(L21) Metabolism of acylglycerol and sphingolipids
1/29
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
30 Terms
acylglycerols
-constitute majority of lipids in body
-play important role in lipid transport and storage and in various diseases such as obesity, diabetes, and hyperlipoproteinemia
triacylglycerols (triglycerides)
major lipids in fat deposits and in food
-glycerol is a simple, 3-carbon chain molecule with hydroxyl group bonded to each carbon atoms
-hydroxyl groups of glycerol react with carboxylic acid groups of fatty acid chains to form acylglycerols
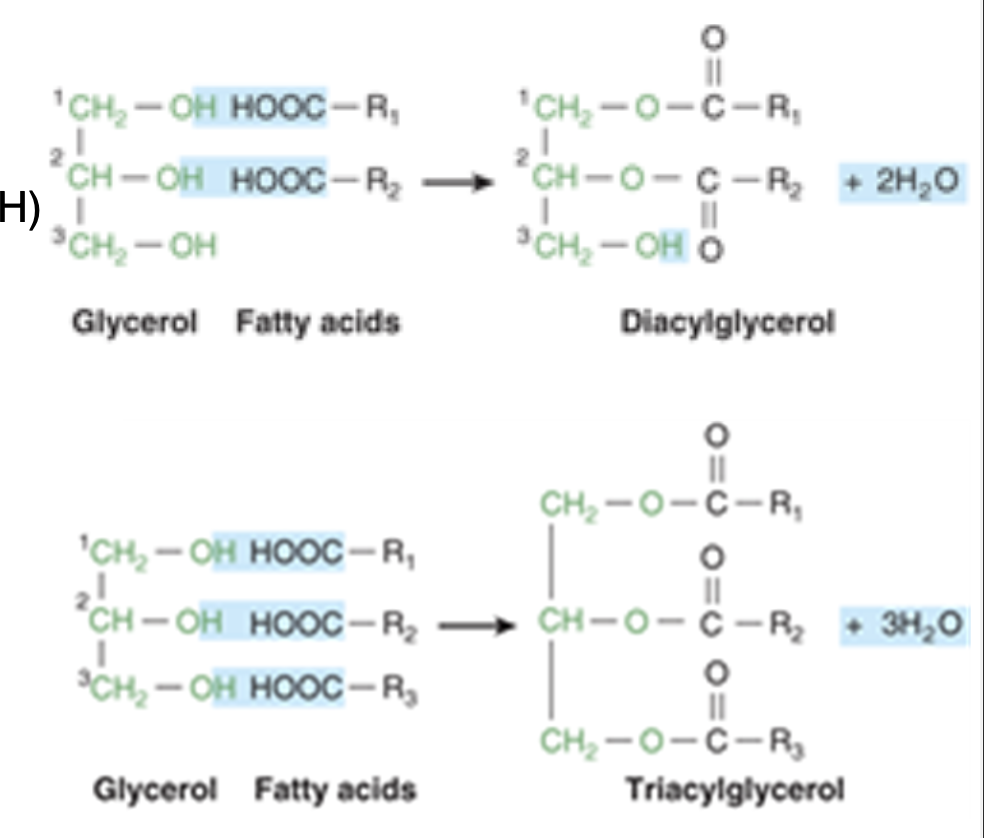
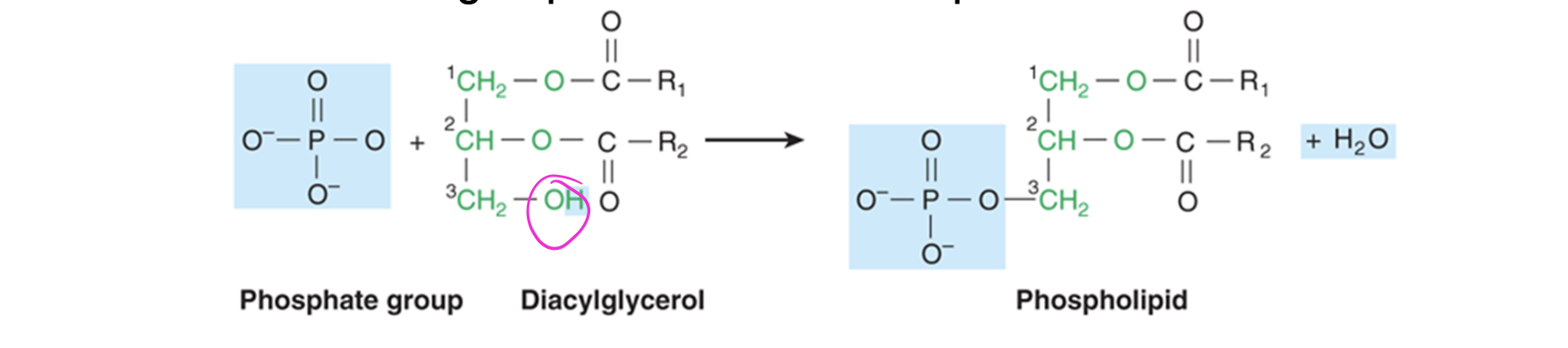
phospholipids
major constituents of biological membranes
-phosphate group attached to third glycerol carbon
phosphoglycerol/phosphoglyceride + fatty acid = phospholipid
-usually, additional molecule is attached to phosphate moiety (resulting in final head group of lipid molecule—> usually charged creating hydrophilic part)
triacylglycerol synthesis
