5. Epiģenētika, Imprintings - Silver-Russell sindroms, ...
1/79
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
80 Terms
Epiģenētika - ko tā pēta?
Epiģenētika pēta gēnu ekspresijas iedzimtību, kura nav saistīta ar DNS primārās struktūras izmaiņām.
Epiģenētiskā regulācija izmanto:
»DNS metilēšanu;
»kovalento histonu modifikāciju;
»nekodējošās RNS.
DNS metilēšana
= dinamisks process, kas norisinās daudzšūnu organismu visas dzīves laikā un nodrošina normālu gēnu ekspresiju
nodrošina:
»X-hromosomas inaktivācija sievietēs;
»svešu ģenētisko elementu inaktivācija.
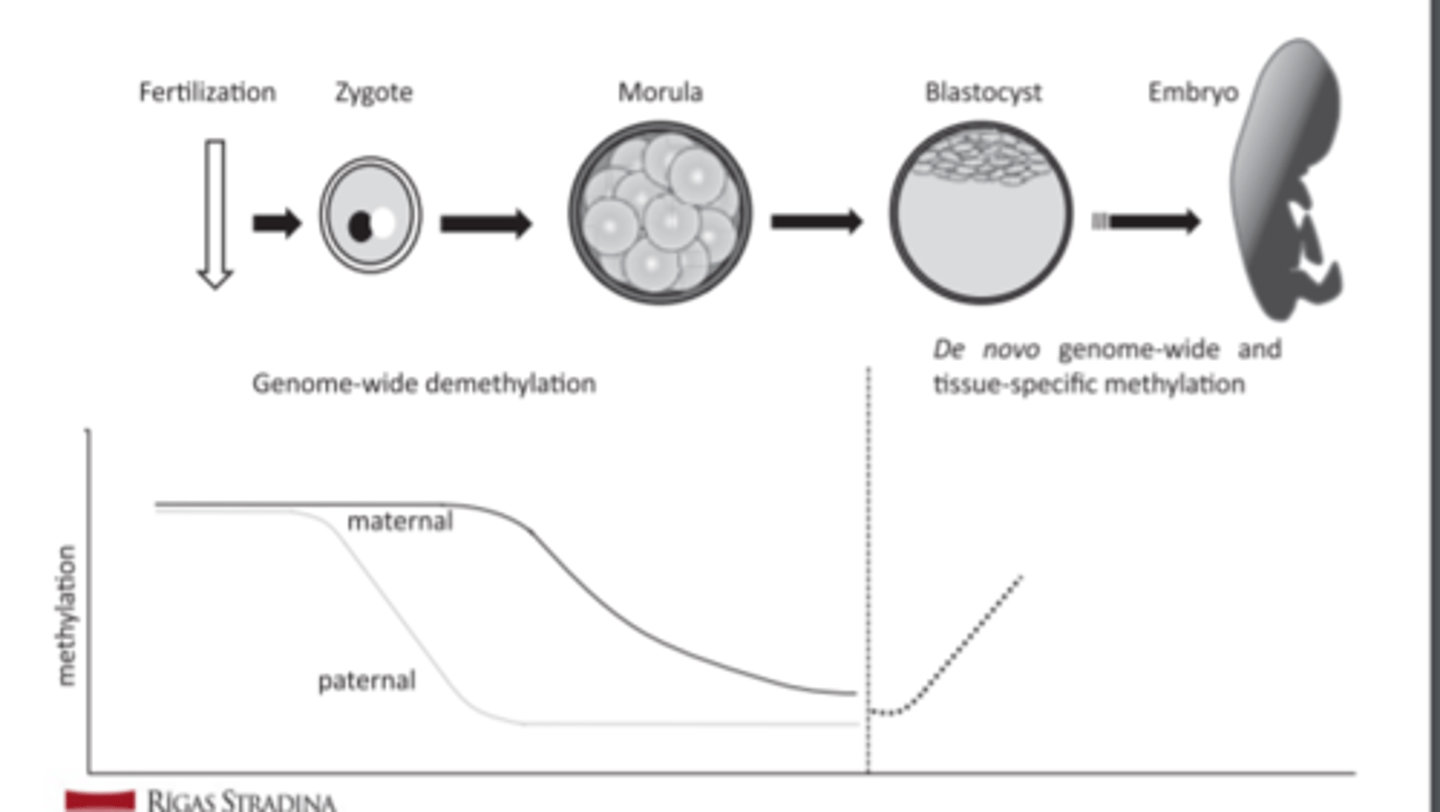
DNS metilācija embrionālās attīstības laikā
- maternālā un paternālā (pamatā) metilēšana tiek dzēsta
- sākot ar blastocistas stadiju, tiek sākta genoma de novo metilēšana
- tad arī audu specifiskā metilēšana
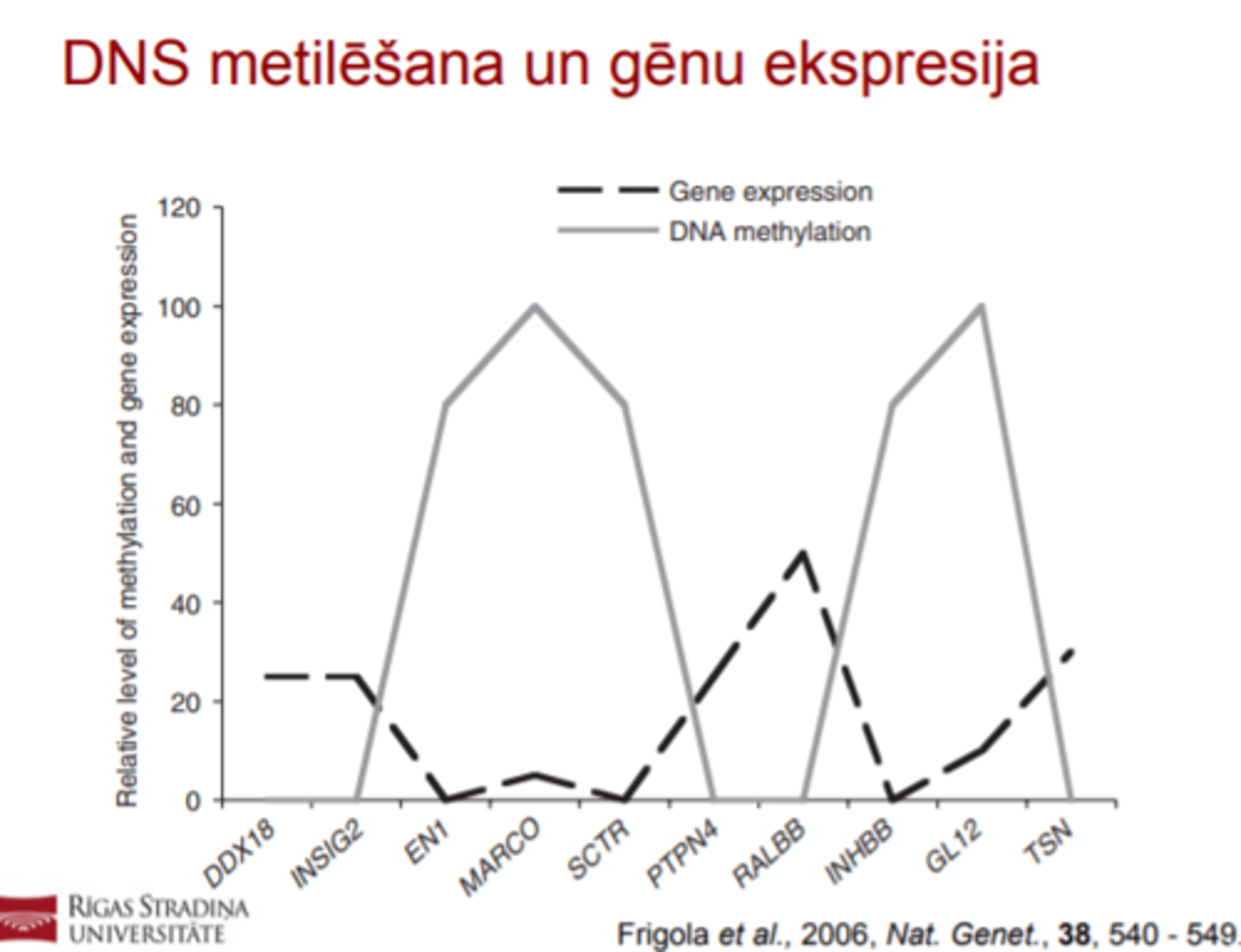
DNS metilācija
- Cik % no cilvēka genoma ir metilēts?
- Kur parasti notiek metilācija?
! Apmēram 1% cilvēka genoma ir metilēts, parasti tiek
metilēti reģioni ar lielu CG sastopamību, tā sauktās
CpG saliņas.
! Lielākā daļa CpG saliņu atrodas promotora reģionos.
! Metilēšanai ir liela nozīme šūnu diferencēšanā un audu
specifiskā gēna ekspresijā.
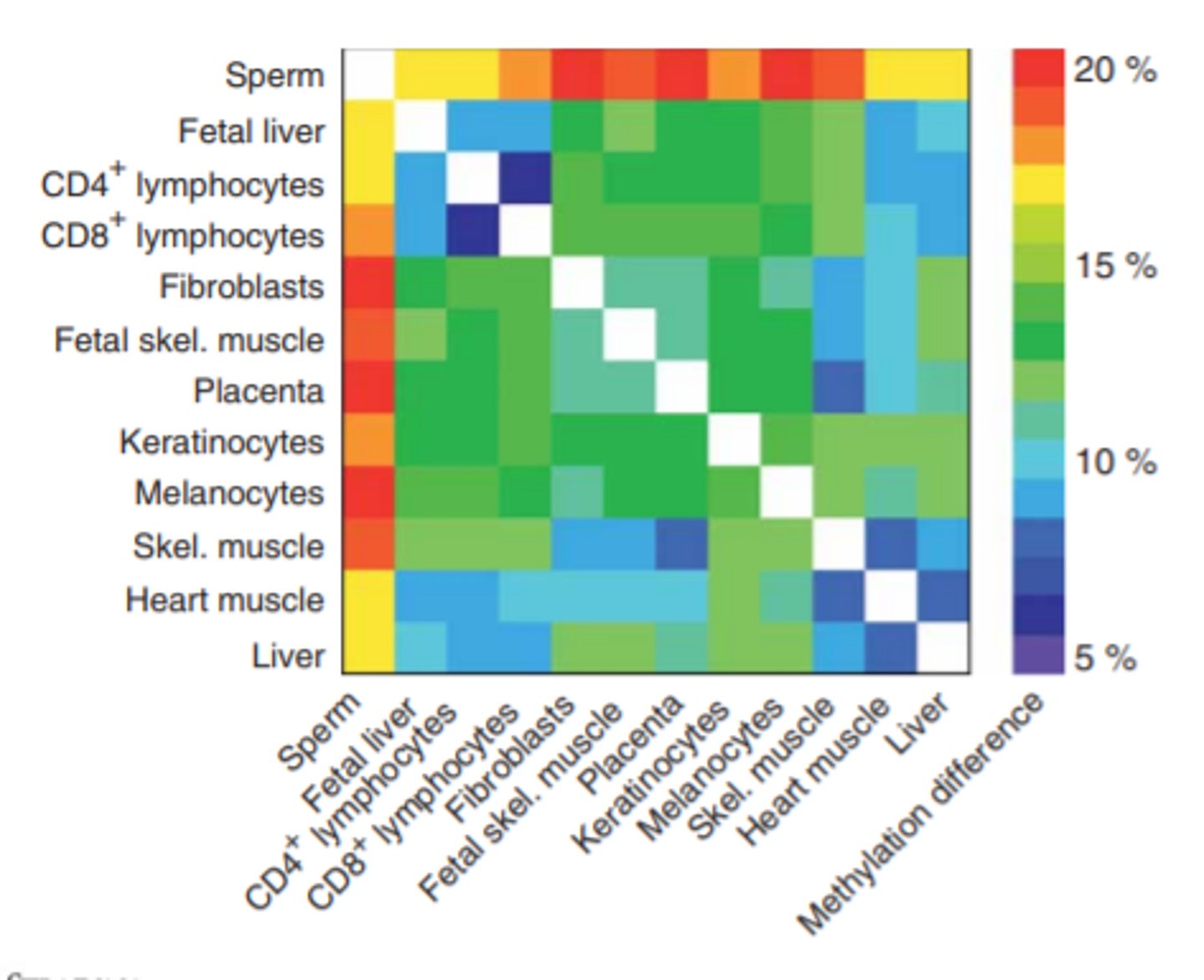
DNS metilēšana dažāda tipa šūnās
jo radniecīgāki audi, jo mazāk metilēšanas atšķirību
Atšķirības metilēšanā - Kādos audos notiek lielākas metilēšanas izmaiņas, cilvēkam novecojot?
tajos audos, kuri ir pakļauti lielākai mijiedarbībai ar ārējo vidi (piem. plaušas, resnā zarna)
Cilvēka patoloģijas, kuras izraisa izmaiņas metilēšanā vai hromatīna struktūrā (epiģenētiskās izmaiņas)
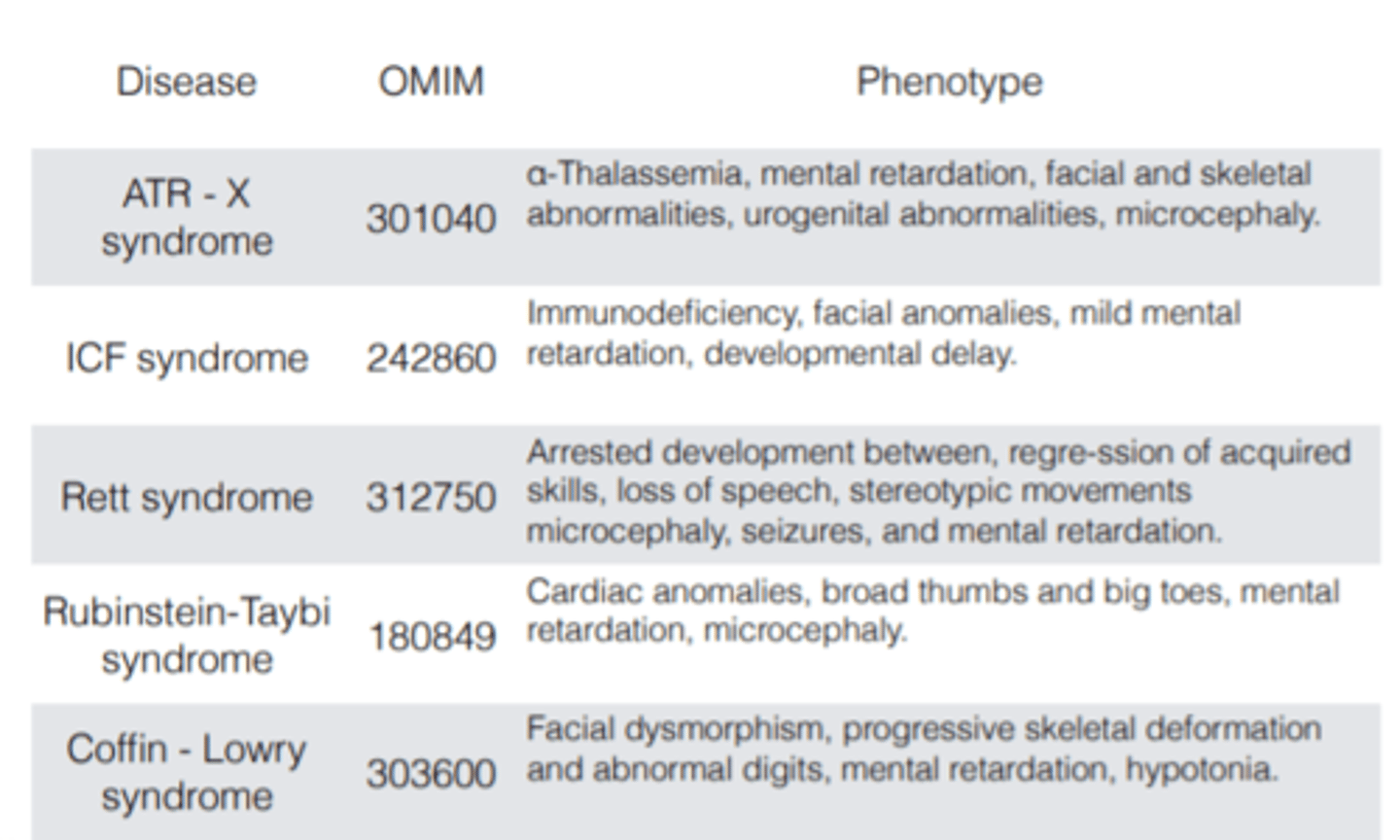
ICF sindroms (kā izpaužas?)
! Imūndeficīts, centromēru nestabilitāte un sejas
dismorfisms (Immunodeficiency, Centromeric
instability, and Facial dysmorphy, ICF) ir reta
autosomāli recesīva patoloģija.
! Pacientiem ir atkārtotas respiratoras un/vai
gastrointestinālas infekcijas, sejas dismorfisms un
dažādas pakāpes garīgā atpalicība.
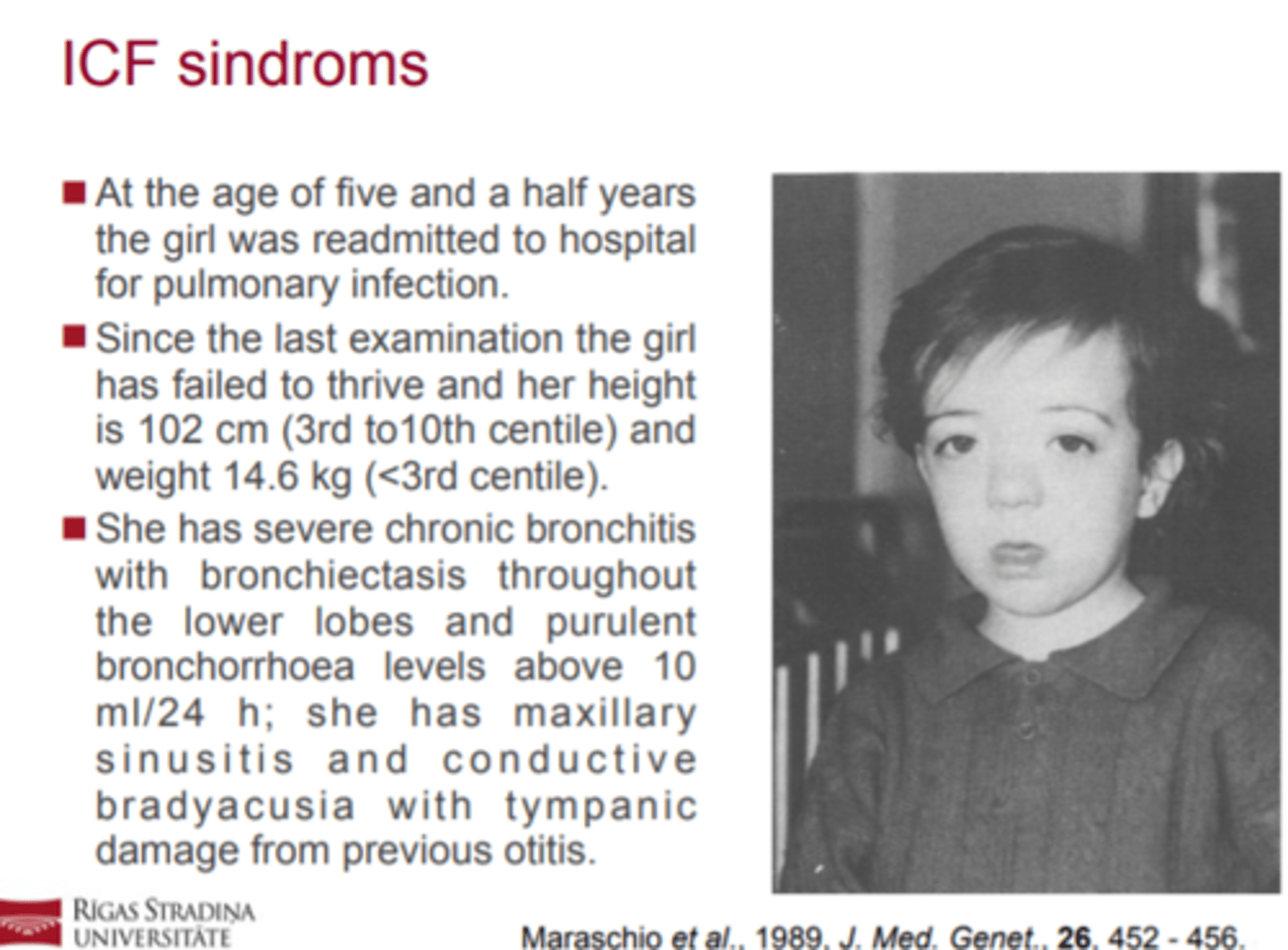
ICF sindroms (kāds alēl. variants? + kādas izmaiņas heterohromatīnā?)
