Hemostasia
1/44
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
45 Terms
Definición
Mantener la sangre en su lugar
Es el concepto contrario a la hemorragia
Qué mecanismos tenemos para la hemostasia? (para mantener la sangre?)
Espasmo vascular (clot/clotting)
Platelet plug
Coagulation pathway
Tenemos unos vasos que la mantienen pero cuando se rompen tenemos los mecanismos de coagulación sanguínea. Cuando la sangre coagule, esta se convierte en sólido. Otro mecanismo es la vasoconstricción. El cuerpo regula el flujo de sangre en las zonas en las que se está perdiendo la sangre. El sistema tiene un límite y no es muy grande. Dependiendo de la pérdida sanguínea que tenga, el sistema funcionará o no.
Espasmo vascular (clot/clotting)
Es el primer elemento que limita la cantidad de sangre que circula por un vaso dañado
Cuando nos hacemos una herida:
Hay una contracción (reflejo muscular al daño)
Esta contracción → vasoconstriñe y hace que el flujo disminuya → apoyando a que en las zonas dañadas el flujo sea menor
→ Otro ejemplo es que los electricistas antes tocaban los cables con el dorso de la mano para no quedarse enganchados al cable debido a este reflejo muscular.
EN el espasmo vascular hay:
Una respuesta miogénica al daño tisular
un reflejo nervioso al dolor
la producción de tromboxano A2 por las plaquetas
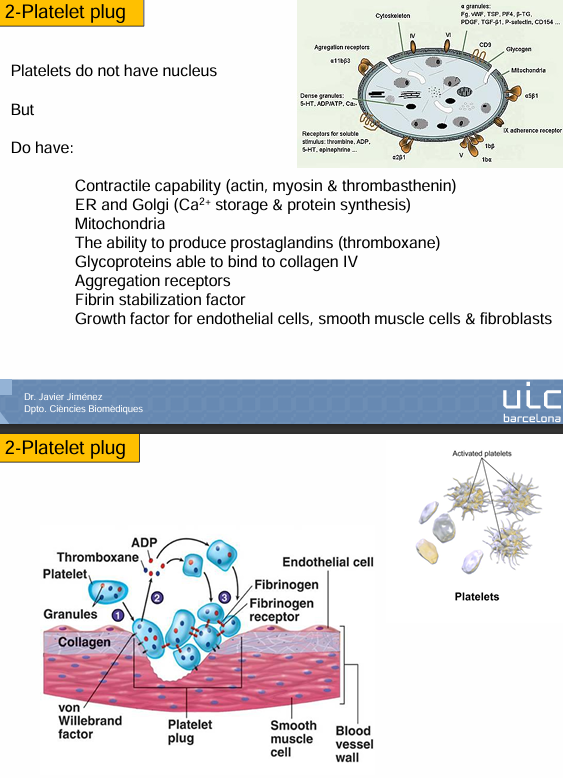
Platelet plug
Plaquetas no tienen núcleo
Son cachitos de células, fragmentos celulares que vienen de la partición de otras células.
→ vienen de:
La descamación de megacariocitos (van sacando vesículas grandes que son las plaquetas)
Tienen capacidad de cambiar su forma o no?
Tienen citoesqueleto contráctill, así que sugiere que sí
También tienen un retículo endoplasmático, mitocondrias y un aparato de Golgi (sirve para producir proteínas y de las que producen muchas van fuera) muy desarrollado
También tienen una importante capacidad de secretar cosas.
Qué son capaces de secretar?
Grandes capacidades de calcio
Asimismo tiene muchas mitocondrias para la producción de energía.
Las plaquetas también tienen la capacidad de producir prostaglandinas.
Moléculas de conexión, mediadores de inflamación, mediadores de otras respuestas mediante otras células…
Para qué nos sirven las prostaglandinas?
Nos sirven para que las plaquetas, cuando se activen, se puedan comunicar con otras (lo hacen mediante las prostaglandinas)
Qué tienen las plaquetas?
