Huntington's Disease
1/27
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
28 Terms
What is Huntington’s disease
Autosomal dominant
Neurodegenerative – loss of function
Hereditary – everyone with a mutated gene will get Huntington’s disease but not all will have a clinical presentation
Probability of inheriting the gene is 50%
Characterised by cognitive, behaviour and motor dysfunction
Patients have involuntary symptoms (uncontrolled jerking disease)
Prevalence
Mostly impacts Western countries in America, Australia and most euorpean countries
Due to family lineage, although there are some de novo mutations, most are hereditary
Prevalence increasing over the past 2 decades
Lower prevalence in Asia and Africa
The faulty gene
Huntingtin (HTT) gene
CAG region repeated many times which gives rise to poly Q repeats towards the N terminus
The actual gene identified in 1983 and then the first genetic test to identify this in 1993
CAG repeats vary in number —> down to the number of repeats which determine the severity and when it will appear in the lifecourse
10-35 repeat individuals will have a relatively normal life
36+ repeats will begin to impact the individual
Anticipation
Relates to each generation having an earlier symptom onset
CAG repeats are prone to errors in DNA replication which leads to more mutation
If this falls within the huntingtin gen we will have more repats added to the gene
Age of onset
Correlation with no of repeats and age of onset
25 repeats – totally unaffected individuals
25-35 – will not develop symptoms but pass on very highly repeated Huntingtin gene to progeny, so children are at risk.
Truly affected individuals have over 36 repeats
36-39 have reduced penetrance —> may have some of the symptoms or may appear later in life
40 or more have full penetrance
In very extreme cases with 60+ repeats not only do you have full penetrance but the age of onset may be a lot earlier
Not an absolute threshold, not everybody will behave according to this model
Prognosis
The time between point of diagnosis and death – usually between 15-20 years, but this does vary between individuals
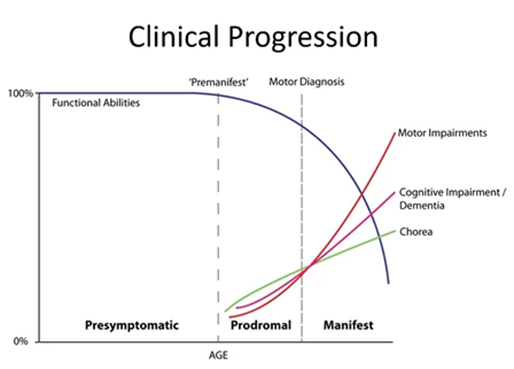
Clinical progression
In every neurodegenerative disease (apart from stroke) we don’t know when the pathology starts
The brain is not something that we can sample easily – reliant on the point which the individual experiences symptoms which is severe enough to consult a clinician
Each aspect of symptoms has a different trajectory
At some point you cross the threshold which this can be clinically diagnosed but all throughout this time the disease has been progressing
Neuron loss of function will preceed neuron cell death but this could be years due to the body’s ability to buffer and won’t manifest into clinical dysfunction
However, from a treatment point of view, this is challenging because when you have crossed the threshold which has allowed you to diagnose it, there’s little you can do to reverse the damage
Huntingtin gene can impact cognitive function, but they can be compensated so they are not noticed by the family.
Symptoms
Occur across 3 main domains: movement, Behaviour and cognitive
Movement and behaviour domains are related to physical symptoms while cognitive symptoms related to planning and thinking
Movement
Main change is the onset of involuntary movement
Usually the first onset
Distinct from other symptoms
Behaviour
Range of behavioural change: apathy, depression, changes in personality, aggression
May be the most significant domain but this can be hard to diagnose from a clinicians perspective
Can really affect family members and those around them
Cognitive
Difficulties with planning and thinking
Can impact day to day life most —> E.g: lose independence
Physical symptoms
Can be affectation to voluntary movements
Motor deficit can include:
jerky or fidgety motor movements
Clumsiness
abnormal eye contact
speech becoming slurred
Weightloss
Incontinence
Cognitive symptoms
Lots of overlap between Alzheimers and parkinsons
Memory and concentration problems
Aggression, demanding, stubborn
Impulsive behaviour
Social isolation
Suicide related mortality
Lack of motivation
Reduced ability to read facial expressions
Hard to plan and think ahead
Normal function of teh basal ganglia
Made up of the:
Caudate and the putamen making up the striatum
Globus pallidus
Usually have the cortex sending Glu (excitatory) projections to the putamen which is inhibitory
Putamen neurons are GABAergic and send inhibitory signals to the globus pallidus (more input from the cortex = more inhibition)
The globus pallidus also inhibitory nucleus and increased inhibition onto an inhibitor (GP) results in more excitation onto the thalamus
The thalamus is an excitatory nucleus which excited the motor cortex and relays directly onto muscles through the spinal chord.