Vairums gadījumos ICF sindromu izraisa mutācijas DNMT3B gēnā, kas kodē DNS metiltransferāzi.
(tā metiltransferāze metilē promotera rajonus)
ICF pacientiem 1., 9. un 16. hromosomu heterohromatīns ir hipometilēts
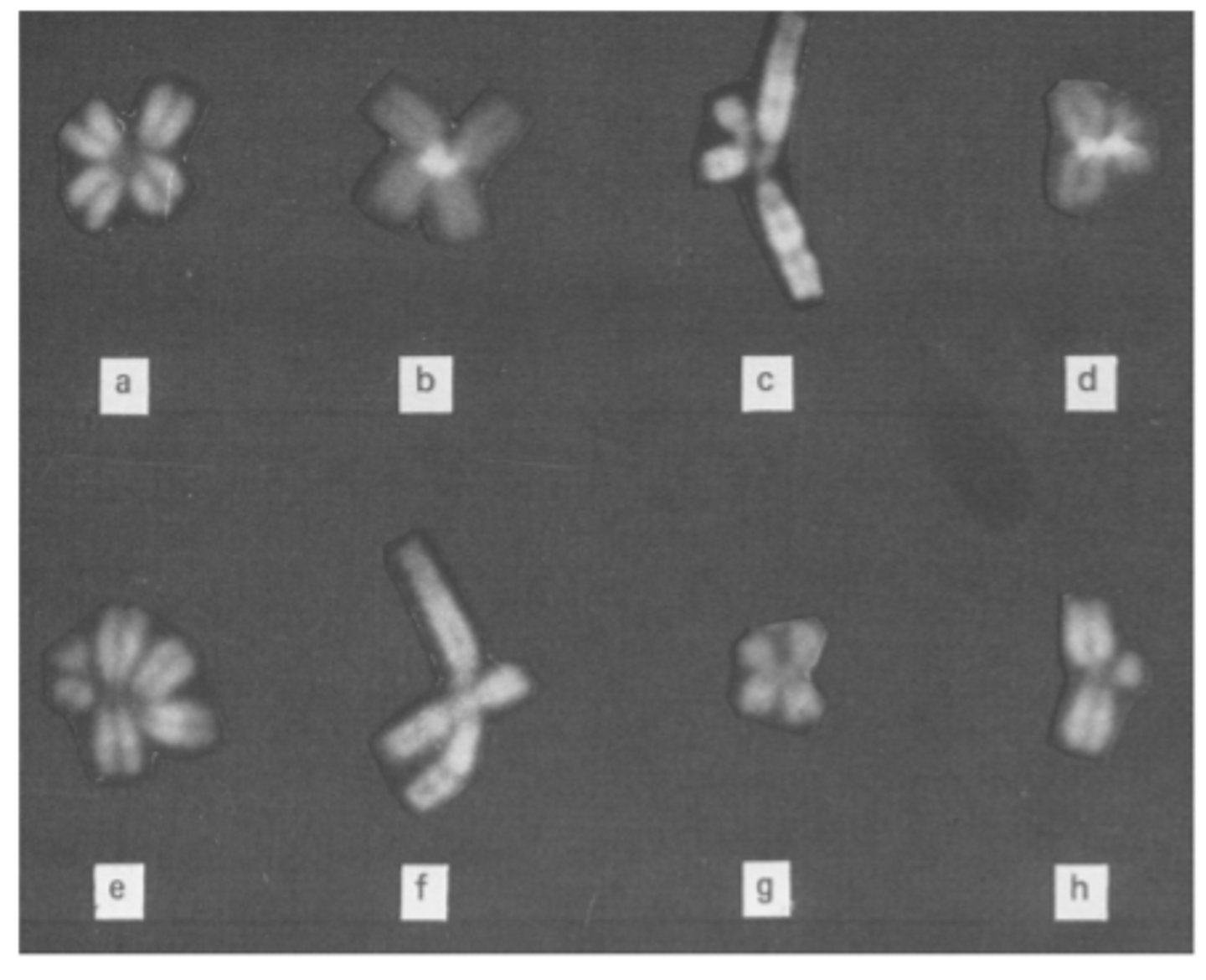
ICF sindroms - hromosomu izskats
! Hromosomu centromēru veidotās asociācijas: 1/1(a,b),1/16(c, d),1/1/16(e), 1/9(f), 16/16(g), 1/16p (h)
tās hromosomas, kuras hipometilētas, veido savdabīgas struktūras (tiek izjaukta normāla gēnu ekspresija)
Coffin-Lowry sindroms
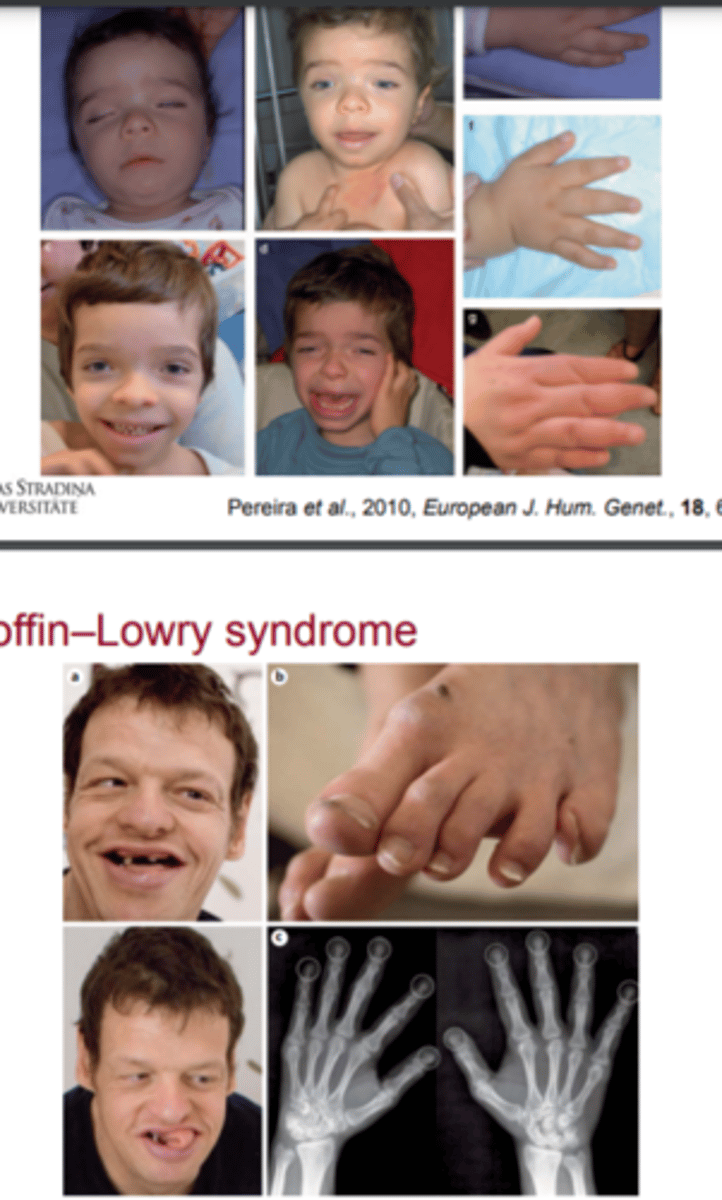
Coffin-Lowry sindroms ir X-semi-dominants sindroms, kas raksturojas ar smagu psihomotoro un augšanas atpalicību, sejas dismorfismu, pirkstu formu un skeleta izmaiņām.
Sindromam ir raksturīga variabla ekspresija
Coffin-Lowry sindroms (incidence un izpausmes heterozigotām sievietēm)
Heterozigotām sievietēm sindroms izpaužas ļoti variabli; no praktiski veselām sievietēm ar normālu attīstību un bez sejas dismorfisma līdz izteiktam sejas dismorfismam un vidēji stipru garīgo atpalicību.
Incidence ir no 1:50 000 līdz 1:100 000 un apmēram 70-80% pacientu ir sporādiski.
Coffin-Lowry sindroms
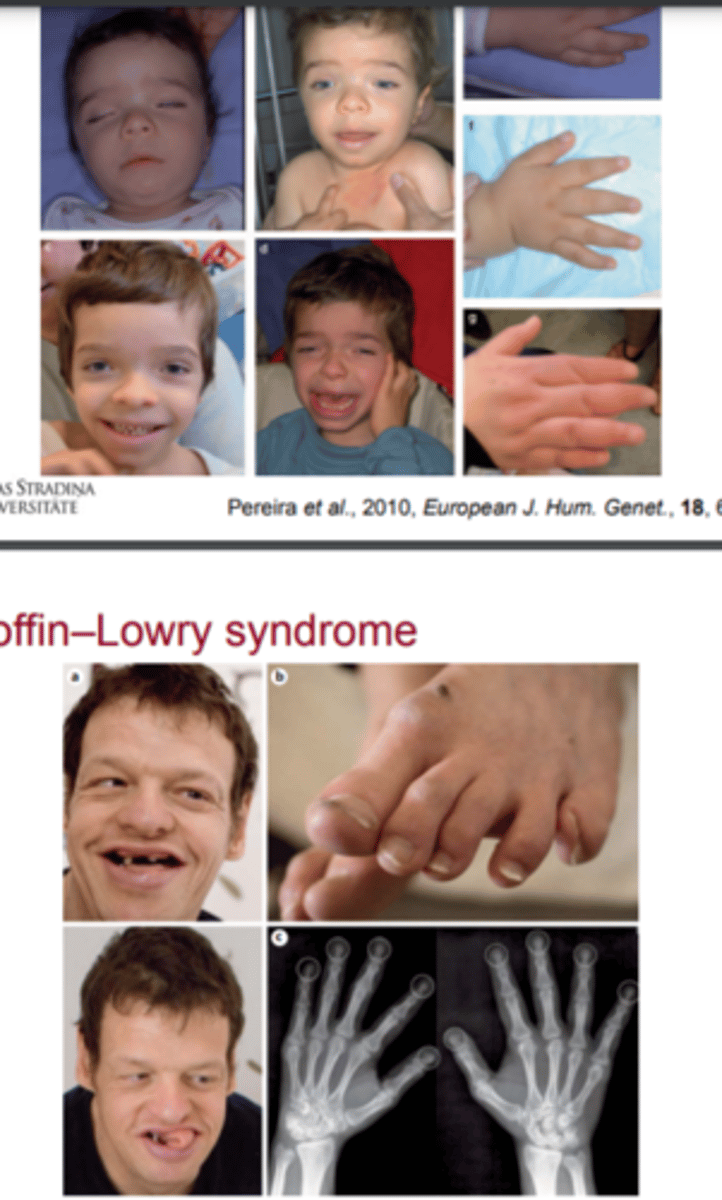
- Kāda gēna alēliskais variants izraisa?
RSK 1,2,3,4 patogēnais alēliskais variants
- gēns kodē kināzi - fermentu, kas fosforilē dažādus substrātus
-nenotiek histonu fosforilēšana = notiek hromatīna struktūras izmaiņas = izjaukta gēnu ekspresija
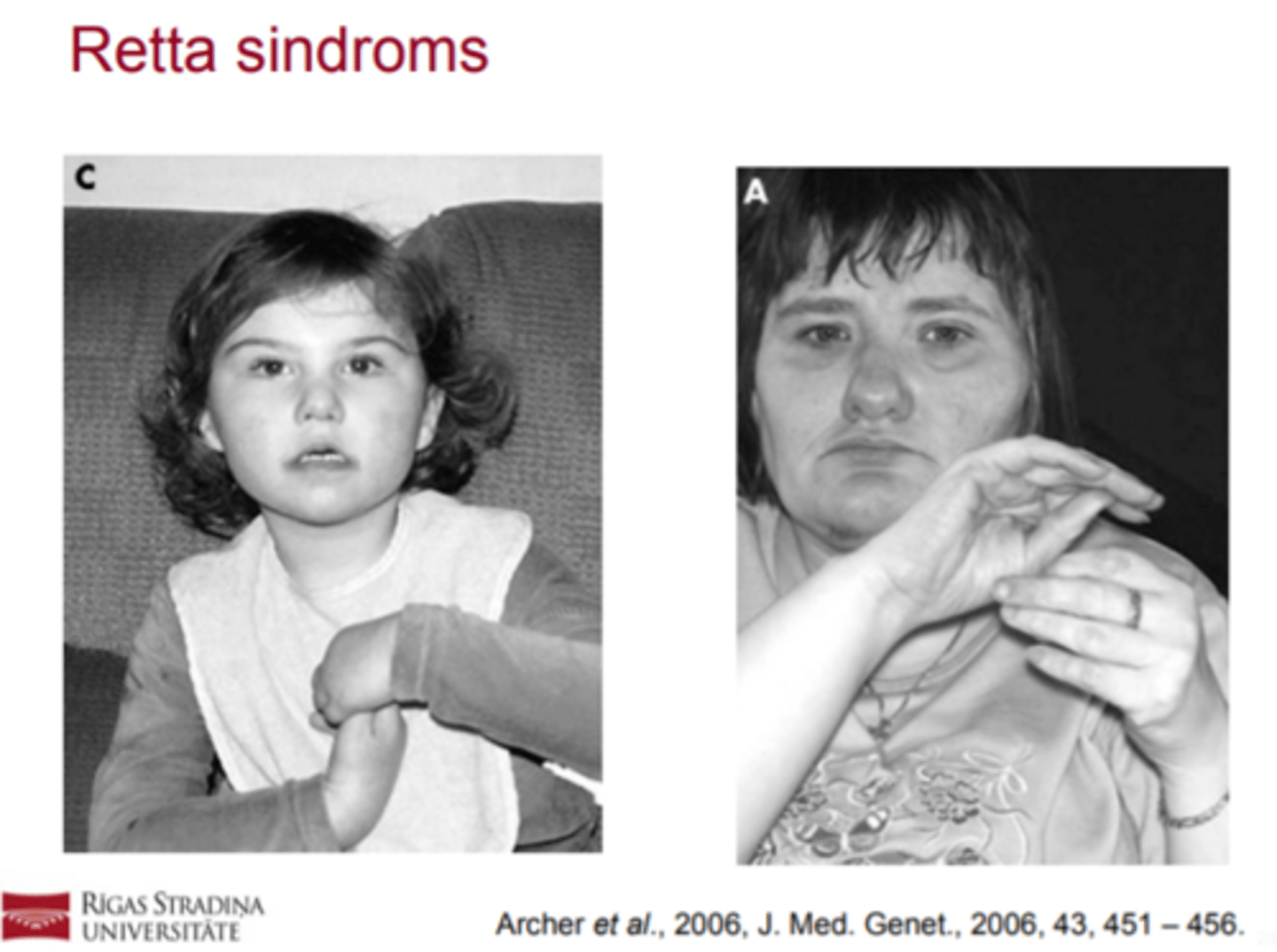
Retta sindroms (biežāk meitenēm, jo zēniem letāls)- izpausmes
! Attīstas stereotipas roku kustības (roku glāstīšana,
virpināšana, savilkšana dūrēs).
! Parādas stīva vai neveikla gaita, vai stāja.
! Normāls galvas perimetrs piedzimstot, bet lēnāka galvas augšana apmēram starp otro mēnesi un 4. dzīves gadu.
! Citu slimību, sindromu vai traumu iztrūkums, kuras izskaidro augšminētās pazīmes.
Retta sindroms (biežāk meitenēm, jo zēniem letāls) - kā sāk parādīties izpausmes?
! Īss periods agras bērnības laikā, kad bērnam ir normāls
progress vai tuvu tam.
! Stagnācijas periods attīstībā apmēram pirmā gada beigās, kurš ilgst līdz sākas regress.
! Regresa periods, kurš sākas apmēram starp 9. un 30. dzīves mēnesi, kad valodas un roku kustību iemaņas samazinās.
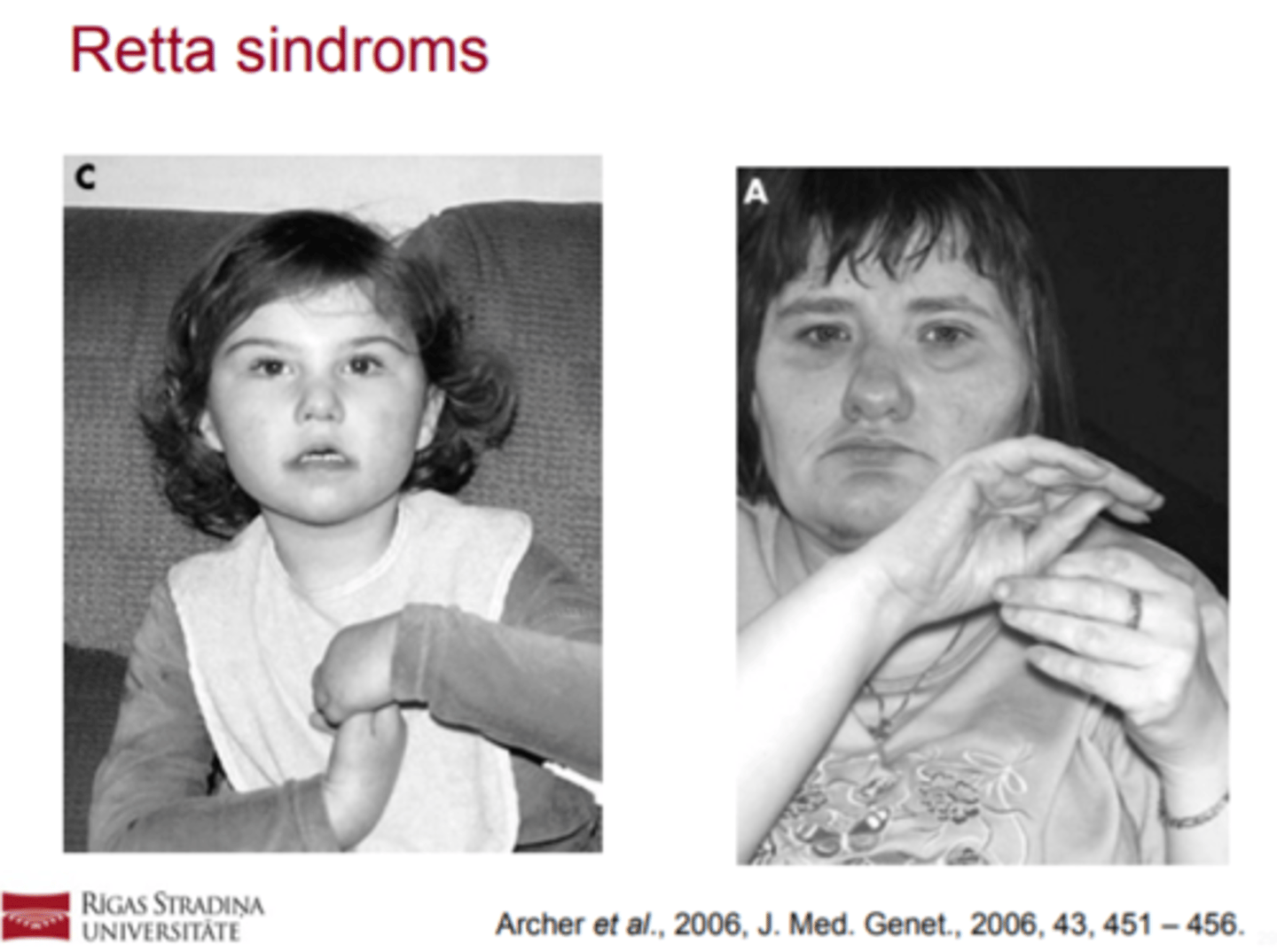
Retta sindroms (biežāk meitenēm, jo zēniem letāls)
Retta sindroms - sakarā ar autismu???