Tienen glicoproteína - estas son capaces de:
Unirse al colágeno
La sangre no tiene colágeno - hay en el endotelio vascular
sangre no tiene contacto con el colágeno
Recordemos que plaquetas tienen receptores para el colágeno
Cuando se rompa el vaso:
Las plaquetas ya tienen receptores que reconocen moléculas que están en la parte de fuera de los vasos
También tienen moléculas de agregación para formar agregación de plaquetas
También tienen elementos que hacen que la fibrina se estabilice y moléculas que ayudan a la fibrina a hacer fibras
Por último, tienen factores de crecimiento para células endoteliales, músculo liso y fibroblastos que ayudan a regenerar el tejido
Qué pasa cuando se rompe el vaso? (endotelio y epitelio vascular)
Cuándo se lesiona un vaso sanguíneo se rompen el endotelio y el epitelio
Ruptura vascular: exposición del colágeno
Dejan expuesto el colágeno subendotelial (especialmente colágeno tipo IV y tipo I)
Este colágeno es normalmente invisible para las plaquetas, pero al quedar expuesto, les sirve como señal de alarma
Adhesión plaquetaria
Plaquetas no se adhieren directamente al colágeno de forma eficiente, necesitan un “puente molecular”, que en este caso será:
el factor de Von Willerbrand (FvW)
Entonces: FvW se une al colágeno expuesto
La plaqueta expresa en receptor GPIb en su membrana
El FvW se engancha entre el colágeno y GPIb, permitiendo que la plaqueta quede firmemente adherida
Que pasaría sin FvW?
esta unión es débil o inexistente → síndrome de von Willebrand, que causa sangrados porque la hemostasia primaria falla
Activación plaquetaria
Una vez adherida, la plaqueta se activa, lo que provoca:
Liberación de gránulos - dos tipos:
Gránulos alfa: FvW, fibrinógeno, factores de crecimiento.
Gránulos densos: ADP, serotonina, calcio
Al activarse, los liberan al exterior, entonces estas:
Reclutan más plaquetas
Activan a plaquetas vacías
Favorecen la agregación
→ libera prostaglandinas:
Plaqueta activa:
La vía del ácido araquidónico
Produce tromboxano A2 (TXA2), una prostaglandina que:
Aumenta la activación plaquetaria
Induce vasoconstricción
Potencia la agregación
Cambio de forma: reorganización del citoesqueleto
La plaqueta en reposo es redonda, cuando se activa:
Reorganiza su citoesqueleto de actina y miosina
Extiende pseudópodos (prolongaciones), adoptando forma de “estrella”
Este cambio:
Aumenta la superficie de contacto.
Facilita la unión entre plaquetas.
Mejora la estabilidad del tapón.
Agregación plaquetaria: formación del tapón
Las plaquetas activadas se unen entre sí gracias a:
El receptor GPIIb/IIIa en su superficie
El fibrinógeno que actúa como puente entre dos plaquetas
Este proceso crea:
El tapón plaquetario
Retracción del coágulo: compactación de la herida
A los pocos minutos (aprox 30min)
Las plaquetas activan su maquinaria contráctil (actina + miosina)
El tapón se contrae, tirando de los borden de la herida
Esto compacta el coágulo y reduce el tamaño del defecto vascular.
Esta retracción es esencial para estabilizar la lesión antes de que la coagulación secundaria forme la malla de fibrina definitiva
Qué provoca la deficiencia de FvW?
Problemas de coagulación
es necesario para la interacción entre el colágeno y las plaquetas.
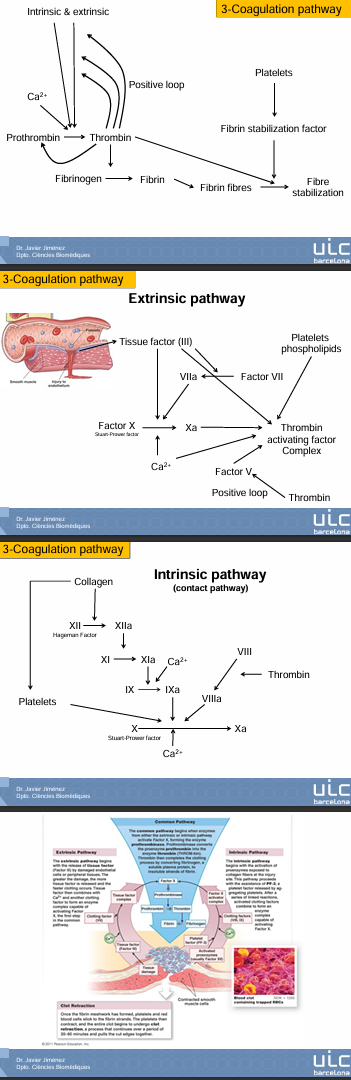
Rutas de coagulación
Qué pasa con la fibrina cuando se activa?
Es capaz de formar fibras cuando se activa
Qué haran estas fibras?
SOn las que formarán la malla donde sustentarán las plaquetas (activadas por las rutas de coagulación)
Qué es un zimógeno?