dietary triacylglycerols are hydrolyzed by lipases to constituent fatty acids and glycerols before further catabolism can proceed
-salivary lipases, pancreatic lipases, hepatic lipases, hormone sensitive lipases mobile fatty acids from adipose tissue
free fatty acid taken up tissues (liver, heart, kidney, muscle, lung, testis, adipose tissue)
-oxidized to obtain energy or re-esterified
-brain does not readily uptake FFA
utilization of glycerol depends on whether such tissues have enzyme glycerol kinase
triacylglycerol and phosphoglycerol biosynthesis
-mainly take place in smooth ER of liver but can also be generated in adipose tissues (small extent in intestinal epithelial cells)
-starting molecule is glycerol-3-phosphate (monoacylglycerol in intestinal)
-enzymatic rxns catalyzed by triacylglycerol synthase enzyme complex
triacylglycerol and phosphoglycerol biosynthesis continued
-in adipose tissue and liver during fed state, glucose taken into adipocytes and used to generate dihydroxy-acetone phosphate (reduced to glycerol-3-phosphate)
-in hepatocytes, glycerol-3-phosphate can be made by phosphorylating glycerol using glycerol kinase (NOT expressed in adipocytes) or from glucose using dihydroxy-acetone phosphate
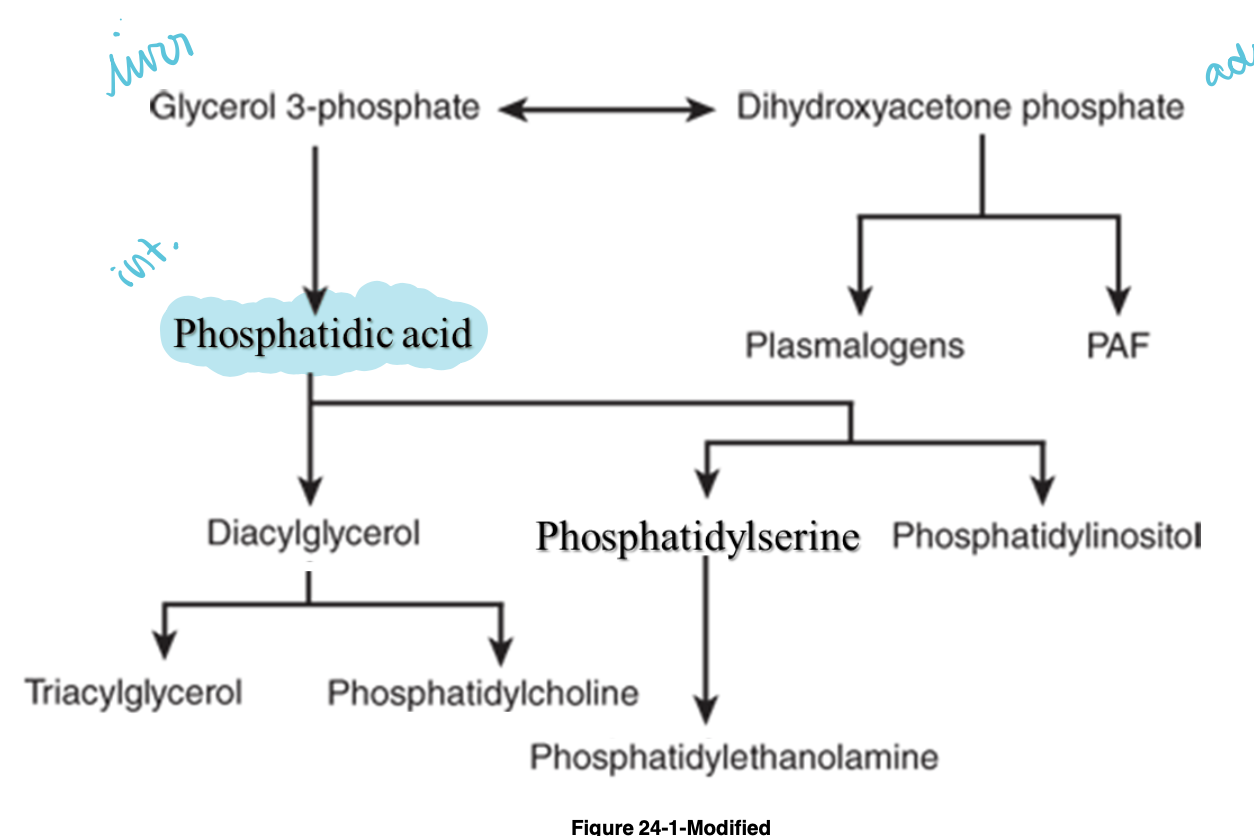
-phosphatidic acid - intermediate common to synthesis
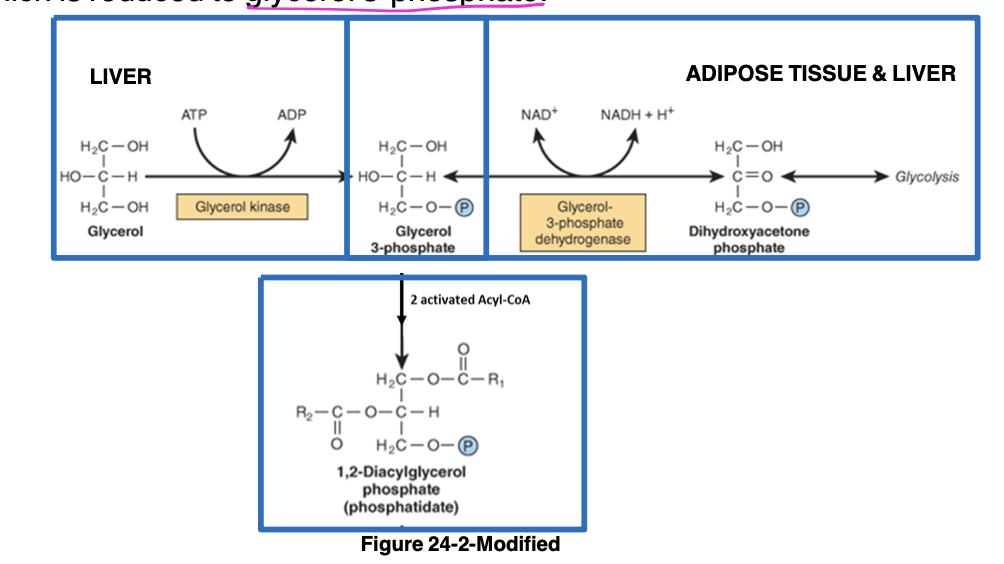
triacylglycerol and phosphoglycerol biosynthesis continued 2
-phosphatidic acid converted to 1,2-diacylglycerol and then triacylglycerol
-in intestinal mucosa, monoacylglycerol acyltransferase converts monoacylglycerol to 1,2-diacylglycerol
-activated cytidine disphosphate intermediate binds at phosphate group of 1,2-diacylglycerol to form CDP-diacylglycerol (branching point)
triacylglycerol and phosphoglycerol biosynthesis continued 3
-cytidine diphosphate in CDP-diaclyglycerol is replaced by serine or inositol to form phosphatidyl serine and inositol
-phosphatidyl ethanolamine is formed by loss of COO- group from serine
-phosphatidyl choline derived from dietary sources by ATP-driven phosphorylation of head group, attachment to CTP, and final attachment to diacylglycerol