! Lai gan daļai meiteņu ar Retta sindromu piemīt autisma iezīmes, ar laiku tās parasite izzūd un pieaugušas sievietes var būt sociāli aktīvas.

Retta sindroms - iedzimšāna
! Retta sindroms ir kompleksa neirologiska saslimšana. Vīriešiem letāla patoloģija (retos gadījumos izdzīvo). Sievietes - 1/10 000
Retta sindroms - kāds alēl. variants?
MECP2 gēna patogēnie alēliskie varianti
Retta sindroms - kāds visbiežākais iemesls?
Tikai apm. 1% gadījumos notiek parmantošana.
Pamatā spontānas de novo mutācijas.
Retta sindroms - retos gadījumos var būt pārmantošana no X hromosomas
Retos gadījumos Retta sindromu izraisa no mātes (nēsātājas) pārmantota X hromosoma ar mutanto MECP2 gēnu.
Retta sindroms - - sporādiskie gadījumi (kāda izcelsme?)
! Sporādiskos Retta sindroma gadījumos, mutācijai gandrīz vienmēr ir paternāla izcelsme netakarīgi no mutācijas veida (aminoskābju maiņa, delēcija).
Vecāku vecumam nav izteiktas ietekmes.
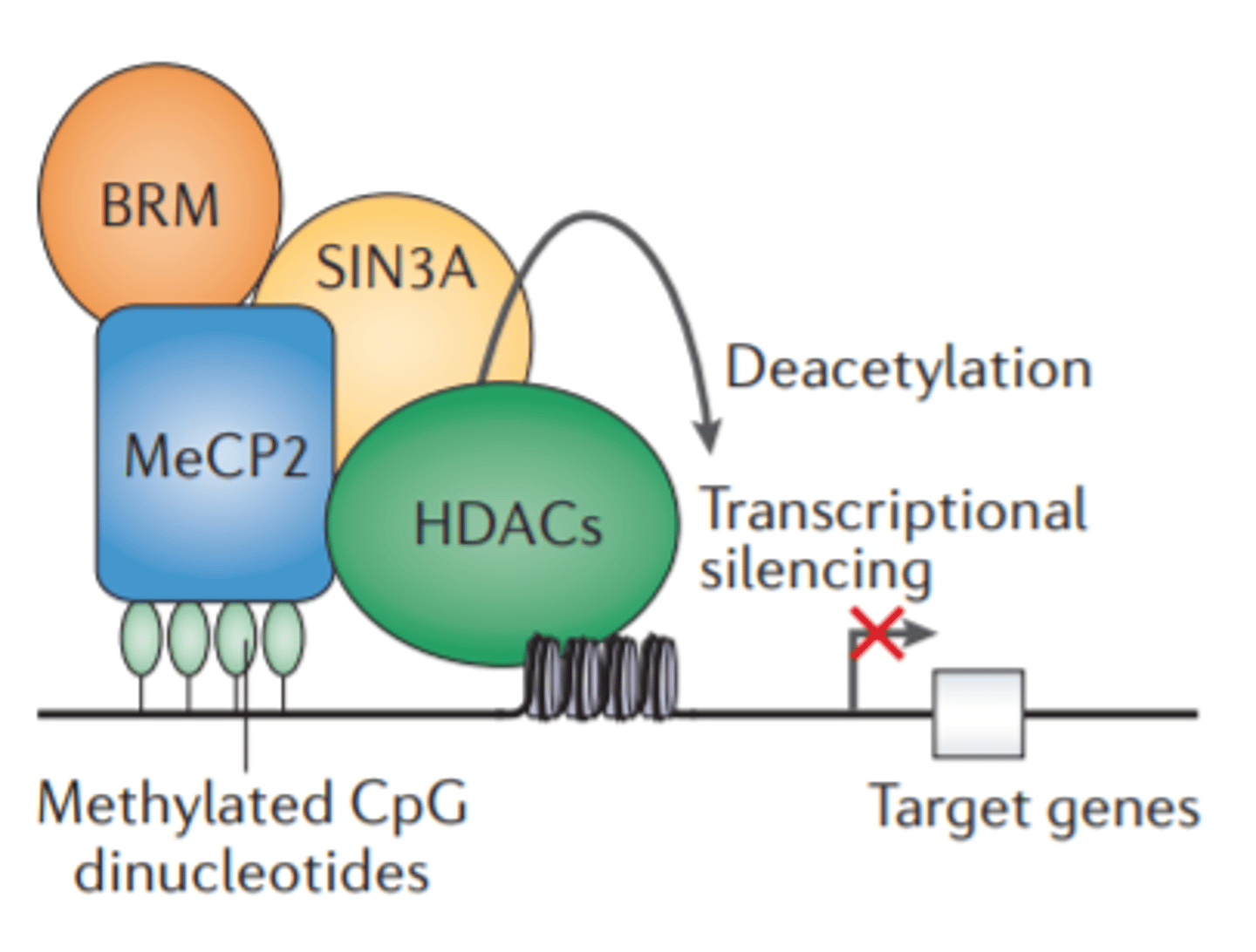
Retta sindroms - to izraisa MECP2 gēna alēliskais variants.
MECP2 gēns kodē metil-CpG-saistošu proteīnu-2.
(tas atpazīst, ka DNS ir metilēta. Piesaista proteīnus (histonu deacetilāzi, piemēram), tiek izraisa histonu hromatīna kondensēšanās, tiek "izslēgta" gēnu ekspresija)
Defekts DNS metilēšanas interpretācijā
Pārmantots Retta sindroms - gadījums
Mātei, I-2, nebija Retta sindroma. Viņa pabeidza mācības 21 gadu vecumā un strādāja par skolotāju. Viņas DNS, kas tika izdalīta no periferālo asiņu leikocītiem, noteica R133Cmutāciju. Praktiski visos leikocītos X inaktivācijai bija pakļauta mutāciju saturošā hromosoma.

Retta sindroms - summary

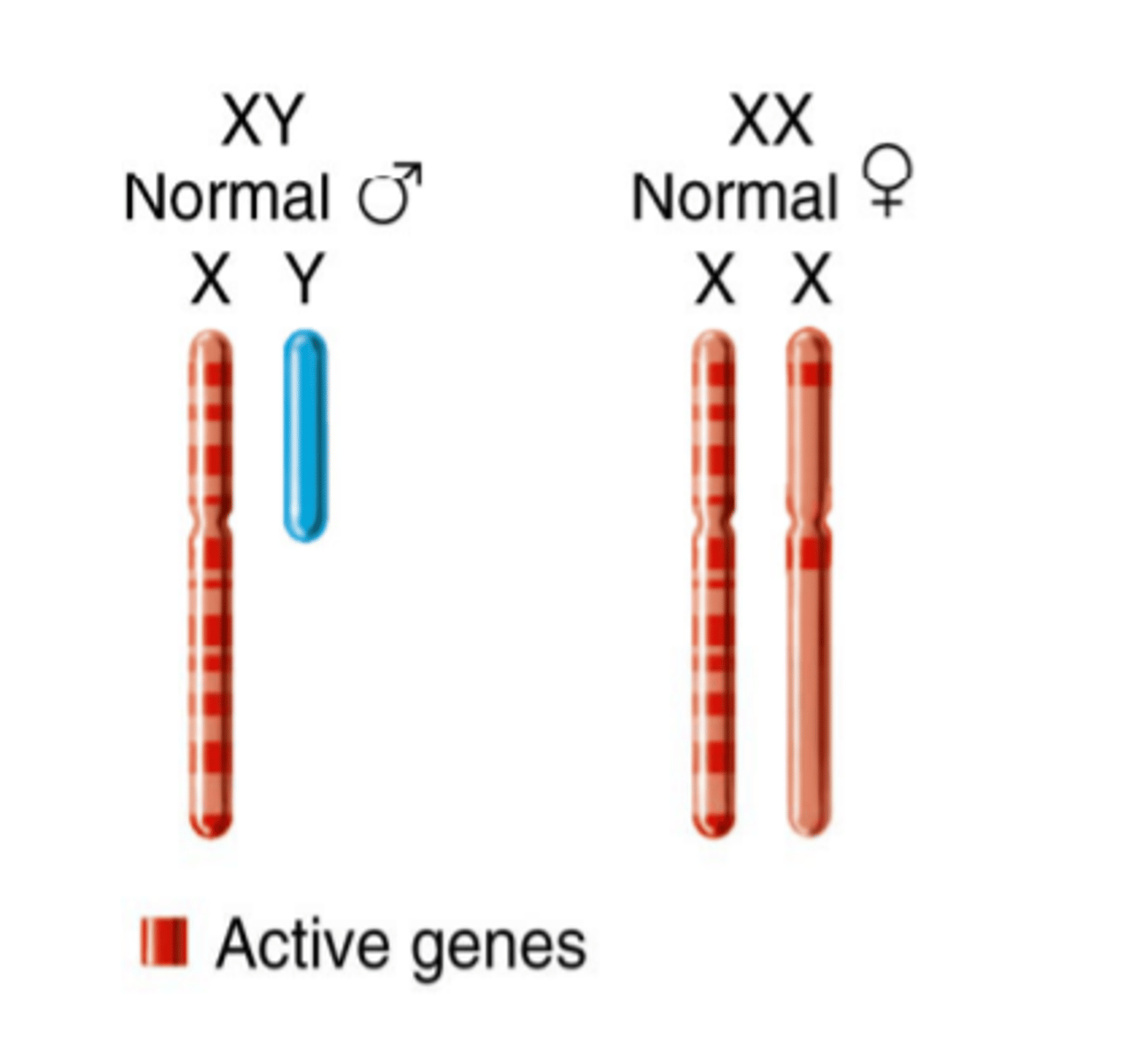
X hromosomas inaktivācija - kāpēc tā ir nepieciešama?
Lai izlīdzinātu ģenētisko nelīdzsvarotību X hromosomu skaitā starp vīriešiem (XY) un sievietēm (XX), viena no X hromosomām sieviešu diploīdās šūnās tiek inaktivēta.
Inaktivējamas X hromosomas izvēle ir nejauša.
X hromosomas inaktivācija - ar kādiem procesiem saistīta?
X hromosmas inaktivācija ir saistīta ar citozīna metilēšanu ar DNS metiltransferāzes palīdzību
X hromosomas inaktivācija - kāds gēns iesaistīts?
Barra ķermenītī tiek ekspresēts gēns XIST (X inaktivācijas specifiskais transkripts), kas kodē lielu nekodējošu RNS molekulu.
Šī lielā XIST kodētā RNS molekula apņem inaktivēto X hromosomu
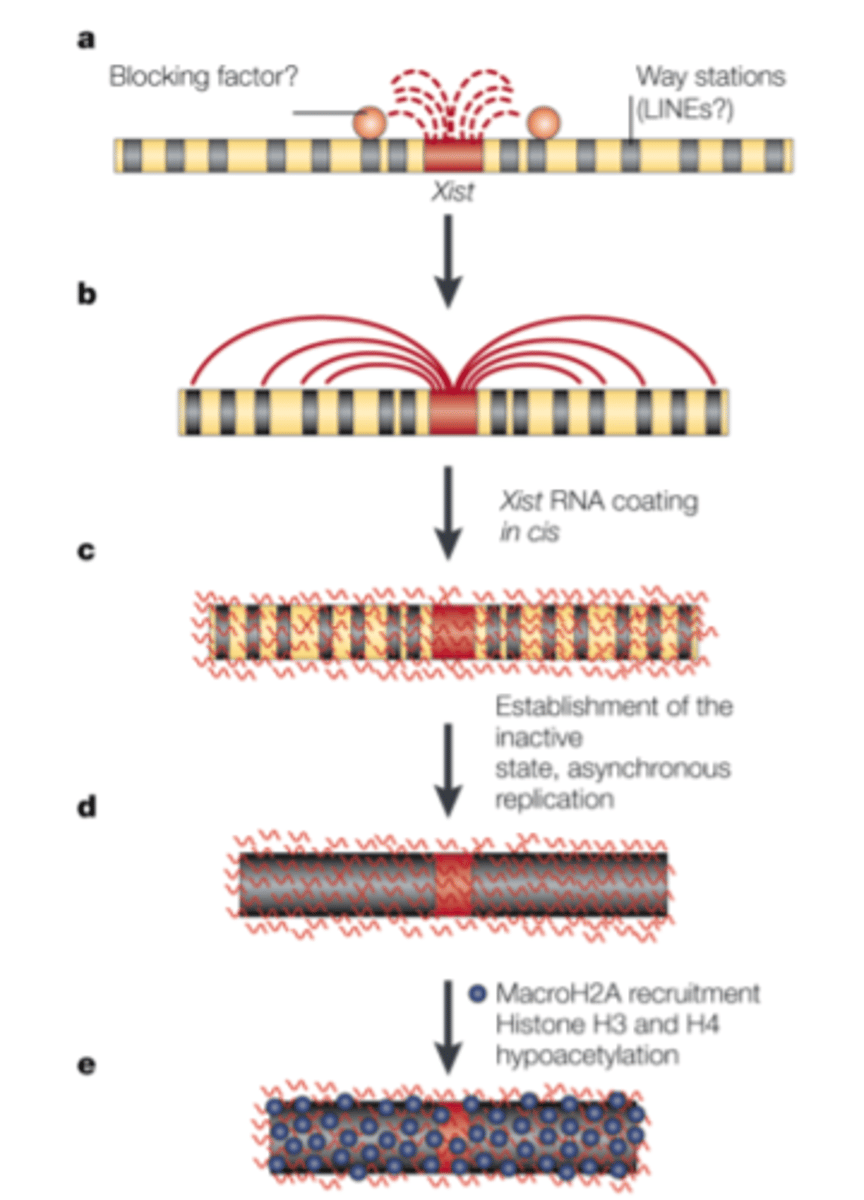
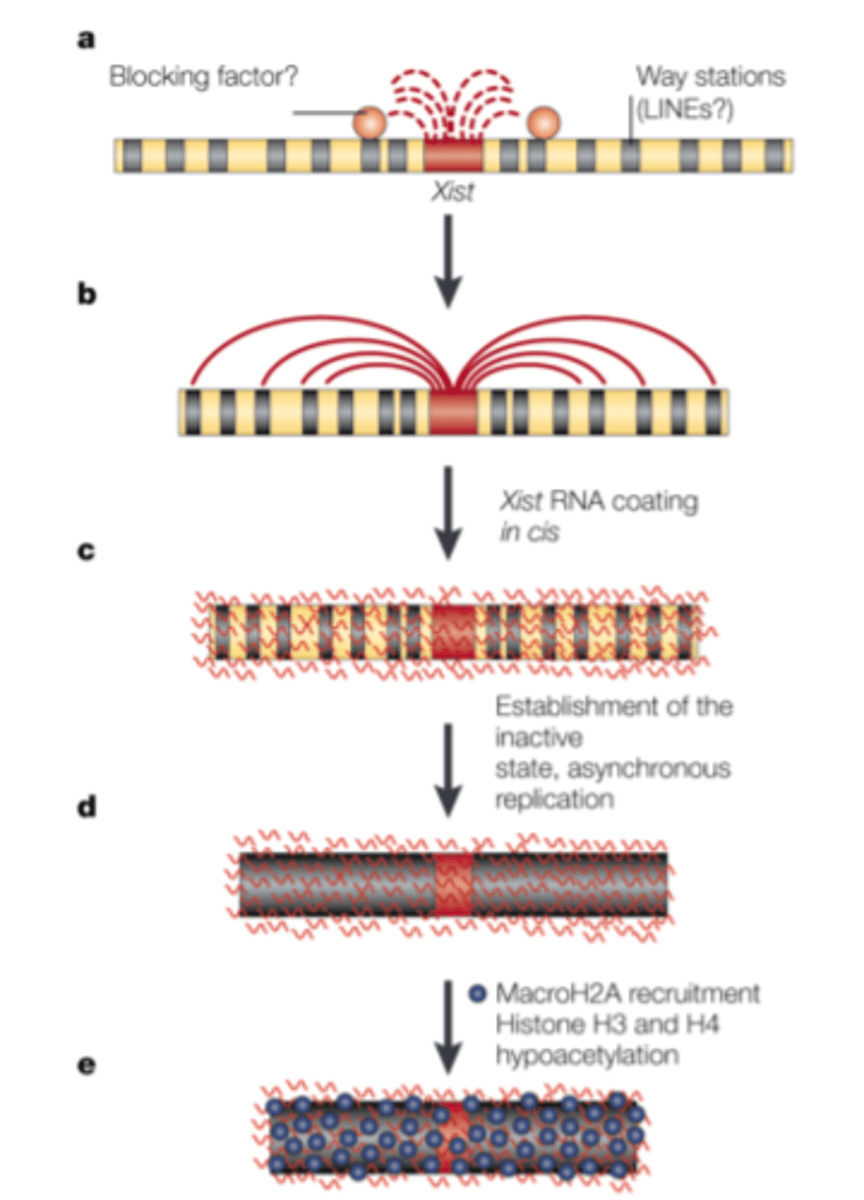
X hromosomas inaktivācija - soļi īsumā
1. XIST gēna ekspresija (nejaušā hromosomā)
2. XIST RNS apņem hromosomu
3. hromosoma tiek inaktivēta
4. tā kondensējas = veidojas Barra ķermenītis
X hromosomas inaktivācija - cilvēkiem tā nav pilnīga.
Tikai 75-85% X hromosomas gēnu ir inaktivēti.
~ 30% Xp gēnu un <3% Xq gēnu netiek inaktivēti.
Gēni, kuri ir lokalizēti PAR1 un PAR2 netiek inaktivēti
X hromosomas inaktivācija - dati par normālo populāciju
Normālā populācijā inaktivēto paternālo X hromosomu īpatsvars vs inaktivēto maternālo X hromosomu pakļaujas Gausa sadalījumam. Vairumam sieviešu tas svārstās starp 50:50 līdz 80:20.
Fenotipiski normālu sieviešu ar ar asimetriski inaktivētu X hromosomu īpatsvars svārtās dažādās populacijās no 1% to 33%.
Asimetriska X hromosomas inaktivācija
Sporādiska
Ģimeņu asimetriska X - inaktivācija, ko izraisa gēnu XIST un SKI1 alēliskie varianti.