Una enzima que no se activa por un activador o inhibidor
Se activa porque se corta (corte proteico)
Cuando está entera está inactiva
Fibrina
Está en la sangre entera → es decir, se encuentra inactiva porque es zimógeno
Proteasa la corta y la activa
→ polimeriza y forma fibras
Cuál es la proteasa que se encarga de cortar el fibrinógeno?
La trombina
→ la trombina es también un zimógeno
(pasa de trombinógeno/protrombina a trombina)
La trombina se retroalimenta positivamente (vías intrínsecas y extrínsecas)
Tiempo de sangrado es limitado, por tanto, una vez se activa el sistema, interesa que este vaya rápido
Una vez que tengo trombina se dispara a toda velocidad la vía.
Fibrinógeno: fibrina inactiva.
Trombinógeno (protrombina): trombina inactiva.
Qué pasa una vez que se forman las plaquetas?
Hay conexión entre estas dos
Una vez que se forman las fibras las plaquetas contribuyen a que la malla sea más estable.
Malla de fibrina
Atrapa plaquetas, eritrocitos y proteínas plasmáticas
Refuerza el tapón plaquetario (por sí solo sería inestable)
Evita que el coágulo se deshaga por la presión de la sangre circulante
Da estructura y solidez al coágulo para que soporte horas o días mientras el tejido se repara
Permite la retracción del coágulo, porque las plaquetas se anclan a la fibrina y tiran de ella.
En resumen: la fibrina convierte un tapón blando en un coágulo firme
Formación de la malla:
Cascada de coagulación
La trombina es la enzima clave (necesitamos formación de trombina)
Activación fibrina
La trombina corta el fibrinógeno, que es soluble, y lo convierte en fibrina, que es insoluble.
Polimerización
Las moléculas de fibrina se unen entre sí formando fibras largas, como hilos.
Estabilización por el factor XIII
Cuando se activa (también por la trombina)
Entrecruza las fibras de fibrina
Las hace más resistentes
Forma una malla densa y estable
Cómo se produce la retracción del coágulo?
La fibrina se une con las plaquetas
Cómo?
Receptores de plaquetas se unen a la fibrina
Cuando fibrina se deposita al rededor del tapón plaquetario, plaquetas se anclan a las fibras y el citoesqueleto de actina-miosina de las plaquetas tira de la fibrina, produciendo retracción del coágulo (reduce tamaño de la herida
La fibrina no solo refuerza: permite que las plaquetas ejerzan fuerza mecánica sobre la herida.
Diferencias entre plaquetas y malla de fibrina
Tapón:
Rápido
Reversible
Poco resistente
Dependiente de la agregación
Malla de fibrina:
Irreversible
Muy resistente a la presión sanguínea
Estable durante horas o días
Capaz de integrar células y proteínas
Base para la cicatrización (sirve de andamiaje para fibroblastos)
La ruta de coagulación sin plaquetas no funciona, están perfectamente interrelacionadas, se necesitan la una a la otra
Las rutas
Hay dos rutas moleculares diferentes → Ruta extrinseca e intrinseca. Son rutas que confluyen en generar trombina. Se activan de dos maneras distintas y tienen una única salida que es generar la fibrina.
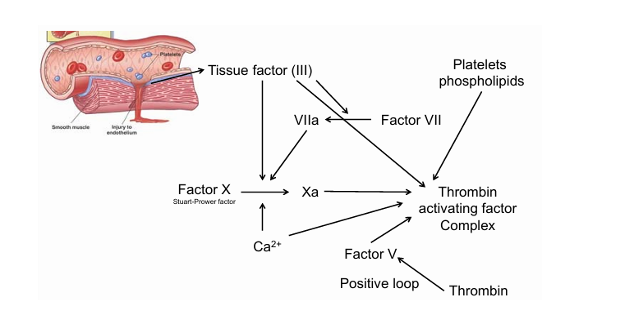
Ruta extrínseca
Cuándo se activa?
Cuando se rompe un vaso
Cuando la sangre entra en contacto con los tejidos
No solo se daña el endotelio, sino que se rompen células del tejido subyacente
Qué contienen estas células?
Factor Tisular (FT) o Factor III
No está en la sangre (Por eso se llama “extrínseca”: el factor que la activa viene de fuera del torrente sanguíneo)
Activan esta ruta
Qué es el Factor III (FT)?
Es una glicoproteína membrana con naturaleza lipídica
Está en:
La membrana de células extravasculares
No circula en la sangre en condiciones normales
Función fisiológica:
Actuar como receptor y cofactor para activar la coagulación
En clase a veces se simplifica diciendo que “no se sabe molecularmente qué es”, pero en realidad sí se conoce: es una proteína llamada tromboplastina tisular.
Qué pasa cuando FT entra en contacto con la sangre?