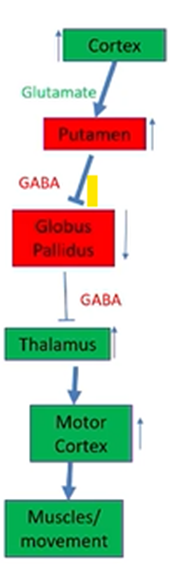
In Huntington’s disease
If there’s degeneration in the putamen then there will be less inhibition, due to decreased neurons to project to the GP
Less inhibition coming into the GP will result in more inhibition coming out of the GP onto the thalamus
Motor cortex will be depressed, muscle movements will be depressed
Not every neuron affected equally
Main neurons affected are medium spiny neurons
95% of the neurons
In contrast with the aspiny neurons which are different morphologically:
Slightly smaller
Spiny neurons are GABAergic
Neuropathology
Can begin to detect with MRI
Enlargement of lateral ventricles
Loss of cortical striatal projection neurons
Striatal atrophy
Severe changes in matter extending to other areas of the cortex, cerebellum, hippocampus in severe cases
Despite the fact that the primary site of dysfunction is the basal ganglia, this will have a knock on effect because the lack of input to the connecting neurons will also result in loss of function and death
Molecular changes
Expansion in the N terminal section of the protein
Normal huntingtin protein will go through normal physiological expression and proteostatsis —> when the protein is not needed to it will be degraded
Poly Q fragments leads to misfolding of protein, leading to intracellular aggregates which disrupts proteostasis
Either:
Can’t be recognised
Can’t be digested and disposed of
These proteins function in many cellular pathways but main part of the pathology is the formation of these large aggregates which molecular mechanisms are ot designed to deal with
The misfolding pathway to explain neurotoxicity
The dysfunctional system can lead to the pathology in different ways:
The disease that is caused by loss of function = the mutant protein no longer able to perform normal function. This occurs in the context of a mix of normal and mutant protein (e.g: sequestering proteins into aggregates away from where it acts, modifying protein interaction making them weaker so binding is lost)
Disease caused by gain of function = extra activity imparted by the mutant protein (expanded polyQ creates protein conformers which are toxic and create new activity/interactions)
So we have lost the normal functioning Huntingtin protein and as a result of this loss of function, it has been replaced by a new protein which has gained new functions.
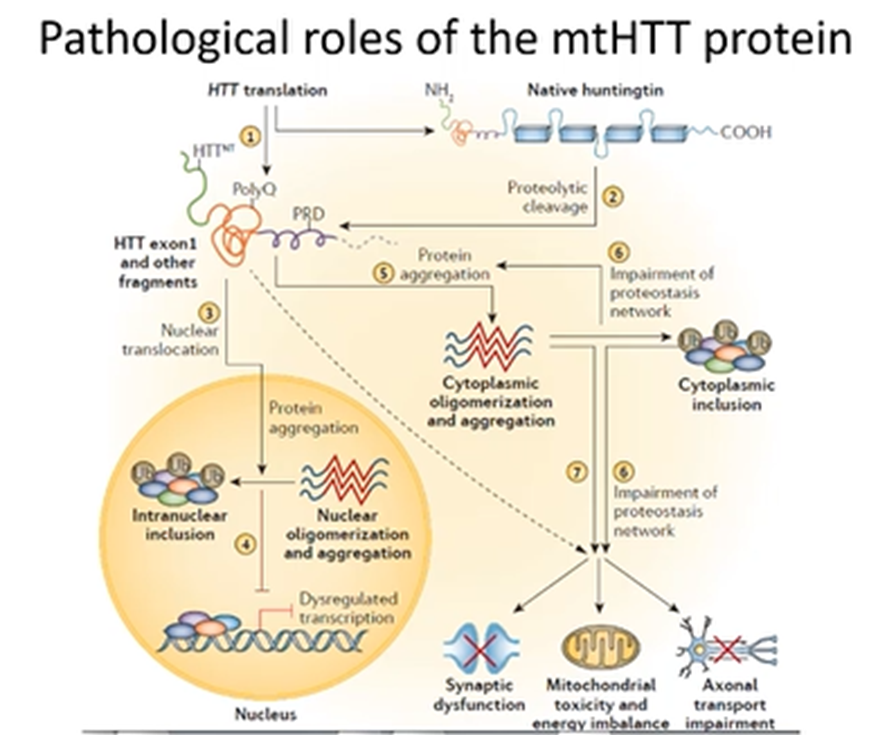
Pathological roles of the mtHTT protein
Wt Huntingtin will be cleaved into protein fragments which can be degraded
New protein will aggregate and also have new properties like:
Translocation to the nucleus where it functions in a way the wt did not
Impact the mitochondrial mechanisms
Generation of toxic protein inclusions which impairs the function of the neuron
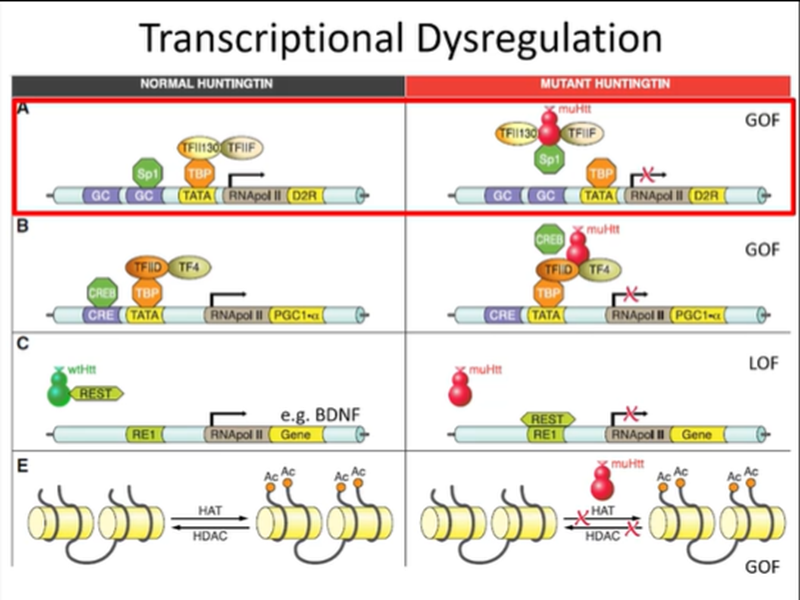
mtHTT impairment of transcription
Impairs 75% of transcription via inhibition of expression of pol2
Could be directly by inhibiting TFs that control expression of pol 2 or could be via histone modifications
A lot of the promoter, transcriptional regulation of the expression of the polymerase is affected by mtHTT which can travel to the nucleus and impact transcription
Usually the TFII130 TBP complex can bind to promoter sections but the binding of mtHTT to these proteins will impair their activation so the polymerase will not be expressed.
It can also bind to other complexes
wtHTT is known to bind to REST which is an inhibitor of transcription of some genes like BDNF and RNA pol2 —> however, the mutated protein cannot complete this function and results in the supressed expression of these genes
Also more generally affecting transcription and epigenetic regulation - mutant HTT blocks DNA acetylation
However, the neuron does have some compensatory mechanisms to survive despite the significant suppression of tranciption
Effect on mt function
Covers a range of functions covered by the mt:
Neurons expressing mutant HTT will have:
increased sensititivity to Ca2+ inducing pore opening and release of CytC
Reduced membrane potential
Decreased Ca2+ buffering capacity
Increase in ROS production
By maintaining membrane potential is the basis of generating energy in the mt so lots of these greatly impact the function of the mt.
E.g: calcium is essential for molecular signaling and altered Ca2+ levele sin the cytoplasm will disrupt these pathways
Increase in ROS is toxic to the neurons
Loss of function driving pathology
Normal Huntingtin blocks procaspase 9 (inhibition)
Procaspase 9 when uninhibited becomes much more available for the engagement of the apoptitic pathway —> still requires this trigger but not as tightly controlled
Degeneration by dying back
Dying back is the concept that the first element of a neuron that is affected is the distal element (E.g synaptic contact) which is trying to buffer the pathology from reaching the cell body
When the synapse is lost there is the retraction of the axon which leads to changes in the body of the neuron that leads to the death of the neuron
Synaptic degeneration before the death of the neuron
Areas which are affected by the disease are often not those where the pathology originates due to the loss of these synapses