Ģimeņu asimetriska X - inaktivācija izraisa hemofiliju A heterozigotos
XIST marķieris
F8 - koagulācijas faktors
I-1 normāli abi tie marķieri
I-2 (vienā hromosomā XIST gēna mutācija notika - tas gēns neekspresējas. Ir aktīvs XIST otrajā hromosomā, bet tajā ir F8 alēl. variants. Tā hromosoma ir inaktivēta tātad )
tas nozīmē, ka tiek nomākta patogēnā F8
XIST un F8 atrodas tālu, tpc liela varbūtība, ka notiks krustmija. Var veidoties hromosoma ar 2 patogēnajām alēlēm (gan XIST, gan F8) - to hromosomu nevar inaktivēt. Meitas saņem tādas hromosomas ;)
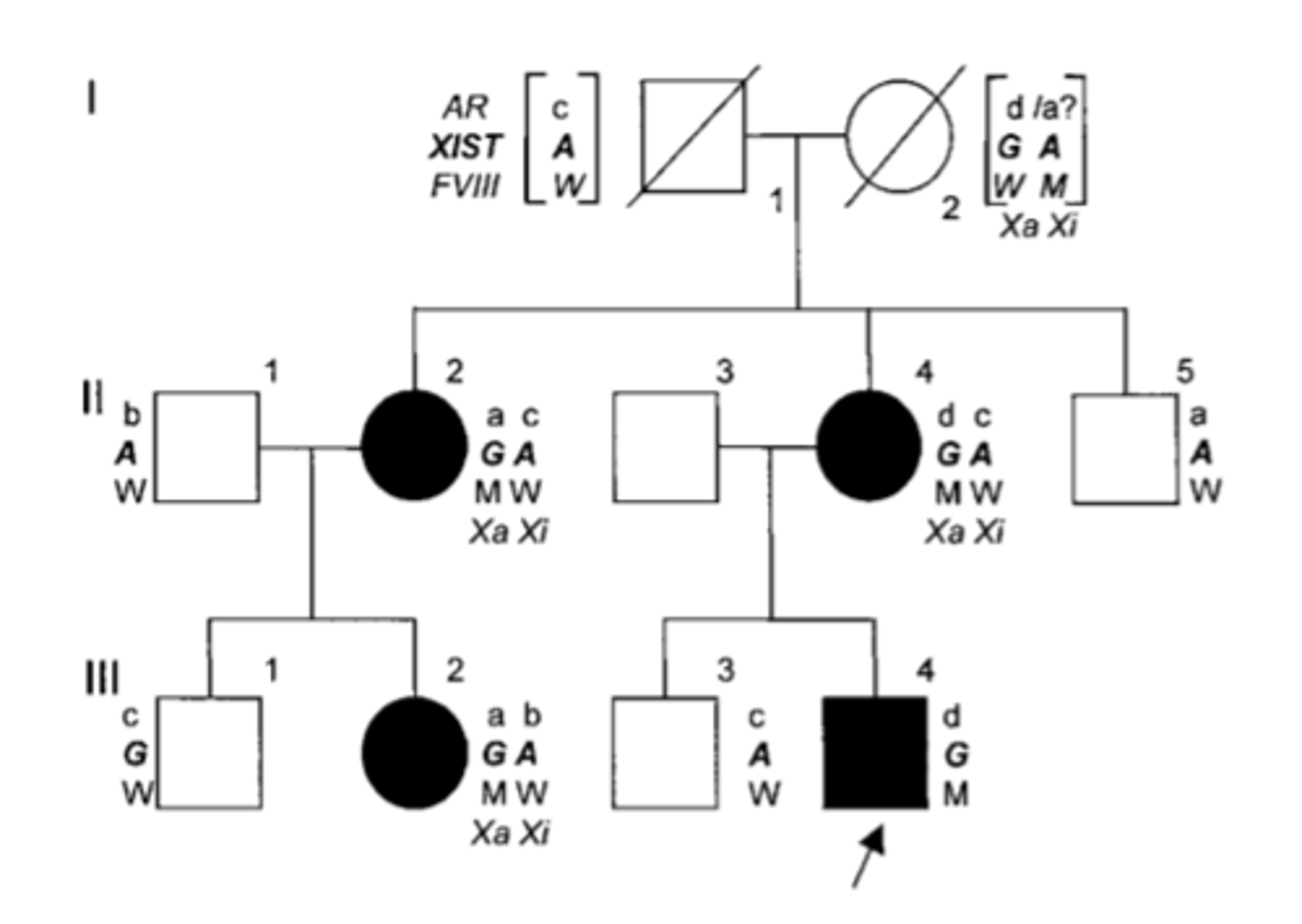
Ģimeņu asimetriska X - inaktivācija izraisa hemofiliju A heterozigotos (papildus)
Visas trīs slimās sievietes bija heterozigotas XIST alēliskā varianta G15944A nesātājas.
Tikai paternālā XIST A alēle ekspresējās no inaktivētās X hromosomas. Līdz ar to, maternālā X hromosoma XIST G alēli un patogēno FVIII alēlisko variantu palika aktīva visās šūnās
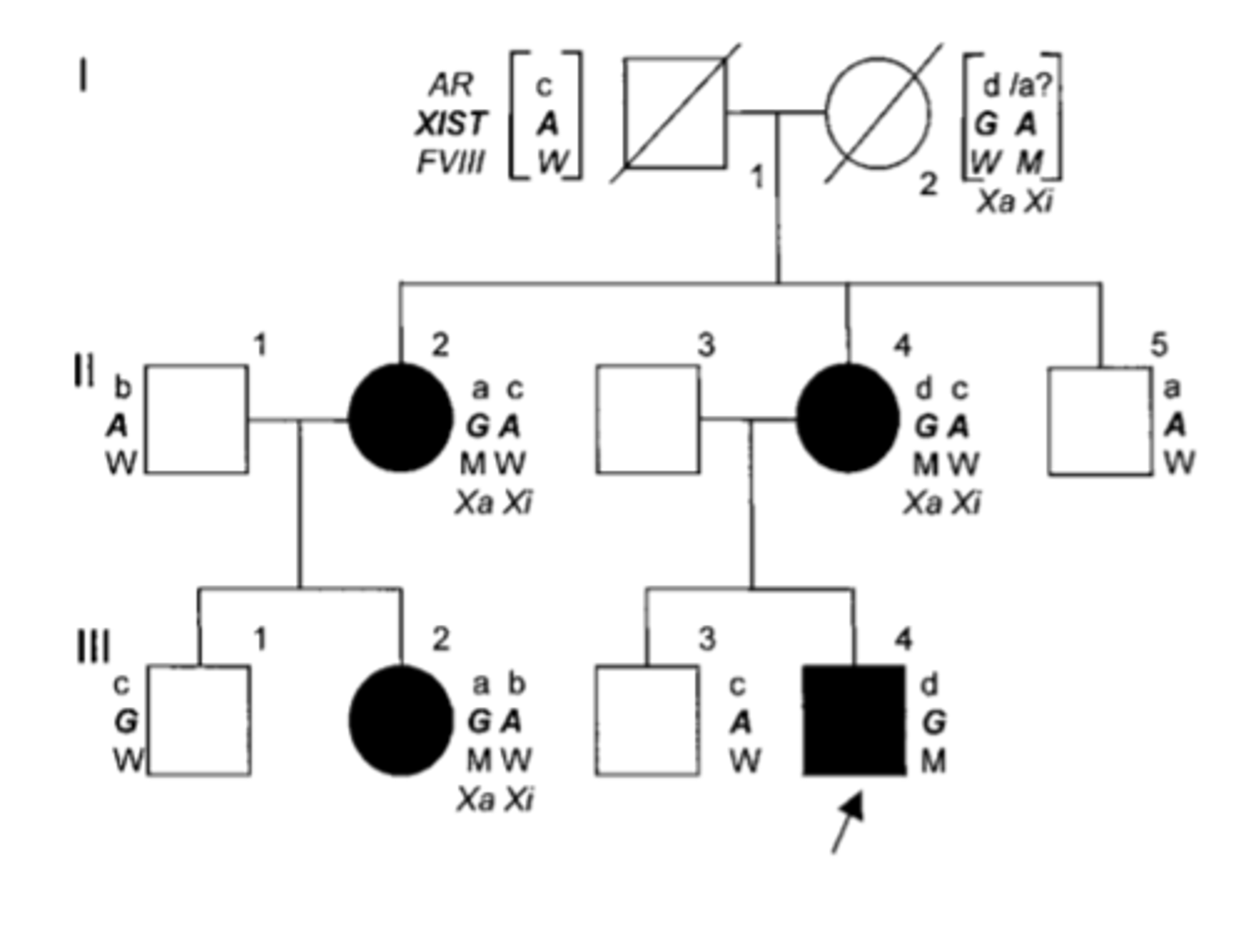
Imprintings
=genoma modifikācija, kuras rezultātā expresējas tikai viena (paternālā vai maternālā) alēle.
Kas nodrošina imprintingu?
Imprintingu nodrošina DNS metilēšana un/vai histonu modifikācija, nevis mutācijas.
Imprintings (skaidrojums)
- ir gēni, kas tiek ekspresēti tikai no maternālās vai tikai no paternālās hromosomas
gadījums= maternāli pārmantotā gēna kopija ir imprintēta (neaktīva)
I-2 saņēma no tēva alēl. variantu (jo mātes kopija ir neaktīva) - varbūtība, ka nodos alēli ir 0,5
sievietes bērni - 2 normāli, 2 veseli nēsātāji (jo mātes kopija patogēnā ir imprintēta. tikai no tēva pārmantotās nav imprintētas)
Mary var nodot tikai patogēno alēli (noklusinātu)
Jims var nodod aktīvo alēli - varbūtība būt slimiem bērniem ir 0,5
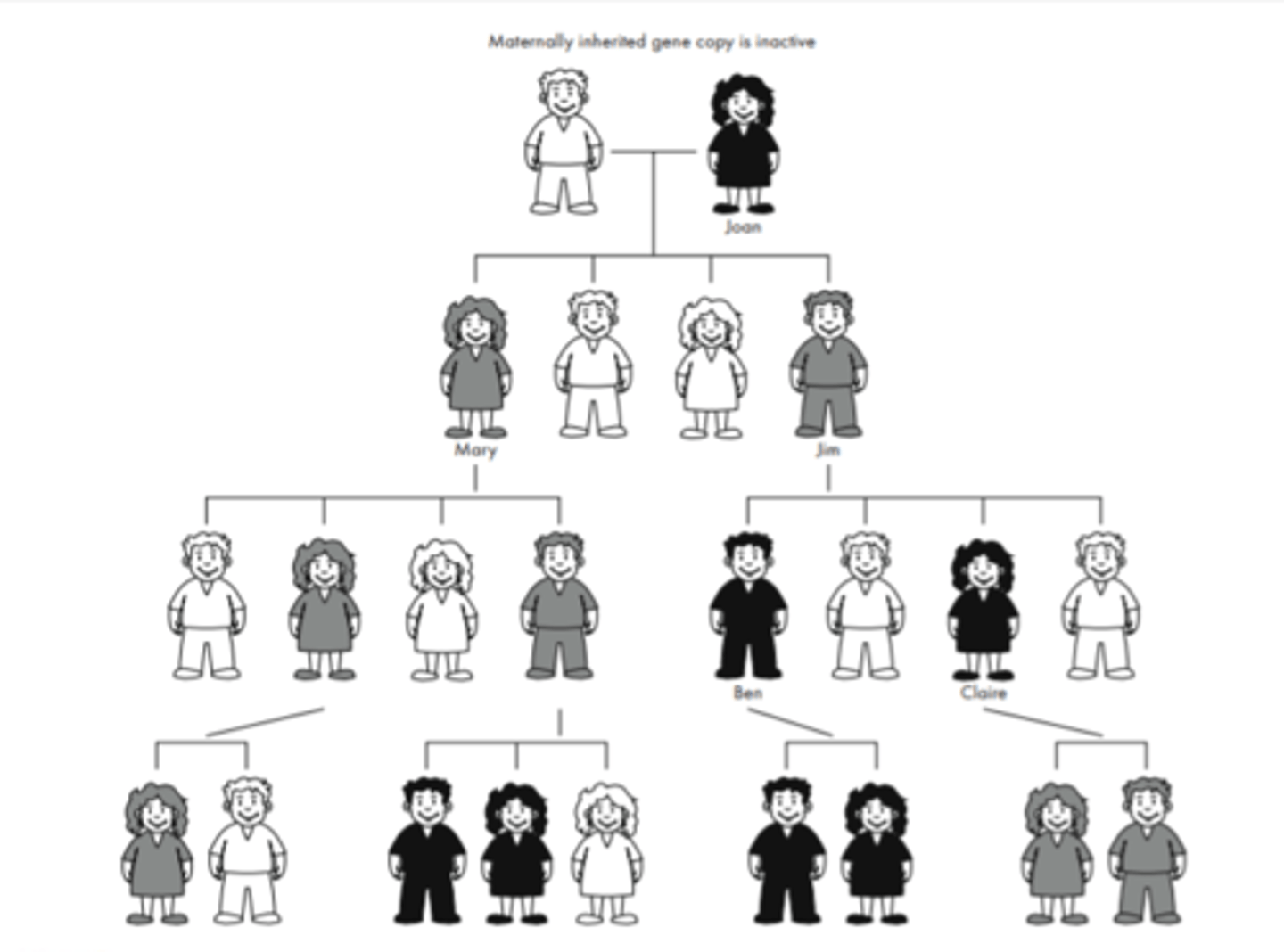
Ciltskoks ar maternāli imprintētu gēnu (pic)
1. paaudze - tēvam ir alēle no mātes (tā ir klusā)
2. paaudze - meita saņem jau aktīvo alēli (jo no VĪRIEŠA, tātad tā nav imprintēta)
4. cilvēks- maternālo saņēma no 2. cilvēka
Cik % no visa cilvēka genoma tiek imprintēts? Un cik ir gēnu, kas ir imprintēti?
apm. 1% no visiem cilvēka gēniem tiek imprintēti
Patreiz ir zināmi apm. 50 imprintēti gēni
Imprintinga sindromi
imprintingu nosaka vecāku dzimums ar to alēli, nevis bērna dzimums
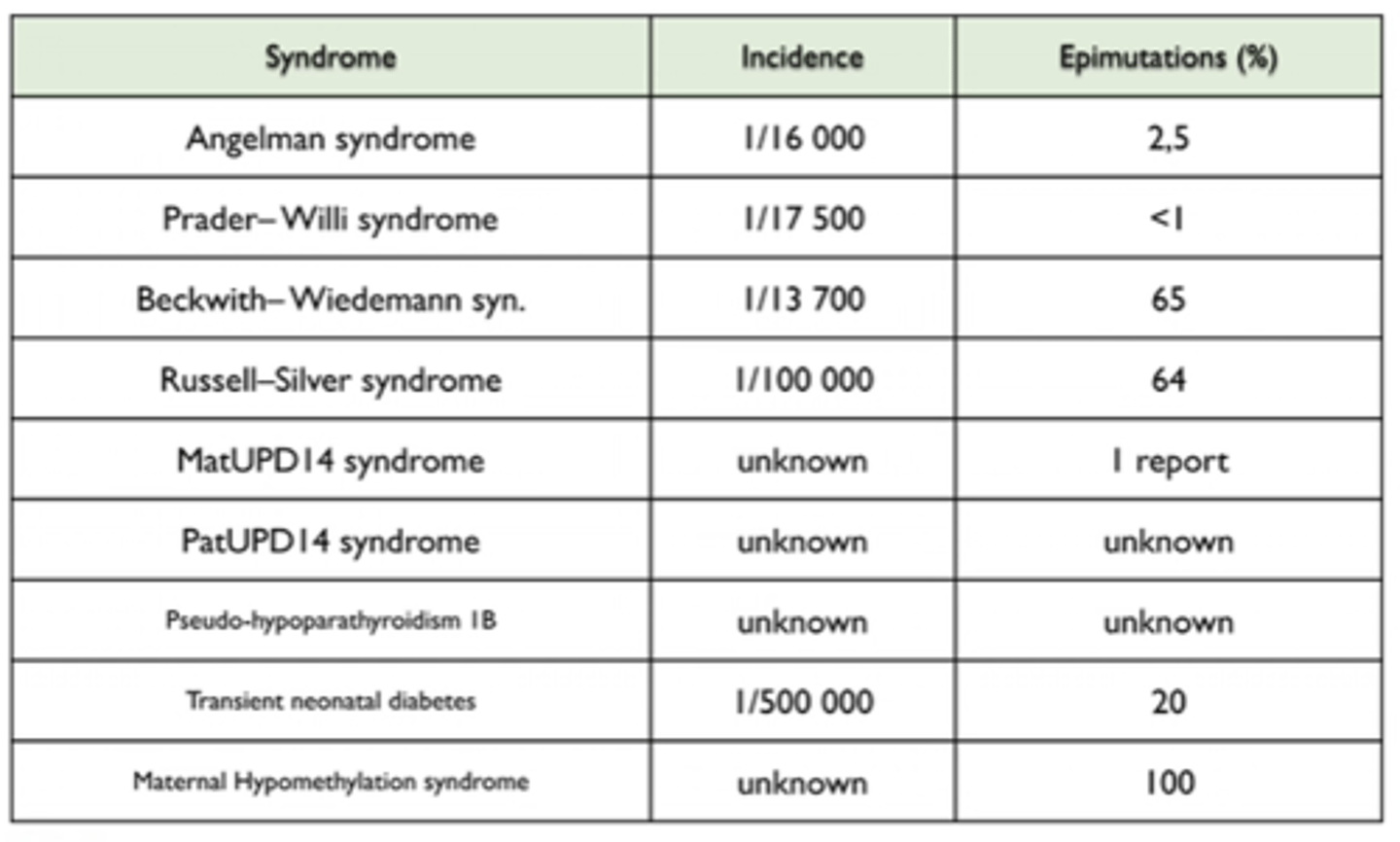
Imprintinga sindromi --- Citi moleklārie mehānismi:
»hromosomālās aberācijas;
»uniparentālā disomija;
»mutācijas.