Se une al factor VII (que circula de forma inactiva)
Complejo FT + VII se activa a FT-VIIa
Este necesita calcio (factor IV) para funcionar
El complejo FT-VIIa-Ca2+ es una potente proteasa que activa directamente el factor X (lo corta)
Este es el paso clave:
FT–VIIa–Ca²⁺ → activa Factor X → Xa
Según los apuntes, el factor 3 corta el factor X, de forma directa o indirecta, activándolo, y este activo corta otros hasta llegar a la trombina
Molecularmente la vía es muy corta y se activa en cuestión de pocos segundos
Una vez que el factor 3 contacta con la sangre se activa la ruta en cuestión de segundos.
Por qué se dice que la vía extrínseca es muy corta?
Porque solo necesita:
FT
Factor VII
Calcio
Factor X
Y en cuanto el Factor X se activa, ya entra en la vía común:
X → Xa
Xa convierte protrombina en trombina
La trombina convierte fibrinógeno en fibrina
Cuanto más daño tisular → más FT liberado → más rápida y potente la coagulación.
Ruta intrínseca
Activada por:
EL contacto con superficies cargadas
Principalmente por la acción de las plaquetas y el colágeno, interacción de células sanguíneas en superficies extrañas.
Se activa cuando:
La sangre toca
Colágeno subendotelial
Superficies cargadas negativamente
Vidrio (por eso se activa en un tubo de ensayo)
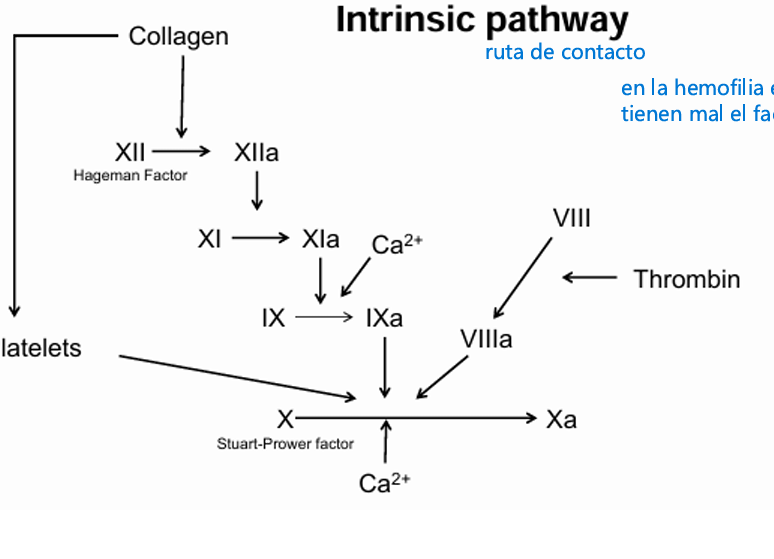
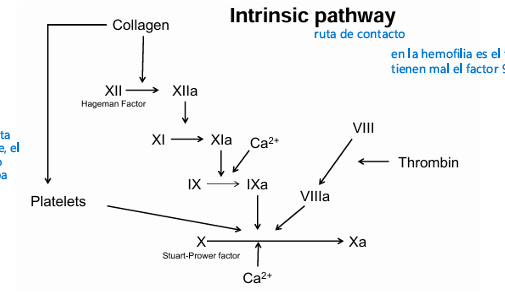
Protagonista → Factor XII (Factor de Hageman)
Qué hace el fator XII?
Cuando la sangre toca colágeno o vidrio:
Factor XII → XIIa (se activa por contacto)
XIIa activa al Factor XI → XIa
XIa activa al Factor IX → IXa
IXa se une al Factor VIIIa (cofactor)
El complejo IXa + VIIIa + Ca²⁺ + fosfolípidos activa al Factor X → Xa
Conclusión:
La vía intrínseca también llega al Factor X, igual que la extrínseca.
Papel de las plaquetas en la vía intrínseca
Qué liberan las plaquetas activadas?
Liberan PF-3, que no es un factor de coagulación, sino:
Fosfolípidos de membrana cargados negativamente
Para qué sirven estos fosfolípidos?
Sirven como superficie para que los factores XII, XI, IX, y VIII formen complejos enzimáticos
PF-3 no activa al factor XII, pero facilita la formación del complejo que activa el factor X
Regla nemotécnica
Extrínseca → depende de un factor que NO está en la sangre (Factor III o FT)
Intrínseca → todos los factores sí están en la sangre
Por eso:
La extrínseca se activa cuando entra FT desde los tejidos.
