomized → the entire cluster of patients within each “practice site” receives the same treatment.
Data analysis team is blinded → single blind
When should you use a cluster randomized trial?
When intervention is delivered at the group level
When there is high risk of cross contamination
Contamination = participants in different groups influencing each other
If patients assigned to both the simple control and complex treatment intervention are patrons of the same pharmacy “practice site,” then they could become aware of the alternative treatment potentially diminishing the magnitude of the treatment effect.
When studying real-world practice settings
What BB have alpha blockage?
Carvedilol and labetalol