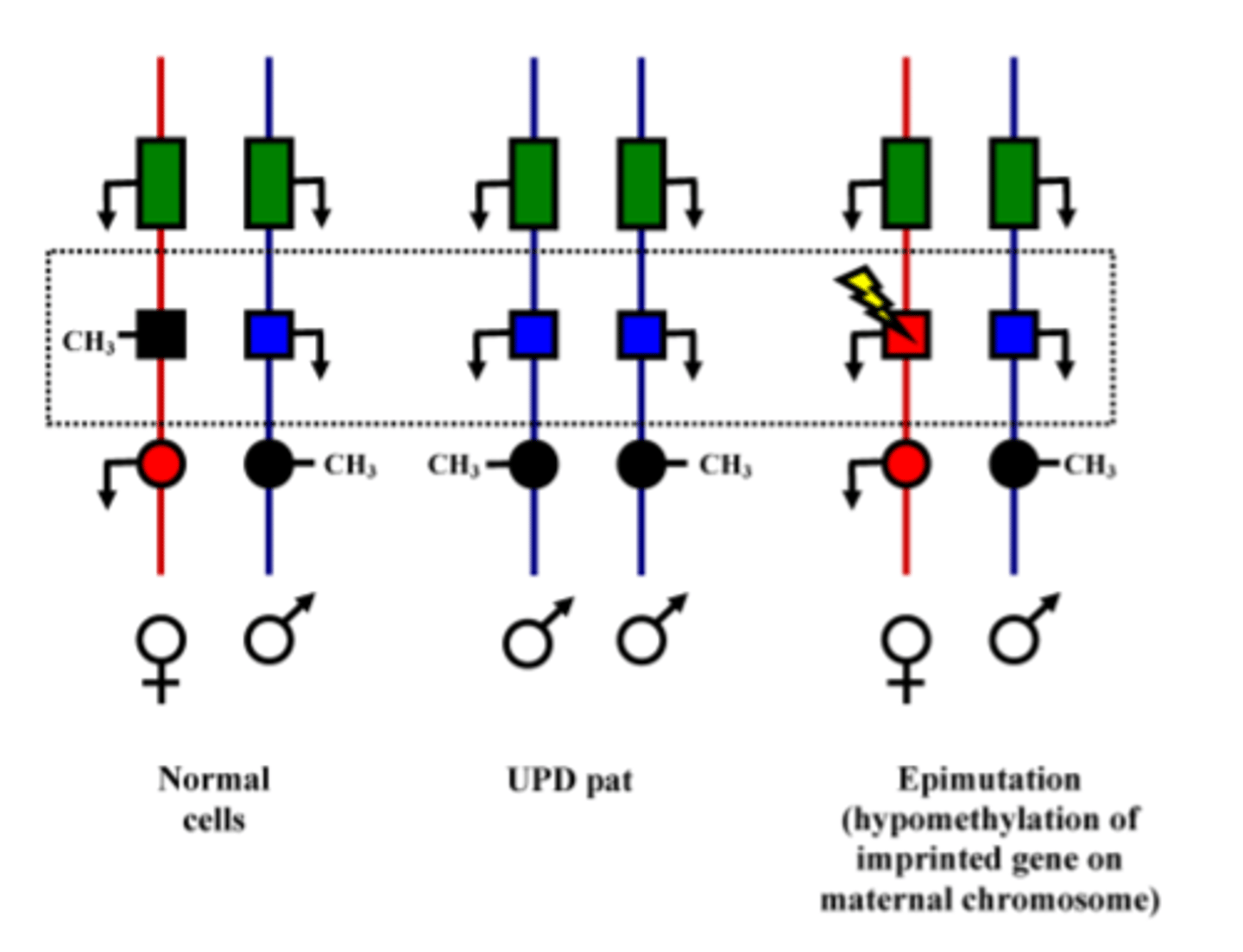
Imprintinga sindromi (pic. skaidrojums)
1. Normālās šūnas - ir paternāls imprintings, otrais gēns darbojas tikai paternāli (zilais. - maternālais imprintēts)
2. Uniparientālā disomija = 2 hromosomas no tēva - divkāršā apmērā ekspresējas zilais gēns, bet neekspresējas vispār sarkanais
3. Ja ir mutācija zilajā gēnā, kas saņemts no mātes, tad fenotipiski nekas nemainās (tas tāpat ir zilais)
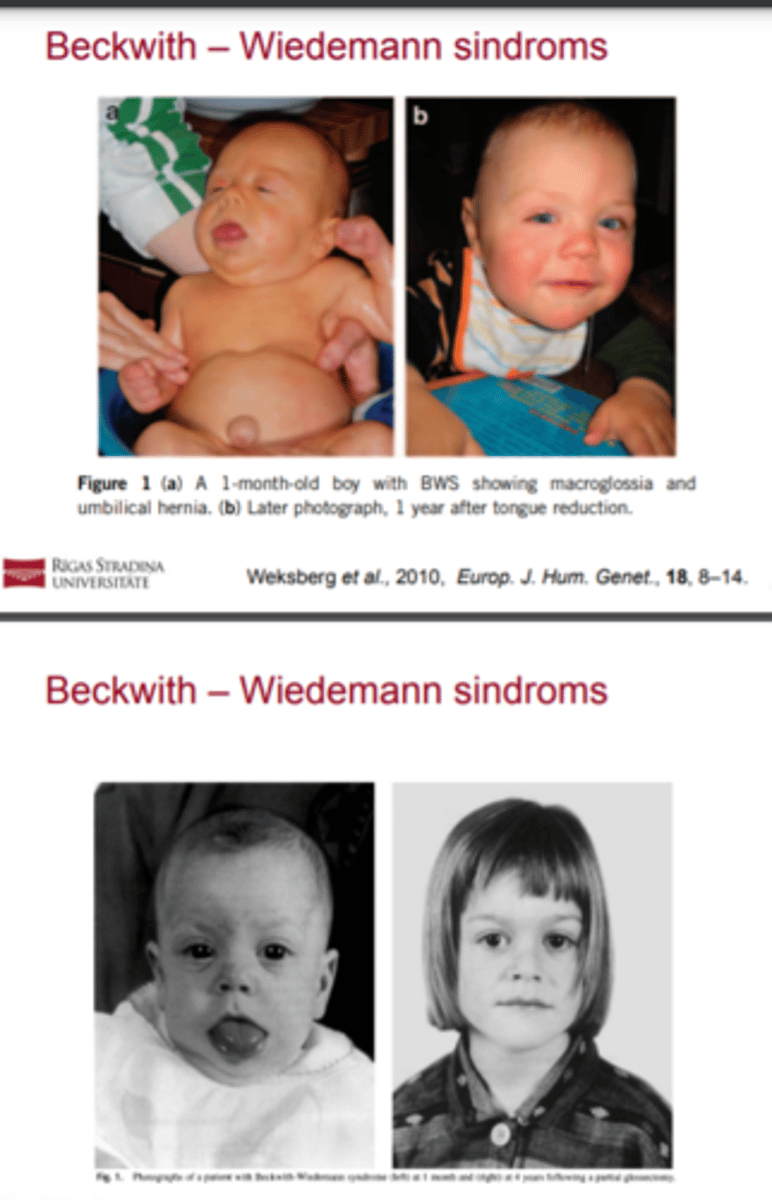
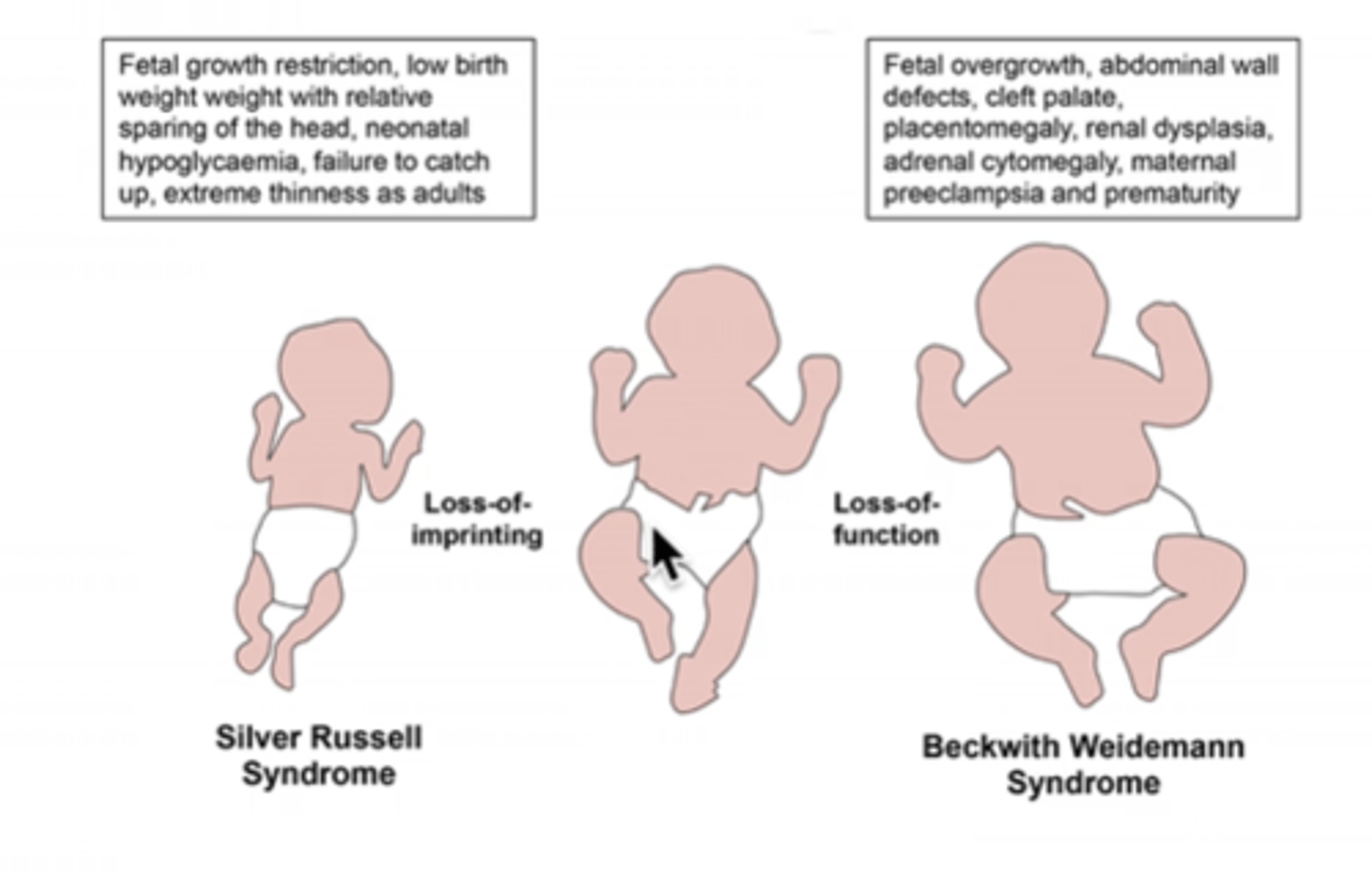
Beckwith - Wiedemann sindroms (izpausmes)
! BWS raksturojas ar makrosomiju un predispozīciju embrionāliem audzējiem bērnībā
BWS izsauc dažādas epiģenētiskās izmaiņas un/vai mutācijas, kuras izjauc imprintēto gēnu normālu ekspresiju 11p15.5 rajonā
Beckwith - Wiedemann sindroms - incidence
BWS incidence ir apm. 1 / 13 700 jaundzimušiem.
Mākslīgā apaugļošana palielina BWS (un citu imprintiga slimību) risku.
Beckwith - Wiedemann sindroms = PAZĪMES (fenotips
! Vēdera priekšējās sienas defekts: omfalocēle vai
nabas trūce.
! Makroglosija.
! Makrosomija (parasti definēts kā garums un svars > 97
percentīli).
! Auss priekšējās daļas krokas un/vai auss gliemežnīcas
padziļinājumi (abpusēji vai vienpusēji).
! Iekšējo orgānu visceromegālija, piemēram, aknu, nieru,
liesas, aizkuņģa dziedzera, virsnieru dziedzeru
palielināšanās
! Embrionālie audzēji bērnībā.
! Hemihiperplāzija.
! Fetālās virsnieru dziedzeru garozas citomegālija,
parasti difūza un abpusēja.
! Nieru patoloģijas, tajā skaitā nieres serdes displāzija
un tālāka nieru policistiskās slimības jeb medullārās
„sūkļa nieres" attīstība.
! Ģimenes anamnēzē BVS.
! Augsleju šķeltne.
Beckwith - Wiedemann sindroms -- iesaistītie gēni
IGF2
CDKN1C
Beckwith - Wiedemann sindroms -- IGF2 gēns
IGF2 kodē insulīnam līdzigo augšanas faktoru 2, kas ir iesaistīts šūnu proliferācijas, augšanas, migrācijas, diferenciācijas un izdzīvošanas regulēšanā. IGF2 ir augstāka ekspresija augļa attīstības laikā, ka arī dažādos somatisko audos pēc tam.
Beckwith - Wiedemann sindroms -- CDKN1C gēns
CDKN1C gēns kodē proteīnu p57, kas ir G1 ciklīna / CDK kompleksa inhibitors un ir negatīvu šūnu proliferācijas regulators
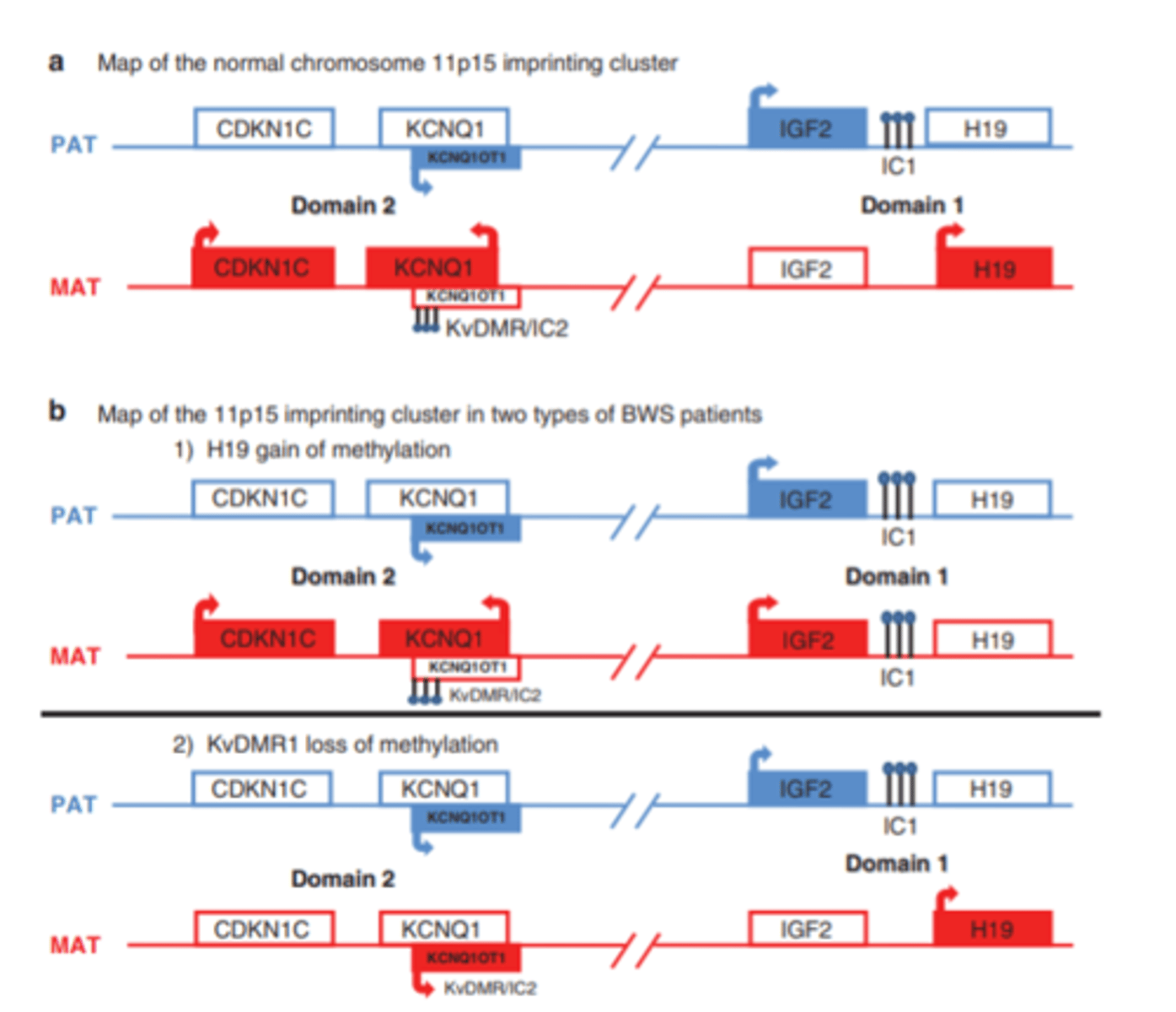
Beckwith - Wiedemann sindroms -- hromosomu attēls un skaidrojums
a - normālās hromosomas ar normālu imprintingu
red-maternal, blue - paternal
gēni, kas ekspresējas=iekrāsoti
IC1 (imprintinga centrs 1) - imprintēts tikai uz paternālās. Uz maternālās nav - tātad ekspresējas H19 gēns (kodē nekodējošo RNS, kas ietin IGF2 gēnu (neļauj tam ekspresēties)
pretēja situācija paternālajā hromosomā - IGF2 ekspresējas
--------
tie gēni ar pretdarbību normā ekspresējas tikai no maternālās
b - 1)
-maternālajā hr ir notikusi mutācija IC1 -> H19 neekspresējas -> IGF2 ekspresējas gan no paternālās, gan no maternālās hr (slikti)
- rezultātā IFG2 rodas dubultā apmērā, bet līdzsvarojošo proteīnu nav pietiekami = pāraugšanas sindroms
b -2)
- IC1 normā abās hr
- imprintinga defekts IC2 - rezultātā normāls IGF2, bet absolūti nav pretdarbības proteīnu = BWS izpaužas
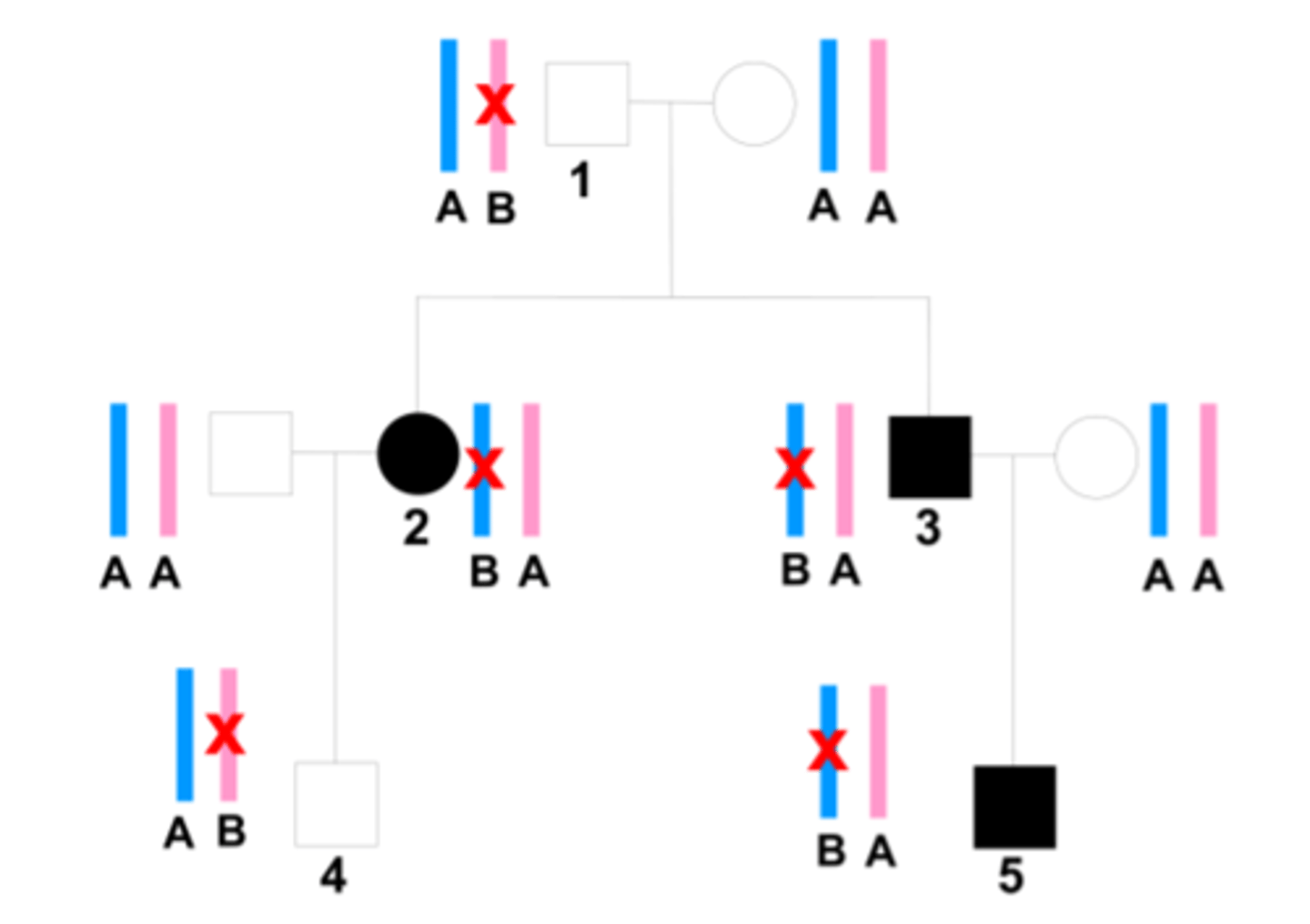
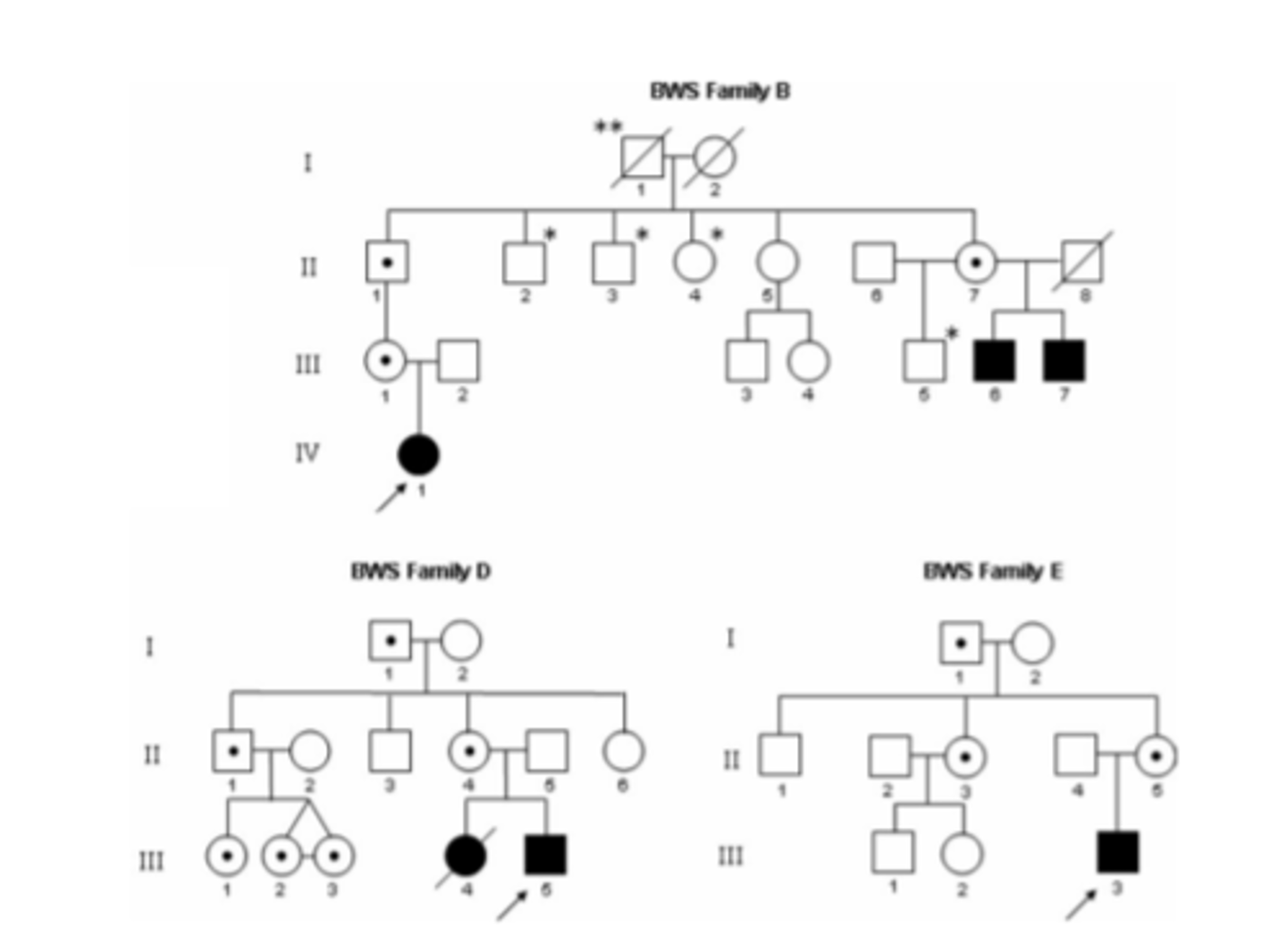
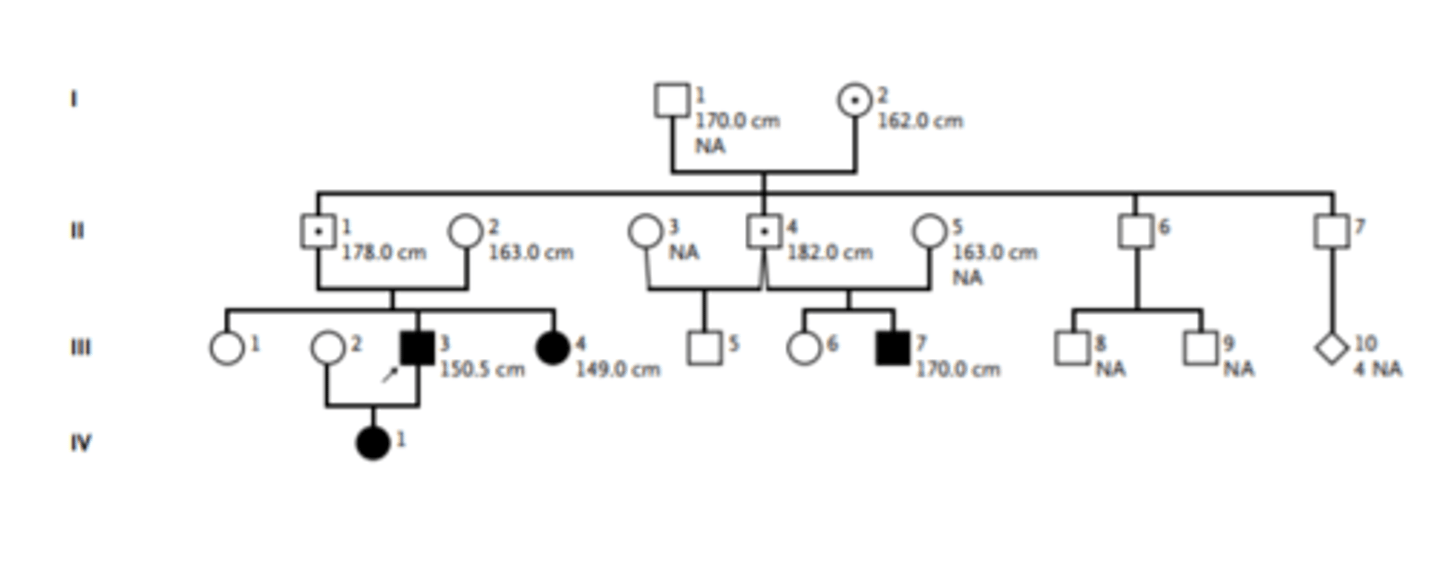
Pārmantotā BWS ciltskoki
I-1 tēvs nodo dēlam II-2 patogēno alēli
dēls nesējs ar normālu fenotipu
saslimt var tikai tad, ja saņem patogēno alēli no mātes
II-1 nodod alēli meitai
meita jau var tālāk nodod savam bērnam (they will have BWS)
Beckwith - Wiedemann sindroms -- summary

Silver - Russell sindroms - fenotips
! Bērni ir salīdzinoši makrocefāli, un viņu sejas ir
trīsstūra formas ar platām pierēm un smailiem, maziem
zodiem.