La intrínseca se activa cuando la sangre toca colágeno o superficies cargadas.
Por qué un paciente sin Factor XII no coagula en un tubo, pero sí coagula cuando se corta?
En un tubo de ensayo (vidrio)
No hay Factor Tisular (III) → no se activa la vía extrínseca.
La vía intrínseca depende del Factor XII.
Si falta XII → no se activa nada → la sangre NO coagula.
En el cuerpo (cuando se corta)
Se libera Factor Tisular (III) desde los tejidos.
La vía extrínseca funciona perfectamente.
La vía extrínseca es tan potente que compensa totalmente la falta de XII.
👉 Por eso los pacientes con deficiencia de Factor XII NO sangran, pero sí tienen tiempos de coagulación alterados en laboratorio (TTPa muy prolongado).
Hemofilia y la vía intrínseca
Hemofilia A → falta Factor VIII
Hemofilia B → falta Factor IX
Ambos pertenecen a la vía intrínseca, por eso:
El TTPa está prolongado
El TP es normal
Sí producen sangrados graves, porque estos factores son esenciales para activar al Factor X en el cuerpo.
A diferencia del Factor XII, cuya ausencia no causa sangrado, la falta de VIII o IX sí es clínicamente grave.
Resumen
Vía intrínseca (contacto)
Se activa por colágeno o superficies cargadas.
Factor XII → XI → IX + VIII → X.
Requiere PF-3 (fosfolípidos plaquetarios).
Todos los factores están en la sangre.
Importante en laboratorio (TTPa).
Vía extrínseca (tissue factor)
Se activa por Factor III (no está en la sangre).
FT + VIIa + Ca²⁺ → activa X.
Es la vía rápida.
Importante en el cuerpo (TP).
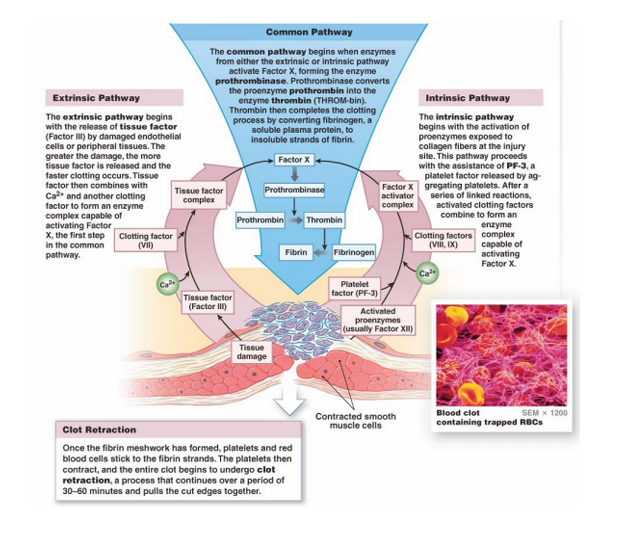
Esquema de las dos rutas:
En la parte común de las dos vías, la activación del factor X acaba formando la protrombinasa, encargada de convertir protrombina en trombina.
Entonces la trombina completa la coagulación convirtiendo fibrinógeno (soluble) en fibrina (insoluble).
Después de coagular…
Una vez que el coágulo está formado y la herida está sellada:
La coagulación debe detenerse para que no siga creciendo
Se activa el sistema anticoagulante
Se activa la fibrinólisis, que disuelve fibrina sobrante
Se activa el plasminógeno → plasmina.
La plasmina corta la fibrina y disuelve la malla.
Coagular es vital, pero descoagular también
El cuerpo necesita:
Coagular rápido cuando hay una herida → para no desangrarse
Desactivar la coagulación cuando ya no hace falta → para no formar trombos
Coagular demasiado poco = hemorragia
Coagular demasiado = trombosis
El cuerpo usa el mismo sistema que lo activa para desactivarlo.
Ejemplos:
La trombina activa la coagulación, pero también activa proteínas anticoagulantes.
El Factor XII inicia la vía intrínseca, pero también activa mecanismos que limitan la coagulación.
Las plaquetas activan la coagulación, pero también liberan sustancias que frenan la activación excesiva.
Es un sistema de retroalimentación → lo que enciende, también ayuda a apagar.
Trombina
Es la enzima clave de la coagulación
Hace dos cosas opuestas pero complementarias:
Activa la coagulación
Fibrinógeno → fibrina
Activa factores V, VIII, XI
Activa plaquetas
Estabiliza el coágulo
Activa la fibrinólisis
Convierte plasminógeno → plasmina (directa o indirectamente)
La plasmina corta la fibrina y disuelve el coágulo
La misma enzima que crea fibrina también activa la enzima que la destruye.