Conserved mechanism across lots of neurodegenerative disorders
Why do MSN neurons selectively die
Activity related specificity of MSNs:
high level of glutamate dependent activity
Selective expression of neuropeptides compared to other neurons
High metabolic rate and high firing rate
Long axon projections
These neurons particularly susceptible to suffer from this pathology
Other neurons affected
Even though the mtHTT is expressed in all neurons, there’s no selection based off the protein present
Its more to do with how the neuron can deal with the metabolic/cellular processes being affected which confers selective vulnerability
Symptomatic treatments
No approved disease modifying treatment
Involves helping the patient with the symptoms but not intervening with the disease pathology
usually targets specific dysfunctions —> E.g: manifestation of psychosis or irritability, or depression/anxiety related symptoms
Very specific for the patient
Lots of the symptoms can be reasonably managed with these treatments —> depends on what is most important to them
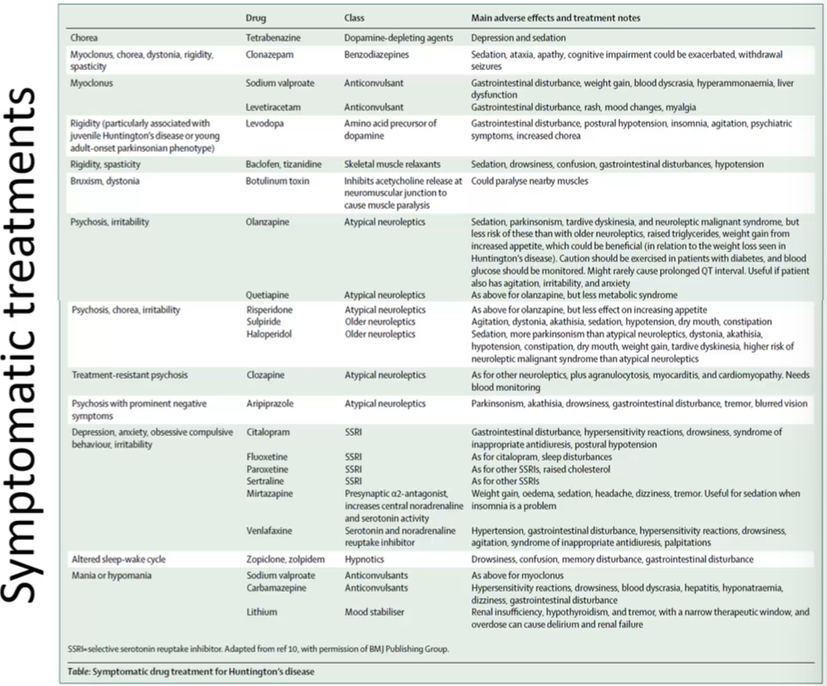
Drugs for treatment
Lots of these drugs are repurposed drugs, initially used for other things
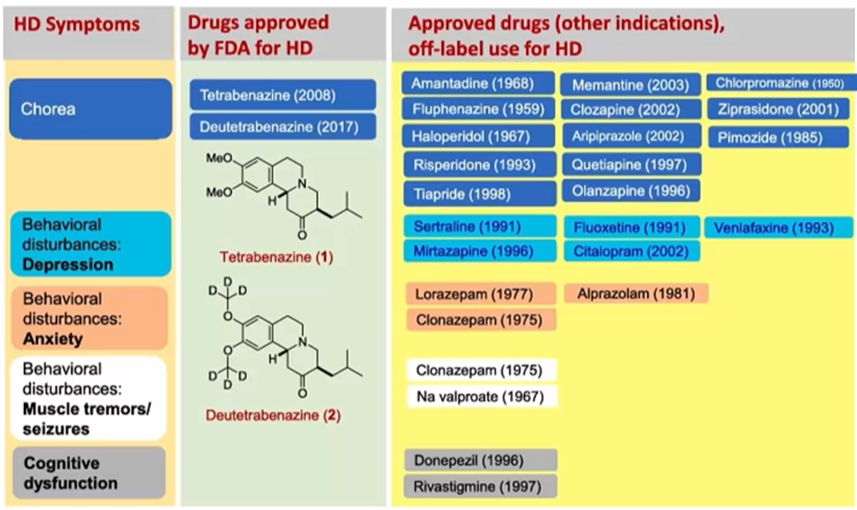
Therepeutic strategies
Try to enhance the removal of HTT
Treatments that help with inflammation (due to activation of glial cells)
Direct lowering of mutant HTT at point of expression (gene therapy)
Gene silencing
Treatment with most potential
As a patient you carry mutated form of given gene —> we know what mutation is, what the sequence is
Can design an antisense oligonucleotide which bind to the expressed mRNA and lead to degradation
Requires you to be very precise at which copy you want to target —> still want the normal allele
Want to design them in a way that leads to effective degradation
Timeline of gene silencing therapy
UK leads these trials, some of the timeline of these trials:
Gene therapy became available in the 2000s
First clinical trial in 2017 —> demonstrated that in a small no. of patients they can reduce the expression of mtHTT
Proved to be safe, so encouraged to phase 3 trial in 2019
By 2021 had to halt the trial due to safety issues
Back to phase 1 and 2 trials in 2025 —> involves surgical delivery to the specific affected nuclei via a viral vector
Currently in submission to FDA approval
Still needs to consider whether there are specifcities within this 12 groups which make them ideal candidates. Also the issue that this is an expensive therapy to roll out for a number of patients
Potential to eliminate this within the next 3 of 4 generations