! Daudzos gadījumos ir novēroja locekļu un ķermeņa
asimmetrica, klinodaktīlija V.
! Fiziskās attīstības aizkavēšanās.
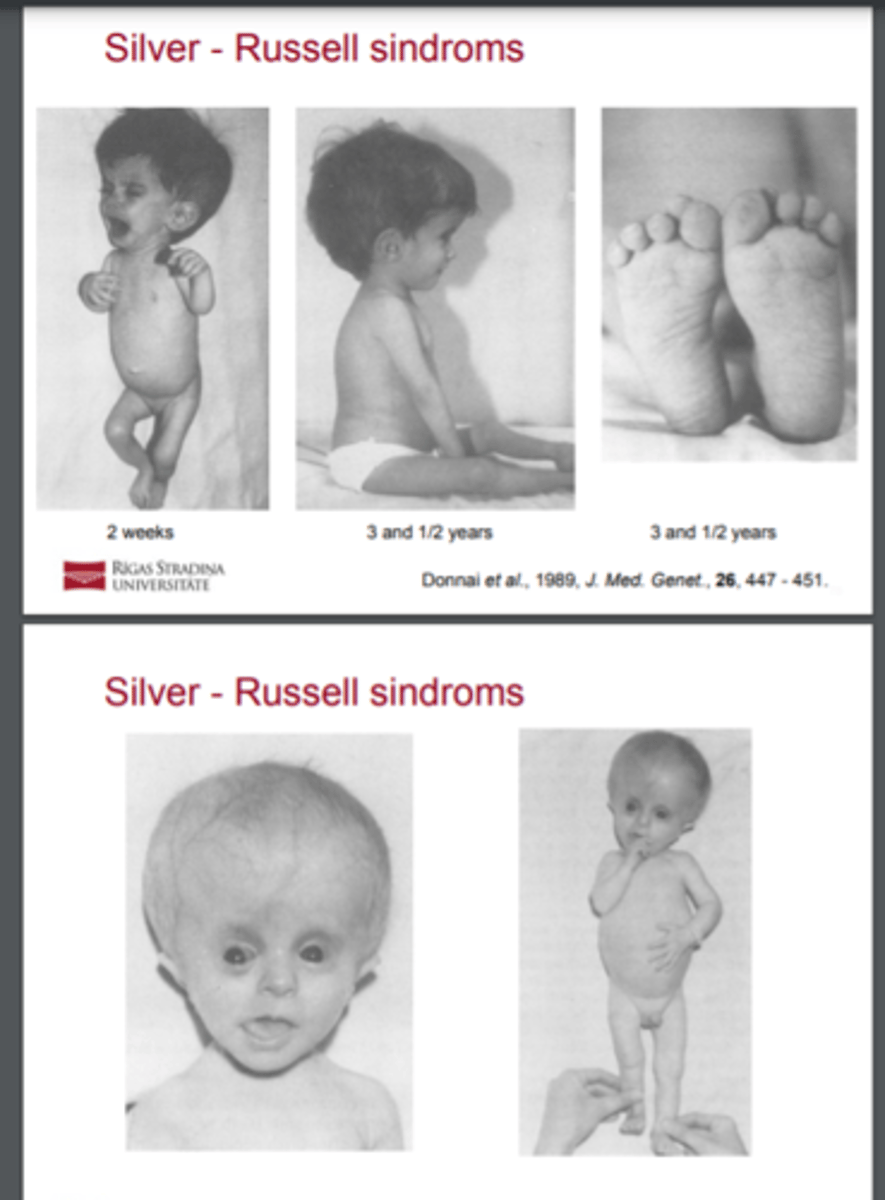
Silver - Russell sindroms
Vairāk kā 40% SRS pacientu ir novērota imprinting centra ICR1 hipometilēšana 11p15 rajonā

Silver - Russell sindroms ---- Genotipa - fenotipa korelācija
atšķirības īsti nav, ja notiek ICR1 hipometilācija vai hromosomas 7 maternālā UPD

Silver - Russell sindroms - attēls ar IGF2 un CDKN1C gēnu
a - norma
b - alēliskie varianti, kas sekmē samazinātu augšanu
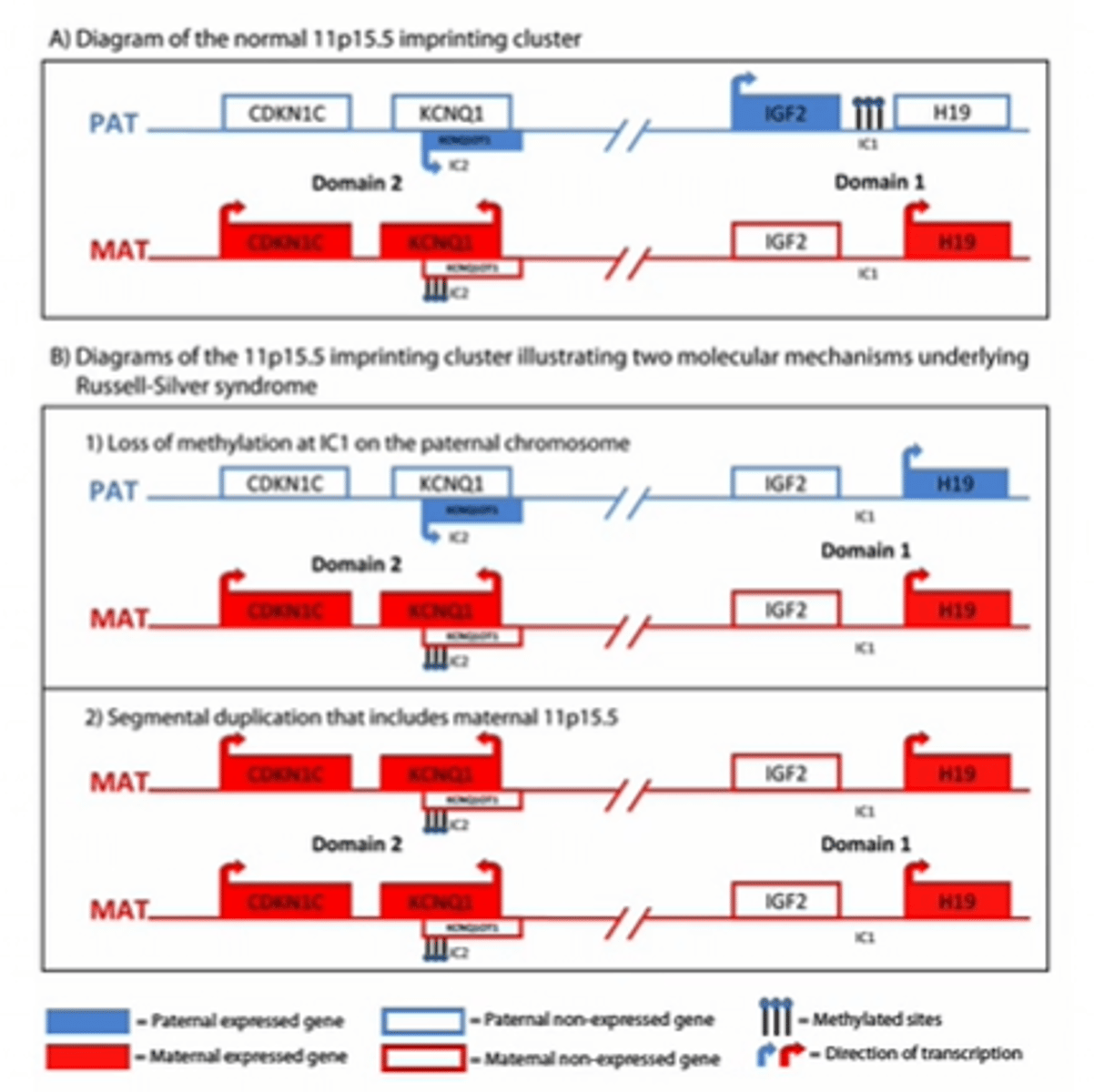
CDKN1C ekspresijas ietekme uz augļa attīstību - salīdzinājums Silver-Russell sindromam un BWS
SRS - imprintinga zudums IC1
BWS - funkcijas zudums
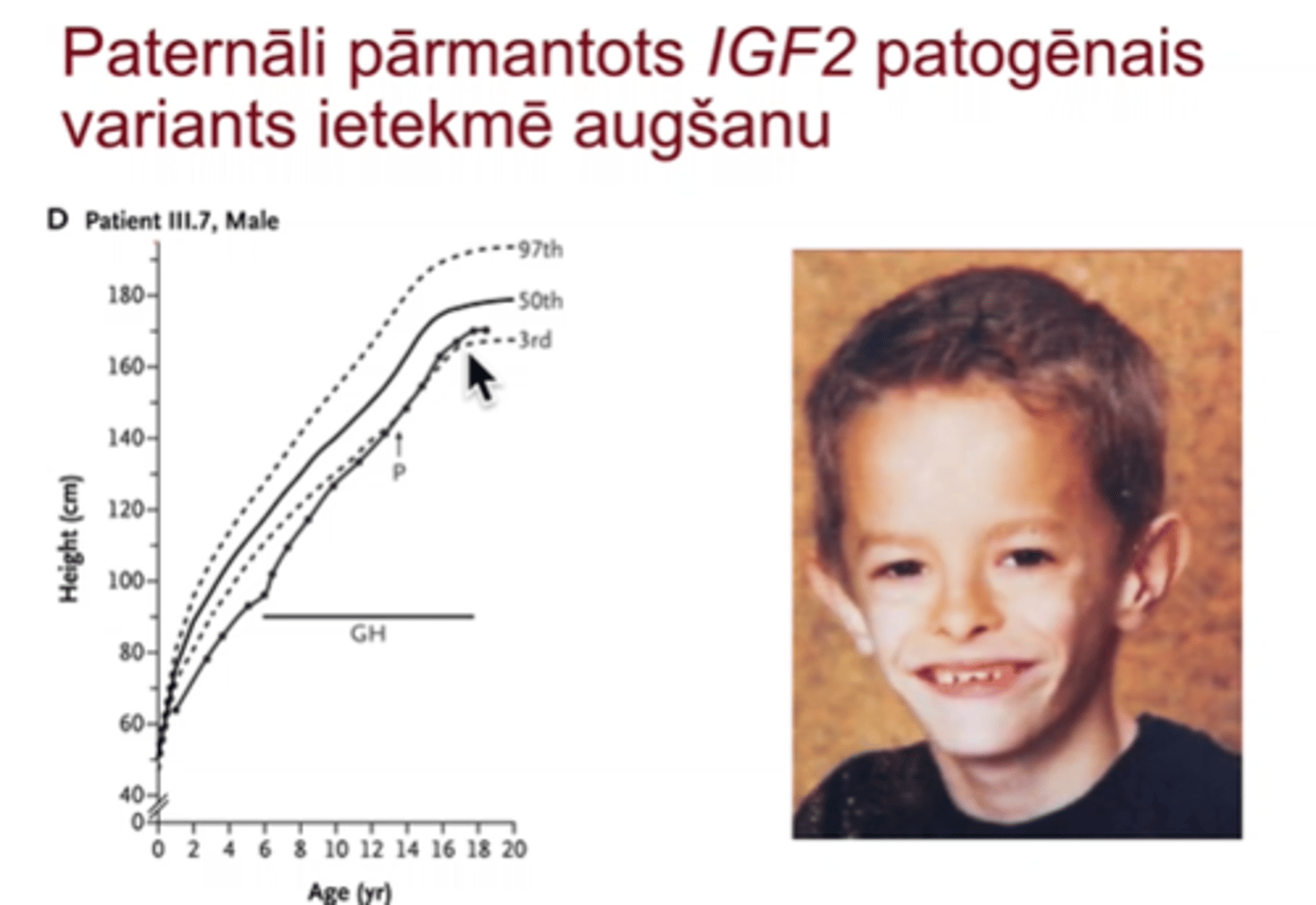
Paternāli pārmantots IGF2 patogēnais variants ietekmē augšanu - ciltskoks
- tas normā ekspresējas tikai no paternālās hromosomas
patogēnā situācija - ja no maternālās hr ekspresējas
--------
II paaudzes dēli veseli, bet saņēma patogēno alēli
varbūtība, ka viņi nodos patogēno variantu ir 50%
Paternāli pārmantots IGF2 patogēnais variants ietekmē augšanu - cilvēkiem var ievadīt augšanas faktorus
Silver - Russell sindroms - summary

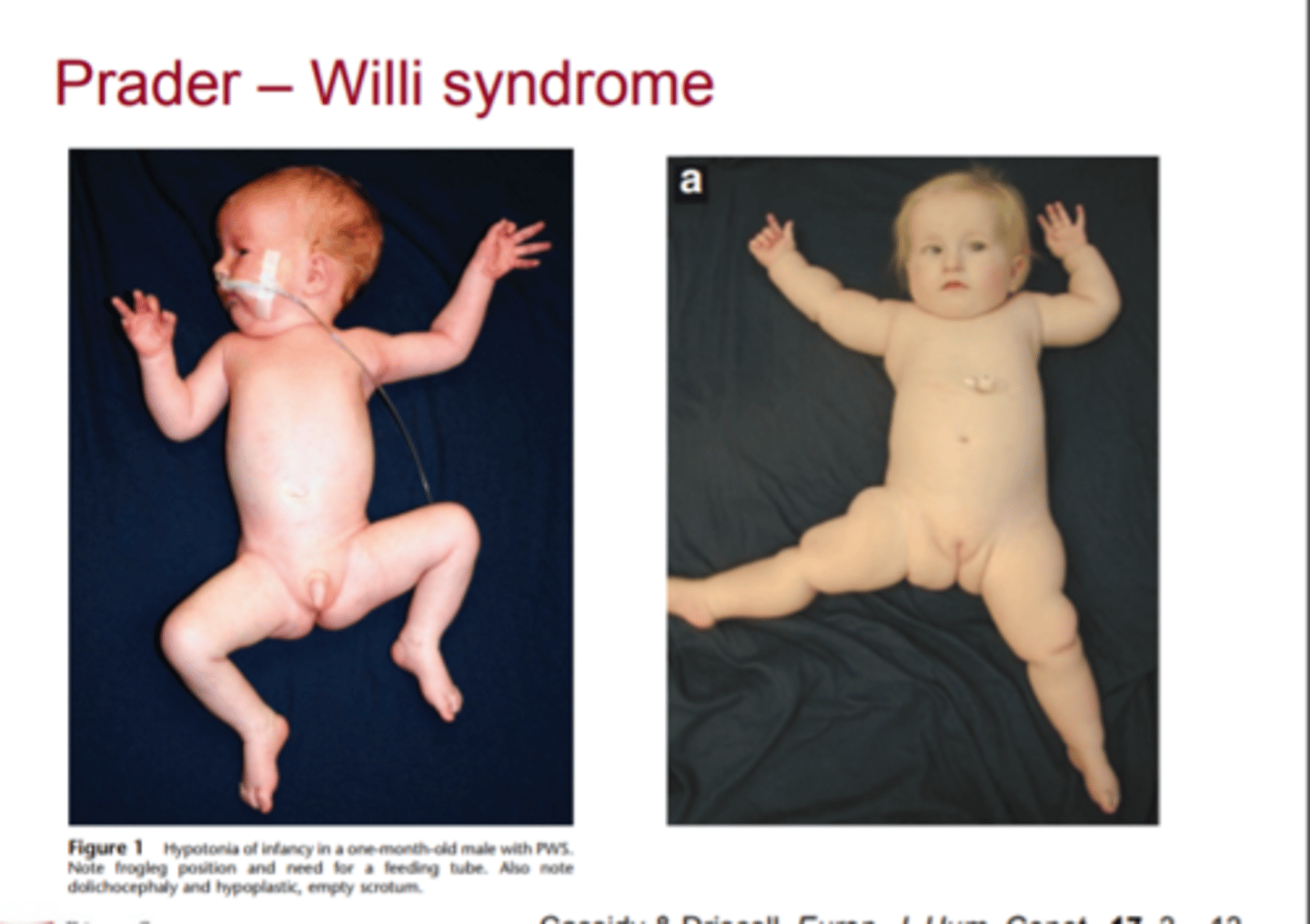
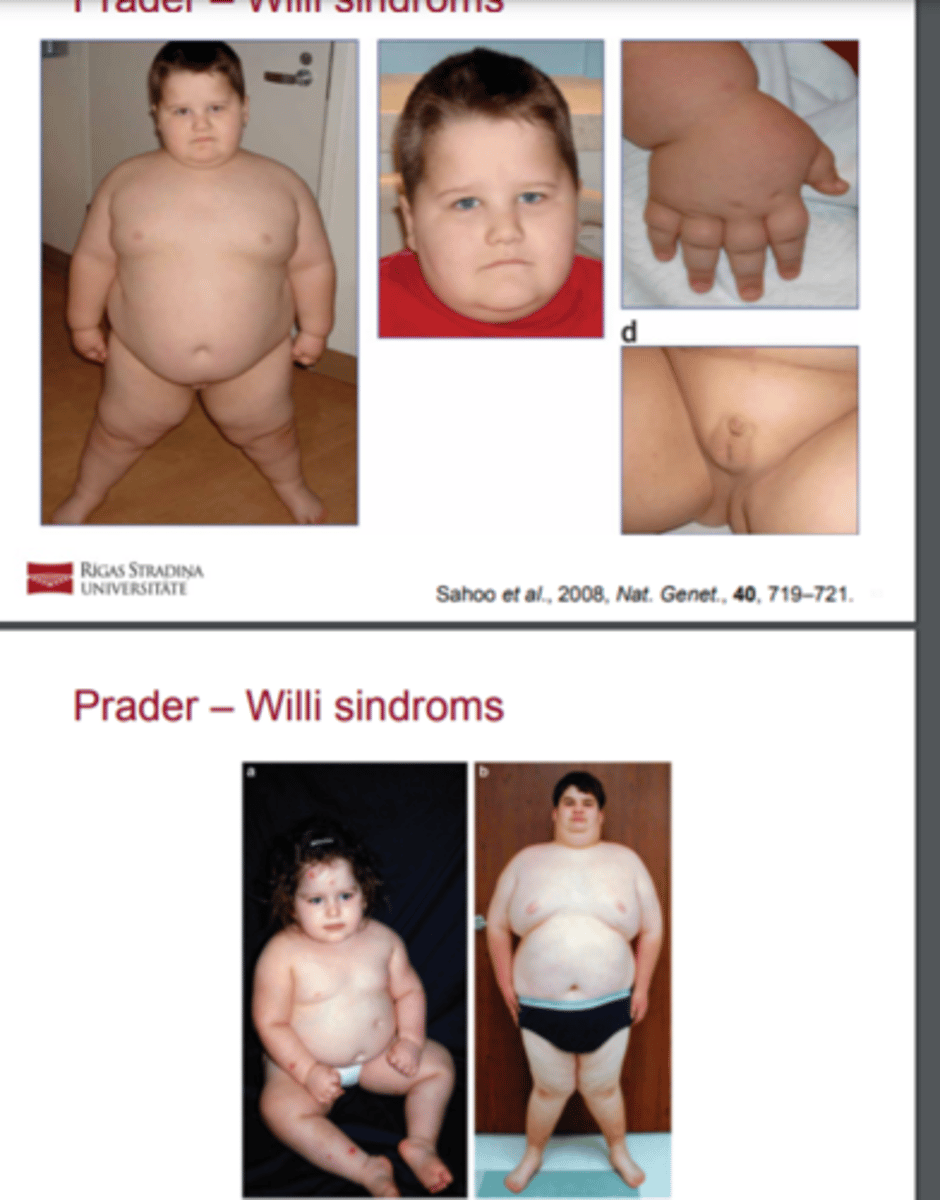
Prader - Willi sindroms - pazīmes
letarģija un hipotonija, kas izraisa ēšanas un attīstības traucējumus, attīstības un intelektuāli traucējumi, hipogonādisms (mazi ārējie dzimumorgāni un pubertitātes nepietiekamība), hiperfāgija, kas noved pie patoloģiskas aptaukošanās, ja tā netiek kontrolēta; īss augums, raksturīgās sejas pazīmes un ķermeņa izskats, un tipisks biheiviorāls fenotips, kurā ietilpst dusmu lēkmes un kompulsīvas iezīmes
Prader - Willi sindroms - Kad parasti izzūd ēšanas traucējumi? Un kad parādās psiholoģiskas + somatiskas problēmas?