Por qué hace falta este equilibrio?
Porque si solo produjéramos fibrina:
El coágulo crecería sin parar
Podría ocluir el vaso
• Se formaría un trombo
Y si solo destruyéramos fibrina:
El coágulo se rompería
Habría hemorragia
Por eso el cuerpo mantiene un equilibrio dinámico:
Al principio domina la formación de fibrina
Después domina la destrucción de fibrina
Si la trombina funciona las dos vías se realizarán correctamente.
Las dos vías están activadas a la vez pero al principio prevalece la producción de fibrina. Una vez el fibrinógeno se reduce, se prioriza la otra vía.
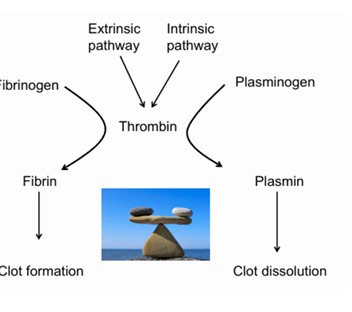
Cómo se hace para regular la coagulación?
Cómo se evita que la coagulación se active cuando no toca?
El cuerpo tiene "2 “candados” principales:
La vía intrínseca sólo se activa si hay colágeno expuesto
El colágeno está debajo del endotelio
Si el endotelio está intacto, no hay colágeno accesible
Por tanto:
No se activa el Factor XII
No se activan las plaquetas
No se inicia la vía intrínseca
Sin daño endotelial, la sangre no coagula.
La vía extrínseca sólo se activa si aparece FT (F.III)
El factor III está dentro de las células, no en la sangre
Sólo aparece cuando las células se rompen
Si no hay rotura celular → no hay Factor III → no se activa la vía extrínseca.
Sin daño tisular, la vía extrínseca no se enciende.
Por qué la fibrina que circula en sangre no se activa?
Porque la trombina queda atrapada dentro del coágulo
Cuando se forma un coágulo:
Las plaquetas se agregan.
La fibrina se deposita encima.
Se forman capas concéntricas.
La trombina queda encerrada en el núcleo del coágulo.
Entonces:
Muchísima trombina dentro del coágulo
Poca trombina en la periferia
Casi ninguna trombina en la sangre circulante
→ si no hay trombina fuera del coágulo, el fibrinógeno que circula no puede convertirse en fibrina
Si la trombina escapara al torrente sanguíneo, activaría fibrinógeno en todas partes
Se formaría fibrina en la sangre
Se producirían trombos masivos
Por eso la coagulación se limita al microambiente de la herida.
se genera un gradiente que cuando más lejos de la heridas estemos menos fibrina habrá porque menos trombina estará accesible. Entonces la fibrina se acaba en el microambiente de la herida, no en la sangre.
Mecanismos adicionales que frenan la coagulación
Thrombomodulin
Proteina C + Proteína S
Fibrina secuestra trombina
Antitrombina III
Sistema plasminógeno → plasmina
Thrombomodulin (en el endotelio sano)
Se une a la trombina
La trombina ya unida ya no puede coagular
→ Activa:
Proteína C
Proteína C + Proteína S
Proteína C =
Thrombomodulin + trombina
La proteína C activada (PCa) destruye;
Factor VIIIa
Factor Va
Recordemos que Factor IXa activa el factor VIIIa
VIIIa + IXa + Ca + fosfolípidos → activan protrombina
Factor Va → activa el factor Xa
Juntos forman el complejo protrombinasa
Va + Xa + Ca + fosfolípidos → activan protrombina
Esto frena la amplificación de la coagulación.
Fibrina secuestra trombina
La fibrina atrapa 85–90% de la trombina producida.
Esto evita que la trombina se difunda por la sangre.
Antitrombina III
Inactiva trombina, Xa, IXa, XIa, XIIa.
Es el “apagafuegos” principal.
Heparina (natural, en baja concentración)
No inhibe directamente.
Aumenta muchísimo la actividad de la antitrombina III.
Por eso la heparina es un anticoagulante tan potente.