Ēšanas traucējumi parasti izzūd ap 6 mēnešu vecumu. Sākot ar 1 2 - 1 8 mēnešu vecumu sākas nekontrolējama hiperfāgija, kas izraisa nopietnas somatiskas un psiholoģiskas problēmas.
Prader - Willi sindroms - alēliskais variants
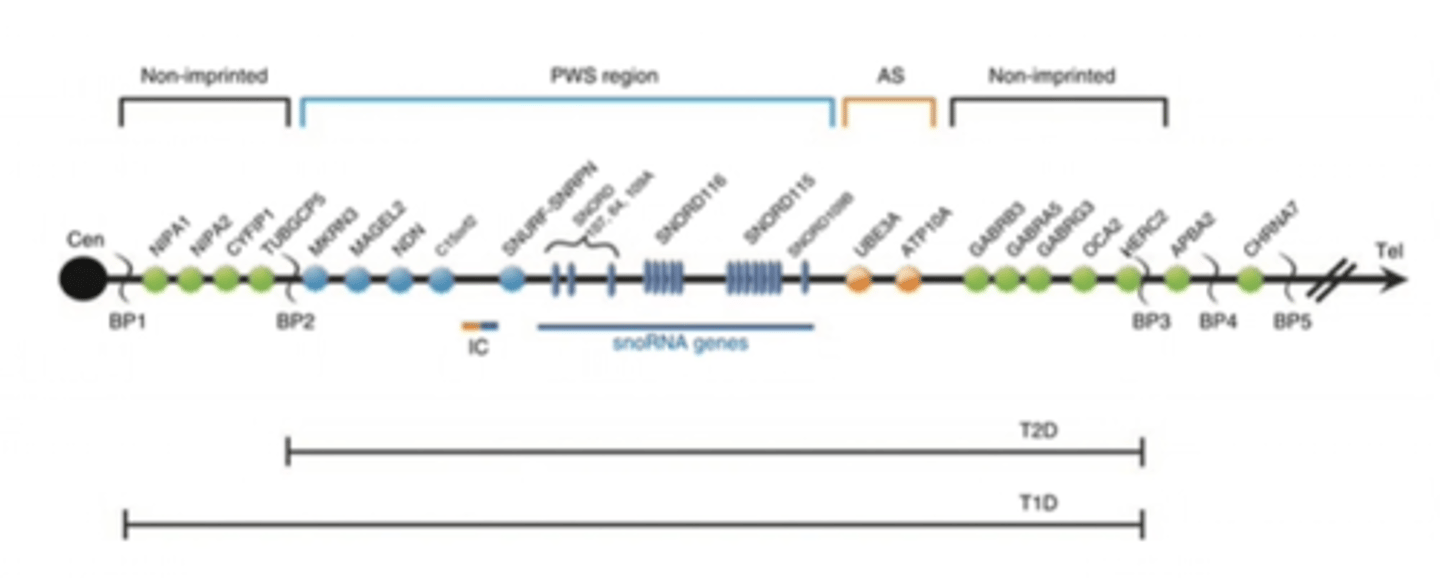
ir PWS kritiskais rajons 15q11.2-q13
PWS rajons - gēni zilie ir imprintēti - tie ekspresējas tikai no paternālās hromosomas
----------
BT - break point - tur notiek delēcijas
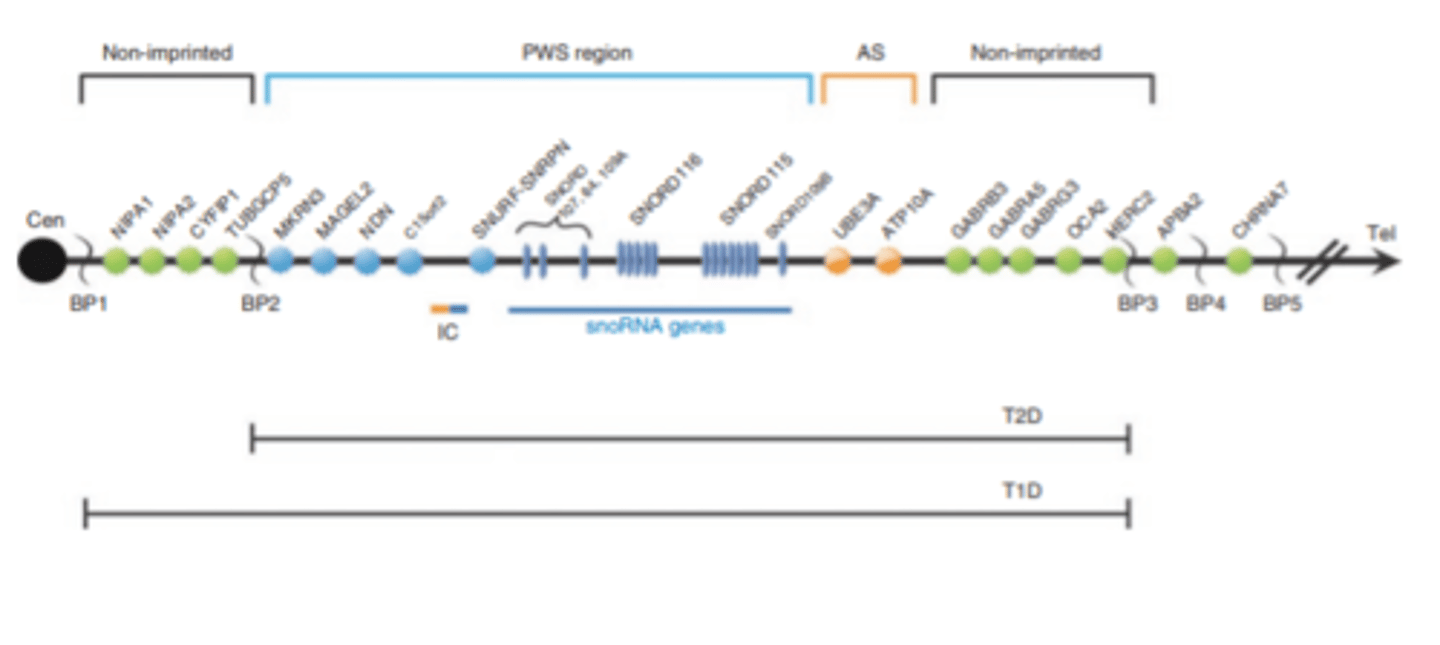
Prader - Willi sindroms - dažiem PWS pacientiem tika noteiktas mazākas atipiskas delēcijas - ko tās ļāva samazināt?
Dažiem PWS pacientiem tika noteiktas mazākas atipiskas delēcijas, kas ļāva samazināt PWS kritisko reģionu līdz 91 kb, kas iekļāva SNORD109A, SNORD116 un IPW20 gēnus.
Lai gan nav zināmi PWS pacienti tikai ar SNORD116 gēna delēciju, peļu modeļi parādīja, ka Snord116 delēcija ir pietiekama, lai izraisītu PWS
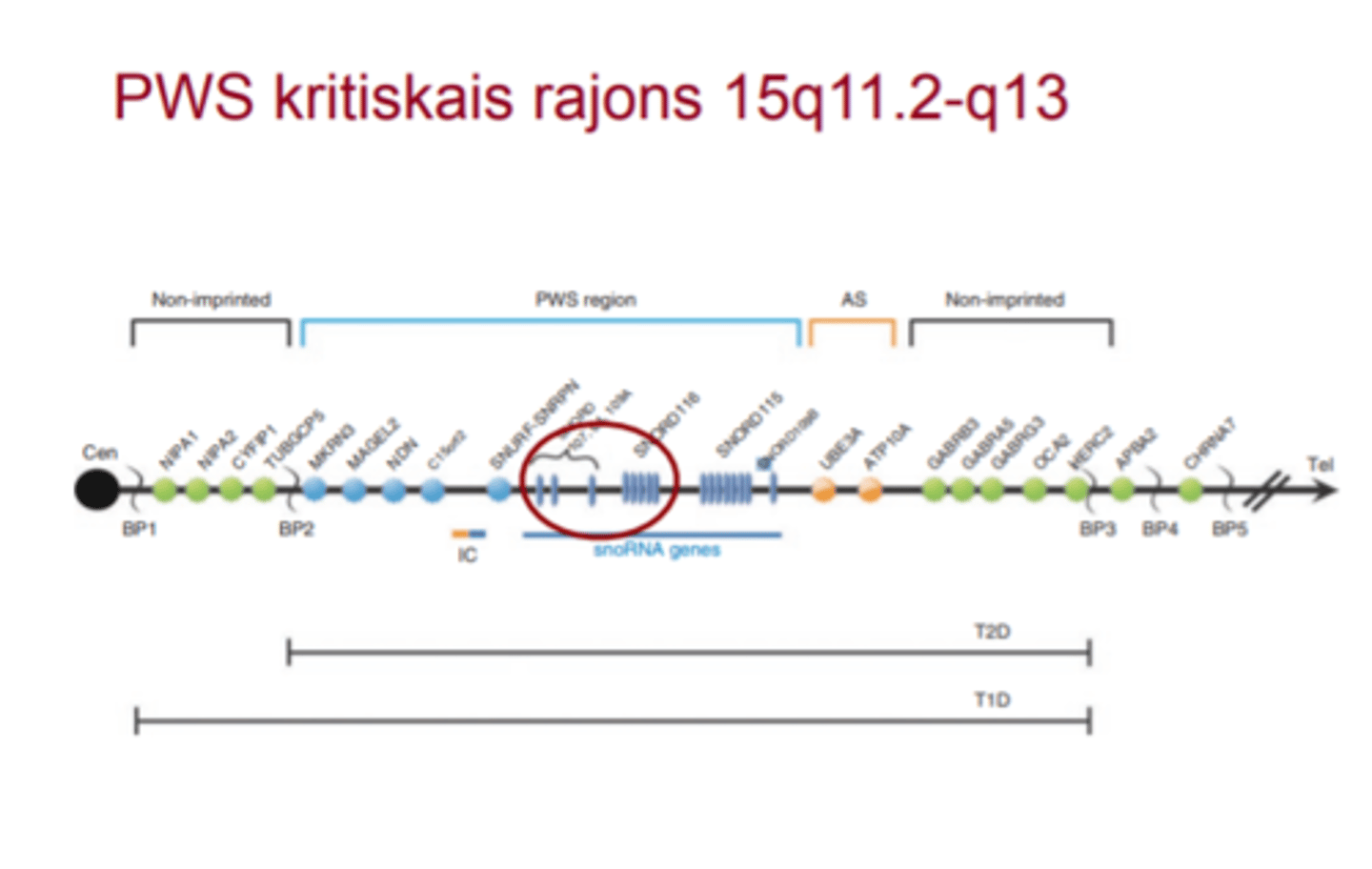
Prader - Willi sindroms - ko kodē snoRNS gēni?
snoRNA gēni kodē mazas kodola RNS, kuras piedalās DNS metilēšanas regulācijā un ir ar augstu ekspresiju smadzenēs
UBE3A - kā darbojas?
UBE3A darbojas gan kā E3 ligāze proteīnu degradācijā, gan kā transkripcijas koaktivators.
UBE3A - - ko izraisa tā funkcijas zudums vai pārmērīga ekspresija?
Ube3a funkcijas zudums vai tā pārmērīgā ekspresija izraisīja dendrītu zarojumu samazināšanos periferālās nervu sistēmas neironos drozofilās.
Paaugstināts Ube3a daudzums negatīvi regulēja glutamāterģiskās sistēmas komponenti Cbln1, kas ir nepieciešama peļu socalizācijai.
Prader - Willi sindroms - 3 varianti, kas gēnu līmenī mainās un izraisa
»5 - 7 Mb delēcija paterāli
pārmantotā hromosomā 15q11.2 - q13 rajonā
»maternāla uniparentāla disomija 15
»Imprintinga defekts paternāli
pārmantotā 15q11.2 - q13 rajonā
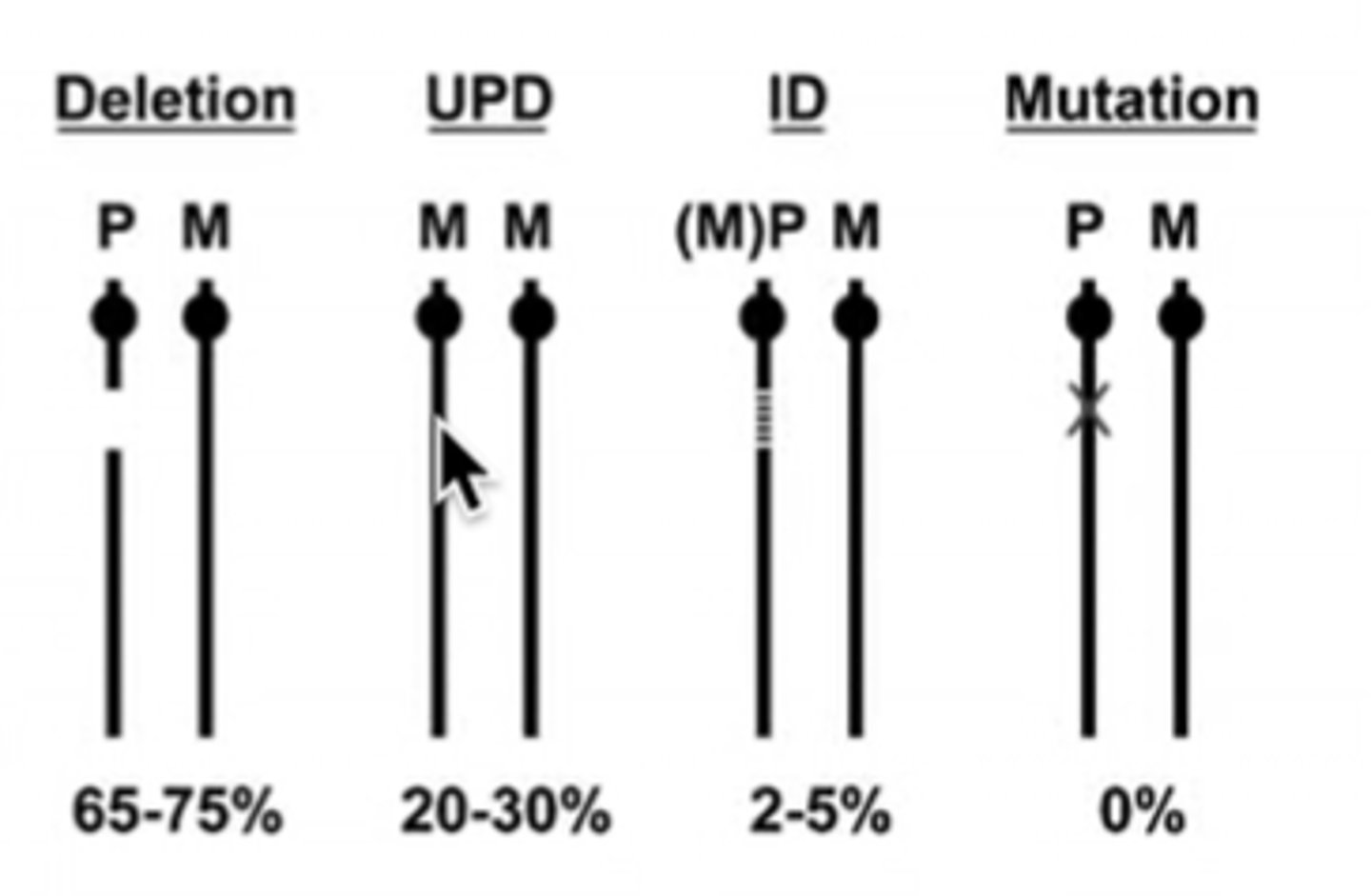
Imprintinga defekts - normal et PWS
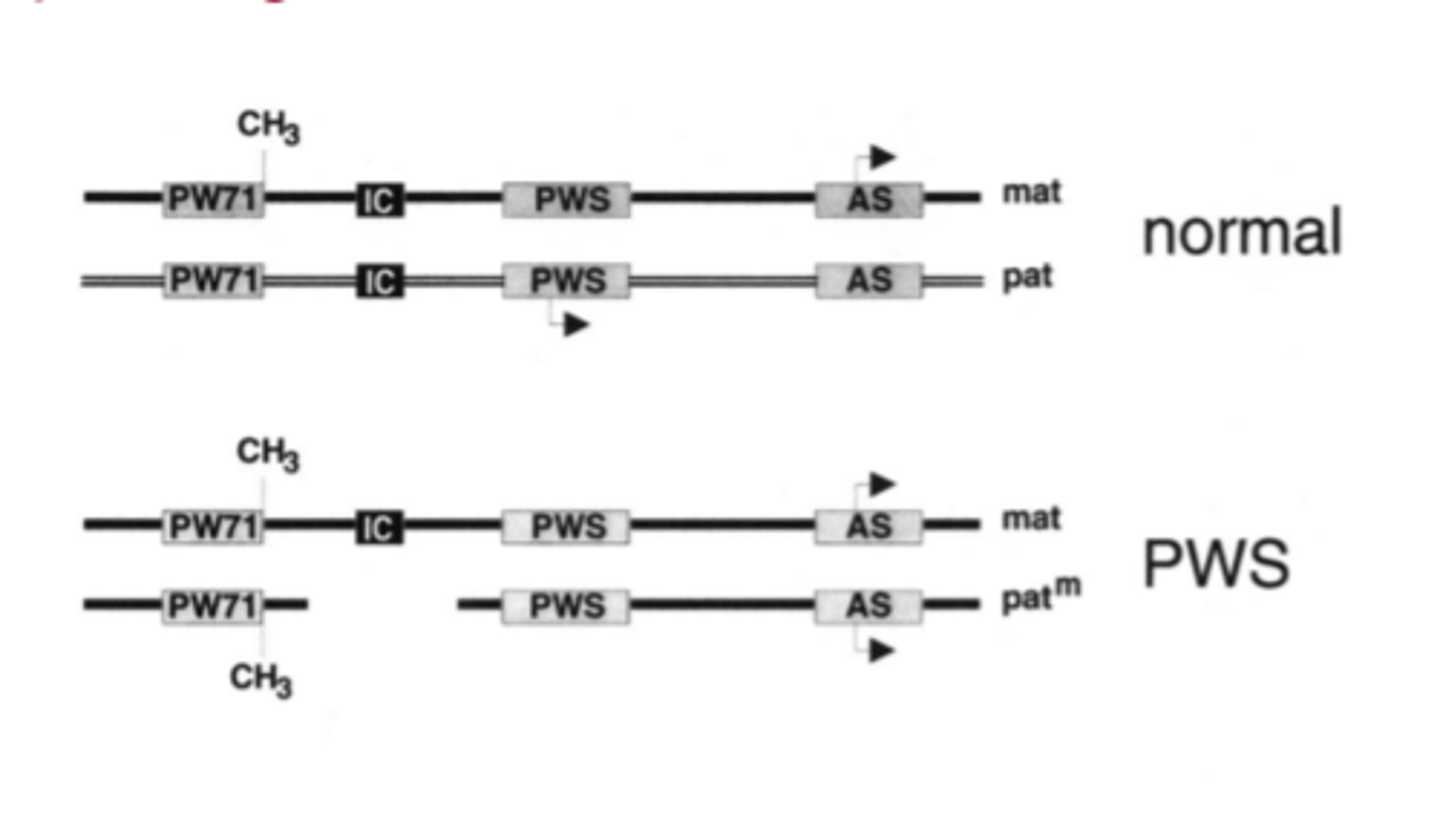
Prader - Willi sindroms summary

Angelman sindroms (PWS pretējs) - izpausmes
Pie AS ir smaga garīga atpalicība, nav runas, ir periodi ar neadekvātiem smiekliem, mikrocefālija, makrostomija, augšžokļa hipoplāzija, uz priekšu vērsts žoklis, neiroloģiskas problēmas ar lellei līdzīgu gaitu, ataksiju un epilepsijas lēkmēm ar specifiskām EEG izmaiņām. Pacientiem ir priecīga un aizrautīga personība.