Sistema plasminógeno → plasmina
El endotelio libera tPA (tissue plasminogen activator)
tPA convierte plasminógeno → plasmina
La plasmina corta la fibrina y disuelve el coágulo
→ esto es la fibrinólisis fisiológica
Heparina
A través de la antitrombina inhibe la trombina
Efecto anticoagulante
Defectos de la coagulación sanguínea
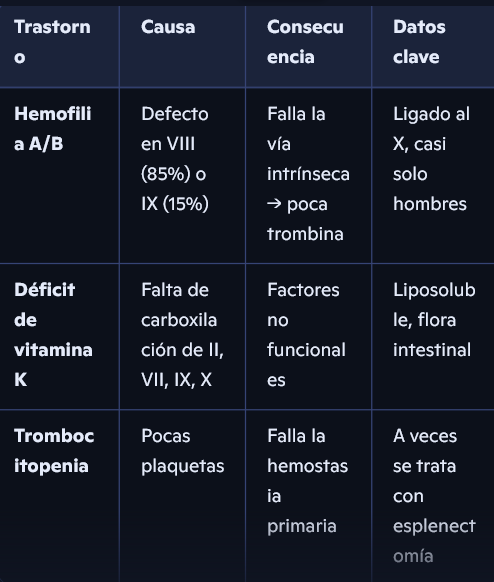
Hemofilia
Deficiencia de vitamina K
Trombocitopenia
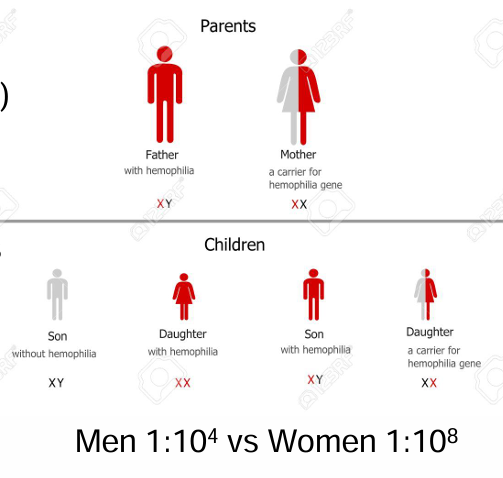
Hemofilia
Defecto de la coagulación debido a:
la falta de un factor esencial de:
La vía intrínseca
Factores afectados:
Hemofilia A
Defecto en el factor VIII
Hemofilia B
Defecto en el factor IX (menos común)
Ambos factores forman parte del complejo tenasa intrínseca, que activa al Factor X.
Si este complejo falla, la producción de trombina es insuficiente → no se forma fibrina suficiente.
Por qué estos defectos permiten sobrevivir al desarrollo embrionario?
Porque:
Los factores VIII y IX no son esenciales para la vida embrionaria.
La vía extrínseca (Factor III + VII) es suficiente para mantener la hemostasia básica durante el desarrollo.
Pero cuando la persona nace y sufre heridas, golpes o microtraumas:
La vía extrínseca no basta
La vía intrínseca no funciona
→ hemorragias prolongadas
Recordemos que afecta al cromosoma X y que por eso no hay casi mujeres hemofílicas
Deficiencia de vitamina K
La vitamina K es esencial para que el hígado produzca varios factores de coagulación:
II (protrombina)
VII
IX
X
Proteína C y Proteína S (anticoagulantes)
→ todos estos necesitan la vitamina K para la γ-carboxilación, que les permite unirse al calcio y funcionar
Qué pasa cuando falta vitamina K?
Los factores se producen, pero no funcionan
La coagulación se vuelve muy lenta o ineficaz
Aparecen hemorragias
De dónde viene la vitamina K?
De la dieta (es liposoluble → se absorbe con grasas)
De las bacterias intestinales
Por eso:
Los recién nacidos (sin flora intestinal) reciben la vitamina K al nacer
Los pacientes con antibióticos prolongados pueden tener un déficit
Los problemas de absorción de grasas también la reducen
Trombocitopenia
Qué es?
Una disminución de las plaquetas
Por qué es grave?
Porque las plaquetas son esenciales para:
Hemostasia primaria:
Adhesión
Activación
Agregación
Formación del tapón plaquetario
SI hay pocas plaquetas:
Aparecen petequias, hematomas, sangrado fácil
El tapón inicial no se forma bien
La coagulación secundaria tampoco puede estabilizar nada
Qué se hace?