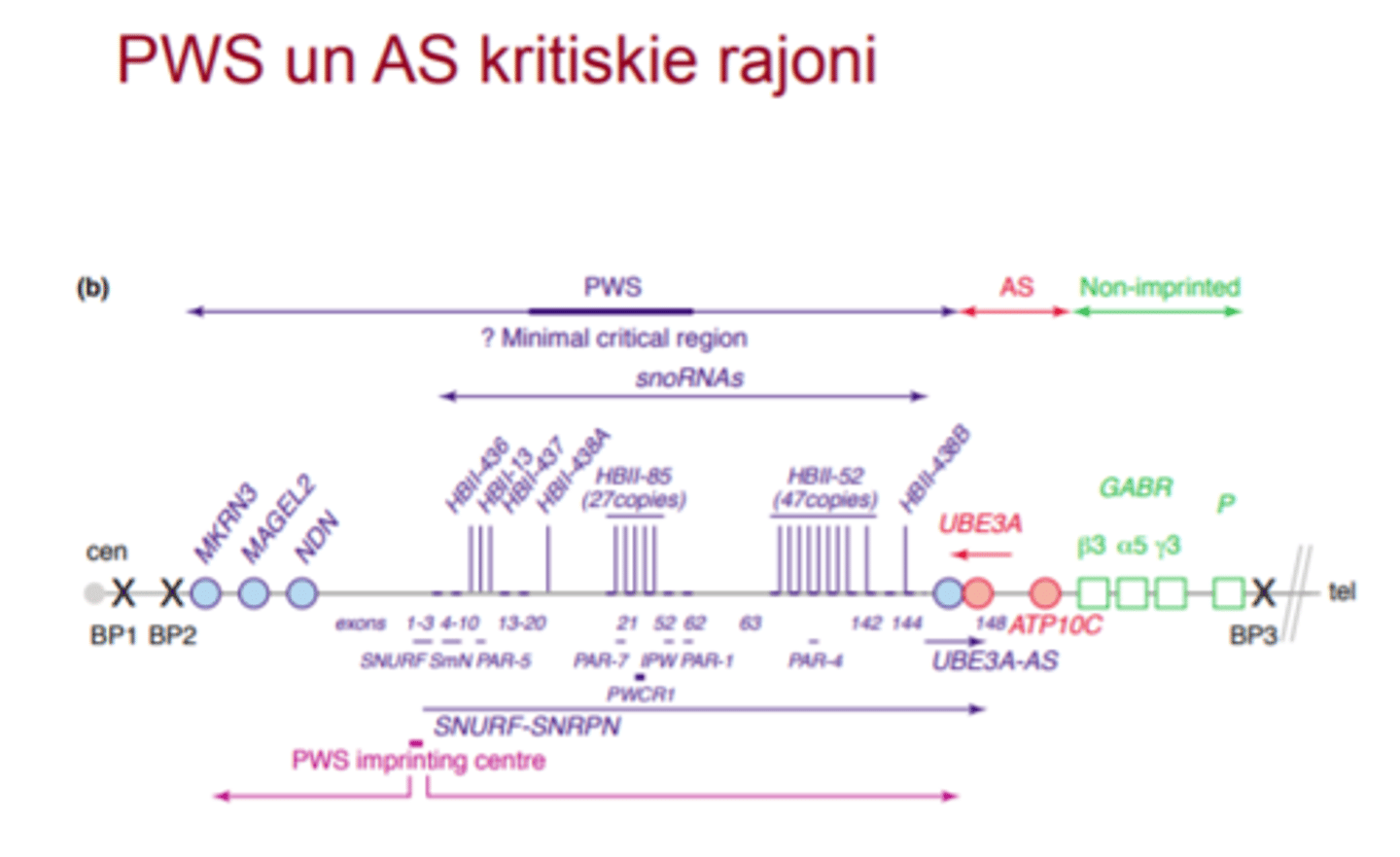
15q11.2-q13 rajona ģenētiskā karte Angelman sindroms
pārmanto no mātes
UBE3A ekspresējas tikai no mātes - ja tas ir deletēts, tad ir Angelman sindroms
UBE3A (atkal)

Rett syndrome and Angelman syndrome - kopīgas īpašības
Rett syndrome and Angelman syndrome share overlapping clinical features with autism including developmental delay, language impairment, seizures and stereotypic behaviors.
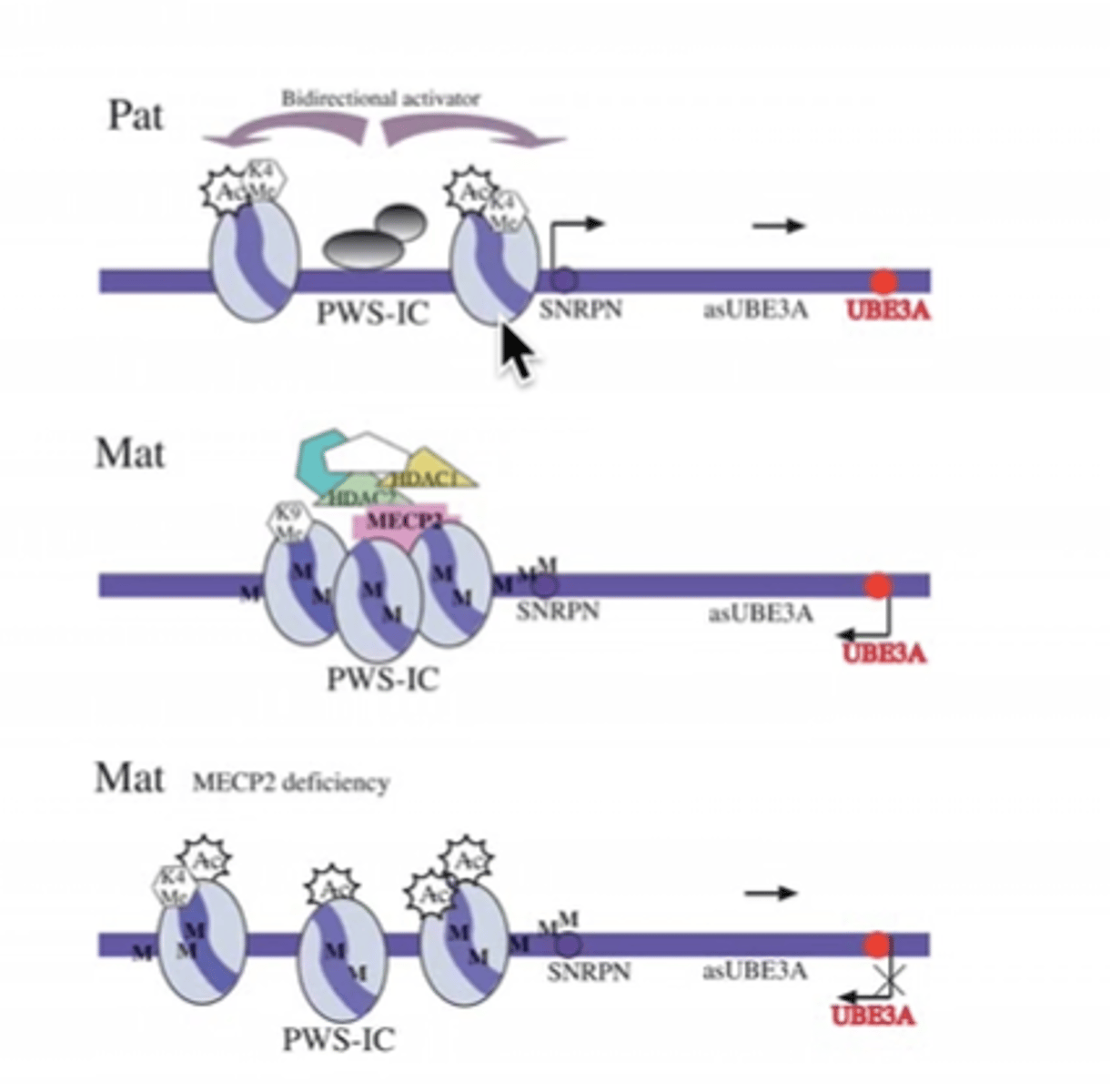
UBE3A blakus ir UBE3A antisensa gēns, kas tikai no paternālās hromosomas ekspresējas un nomāc UBE3A ekspresiju
UBE3A ekspresijas regulācija
UBE3A (tas tikai no maternālās ekspresējas) blakus ir UBE3A antisensa gēns, kas tikai no paternālās hromosomas ekspresējas un nomāc UBE3A ekspresiju
asUBE3A no paternālās ekspresējas - nomāc UBE3A ekspresiju no paternālās.
Maternālajā neekspresējas asUBE3A. Ekspresējas UBE3A. Norm. fenotips.
Angelman sindromu izraisa:
» 5 - 7 Mb delēcija maternāli pārmantotā hromosomā 15q11.2 -q13 rajonā
»paternālā uniparentāla disomija 15
»imprintinga defekts paternāli pārmantotā 15q11.2 - q13 rajonā
»UBE3A gēna patogēnais variants (ja no mātes pārmantots)

Angelman sindroms summary

Ārējas vides ietekme uz epiģenētisku regulāciju
Two different scenarios are illustrated (embryonic/ lactation stages and adult life), in which different factors associated with safe or harmful environment can determine alterations during life development. The effects in adulthood of alterations occurring in early stages are also shown.
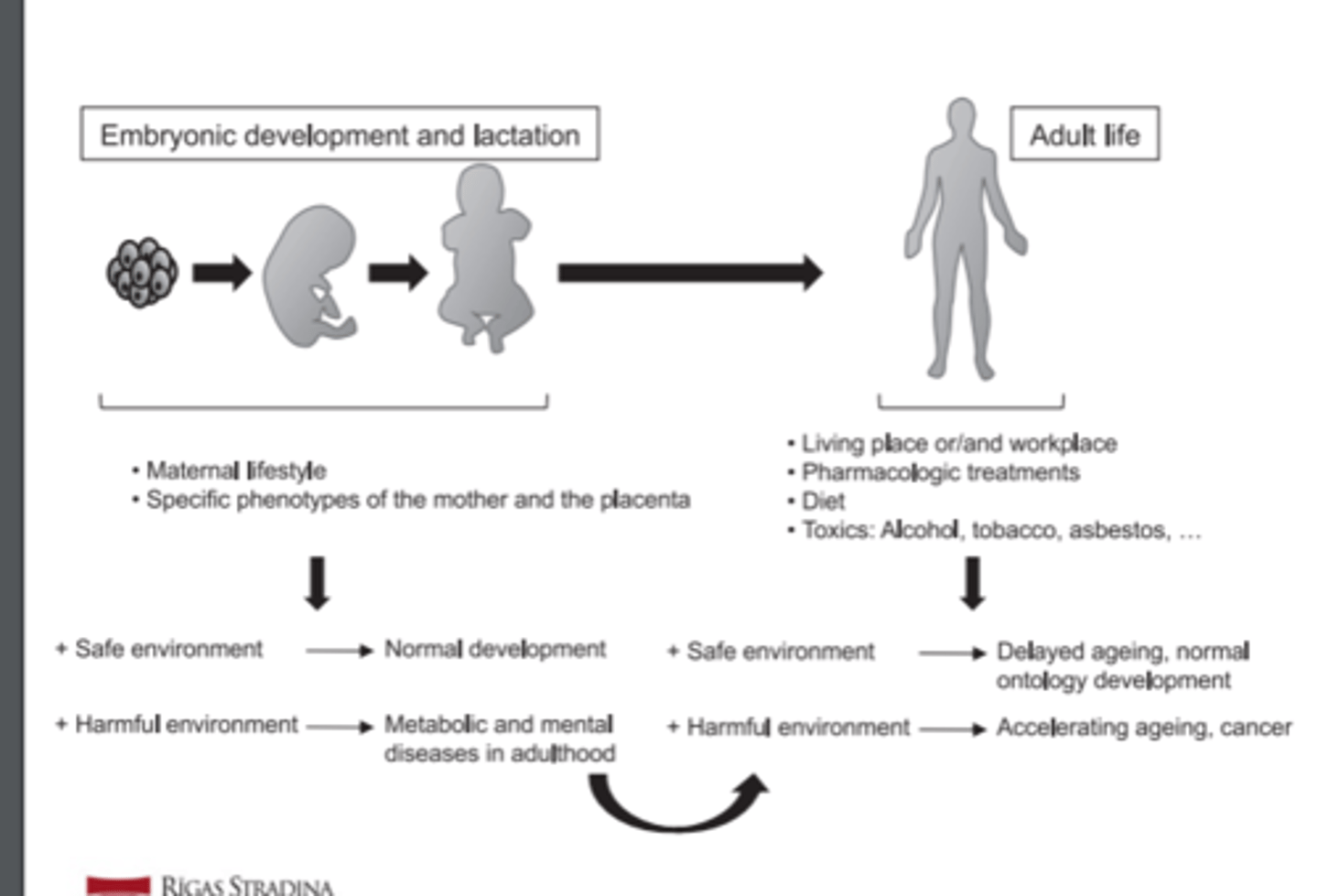
Ārējas vides faktori, kuri ietekmē epiģenētiskās izmaiņas

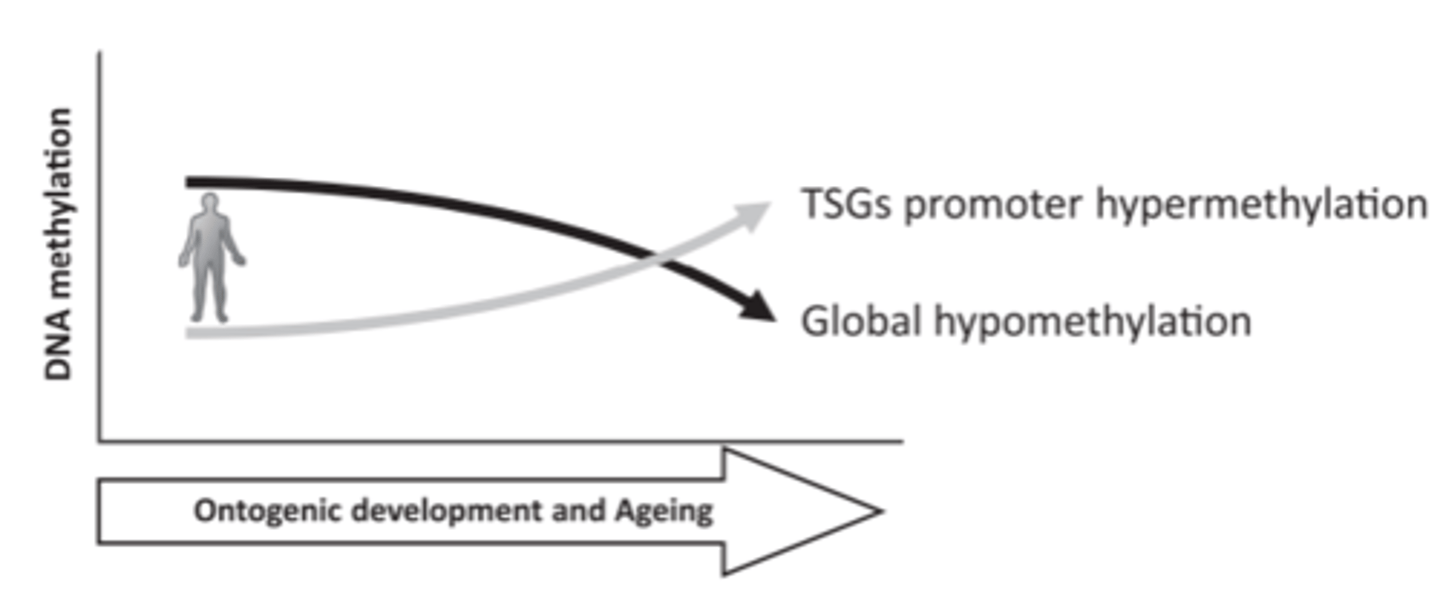
DNS metilēšanas izmaiņas un novecošana
DNS metilācija mainās ontogēnas attīstības un novecošanās laikā. Daudzos audu tipos globālā DNS metilācija samazinās ar vecumu, savukārt vairāki specifiski genoma DNS reģioni kļūst hipermetilēti. TSG, audzēju nomācoši gēni.