Un esplenectomía
El bazo:
Destruye plaquetas
Almacena plaquetas
Puede secuestrar demasiadas en ciertas enfermedades
Si se quita el bazo
Las plaquetas duran más
Aumenta su número en sangre
Mejora la hemostasia
Resumen
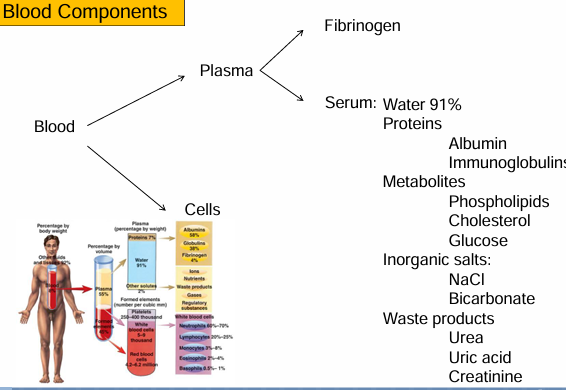
Componentes de la sangre
Plasma:
Parte líquida de la sangre sin coagular
Contiene;
Agua
Sales
Glucosa
Hormonas
Proteínas (incluyendo fibrinógeno)
Factores de coagulación
Nutrientes
Productos de desecho
Para obtener plasma:
Se toma sangre con anticoagulante y se centrifuga
Arriba queda el plasma
Suero:
Es el plasma sin fibrinógeno ni factores de coagulación activos
Cuándo se obtiene?
Después de que la sangre coagule
Para obtener suero:
Se deja coagular la sangre
La fibrina forma una malla y atrapa células
Se centrifuga
El líquido que queda arriba es suero
Convertir plasma en suero:
Plasma aún tiene fibrinógeno
Podemos convertirlo en suero añadiendo trombina
La trombina convierte fibrinógeno en fibrina
LA fibrina forma una red
Se centrifuga
La fibrina queda abajo
El líquido resultante es el suero
Cómo evitar que la sangre coagule en un tubo de ensayo?
Se añade anticoagulante
El más usado en laboratorio es:
EDTA (ácido etilendiaminotetraacético)
Es un secuestrador de calcio
El calcio es esencial para casi todos los pasos de la coagulación
Si no hay calcio → no hay coagulación
Por eso la sangre con EDTA no coagula nunca.
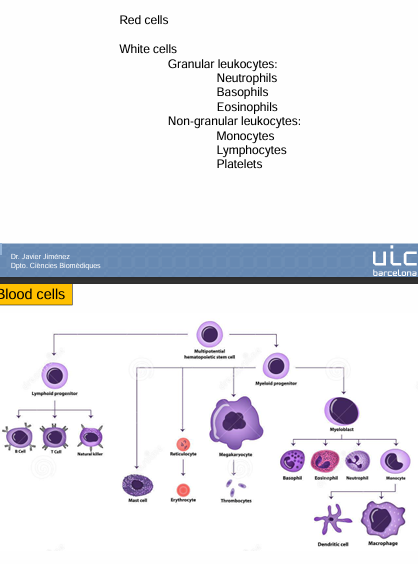
Clasificación histórica de las células sanguíneas
Agranulocitos vs granulocitos
Granulocitos:
Tienen gránulos visibles al microscopio
Neutrófilos
Se tiñen con colorantes neutros
Son los más abundantes
Eosinófilos
Se tiñen con colorantes ácidos (eosina → rosa)
Por eso se llaman eosinófilos
Basófilos
Se tiles con colorantes básicos (azules)
Agranulocitos:
No tienen gránulos visibles
Linfocitos
Linfocitos B
Linfocitos T (y sus subtipos)
Monocitos
Proteínas del suero: albúmina, globulinas, y gammaglobulinas
Cuando se estudió el suero, los investigadores separaron sus proteínas mediante cromatografía en papel o electroforesis.
Las proteínas se van separando mediante la capacidad que tienen de pasar el papel (capilaridad).
Las proteínas se separan según:
tamaño
carga
forma
movilidad
Albúmina
Es la proteína más abundante del suero.
Funciones:
Mantiene la presión osmótica
Evita que salga agua de los vasos
Por eso se usa un shock hipovolémico
Transportador inespecífico
Hormonas, fármacos, ácidos grasos, bilirrubina, iones.
Reserva de aminoácidos
Muy pegajosa → se une a casi todo (por eso se dice que es transportador inespecífico de casi todo)
Globulinas
Todo lo que no es albúmina se llamó globulina.
Son proteínas globulares, muy diversas.
Se dividen en
α-globulinas (alfa)
Producidas por el hígado
Muchas son proteínas de fase aguda
Transportadores específicos
β-globulinas (beta)
También producidas por el hígado
Transportadores específicos como:
Transferrina (hierro)
β-lipoproteínas (lípidos)
Se diferencian de las alfa por su origen genético ya que se sintetizan a partir de otra zona del gen, y movilidad en electroforesis.
→ Las globulinas son el resto de proteínas que no son albúmina. Una globulina es una proteína con forma claramente definida, globular.
Gammaglobulinas
→ sinónimo de:
Anticuerpos
Son las inmunoglobulinas
Producidas por linfocitos B y células plasmáticas
Son esenciales para la inmunidad humoral