glycerol ether phospholipids
-glycerol attached to hydrocarbon chain by ether linkage rather than ester bond
-plasmalogens and platelet-activating factor (PAF) are important examples of this type of lipid
-dihydroxyacetone phosphate = precursor of glycerol moiety
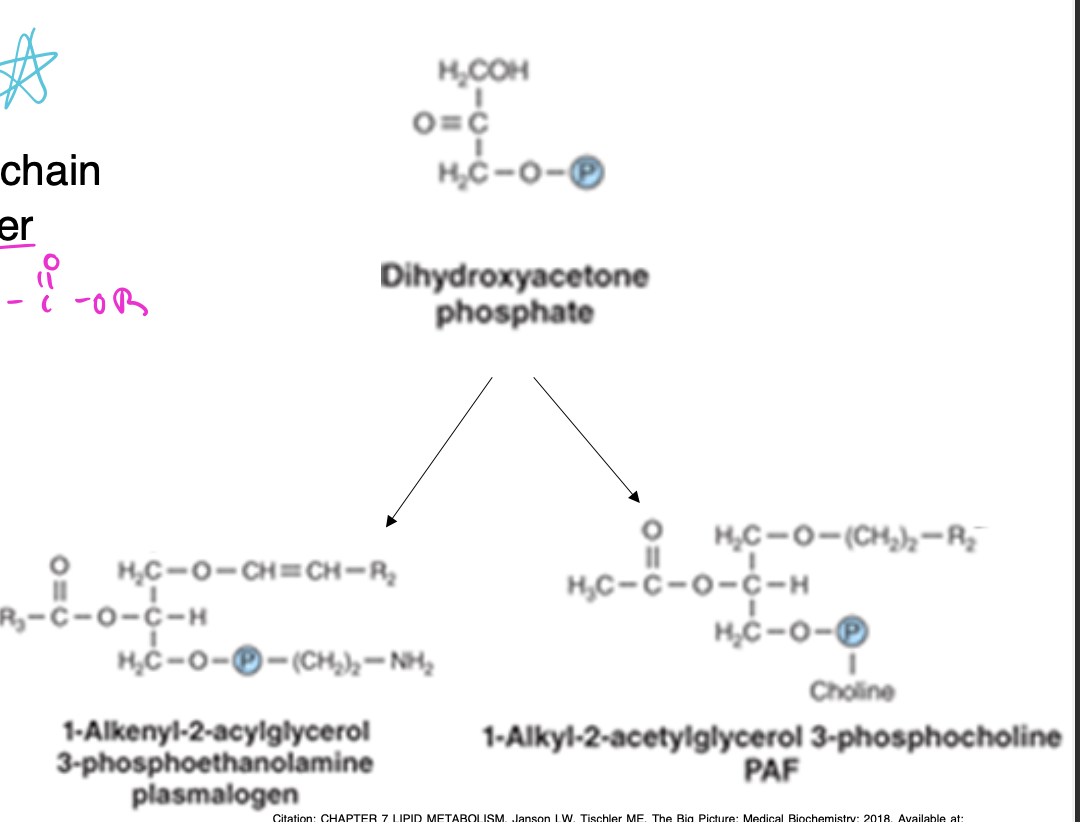
summary of acylglycerol biosynthesis
triacylglycerol breakdown
-hydrolysis begins in mouth and stomach where lingual and gastric lipases (produced in back of mouth and stomach) preferentially hydrolyze short and medium chain fatty acids from dietary triglycerides
-as chyme moves to small intestine, it is emulsified with bile salts from gall bladder (partly ingested, gallbladder stores and concentrates bile secreted by liver)
-pancreatic lipase hydrolyzes fatty acids from carbons 1 and 3 of triglyceride's glycerol backbone (hydrolyze to FFA and monoacylglycerol)
-FFA and monoacylglycerol taken up by intestinal epithelial cells, re-esterified and incorporated into chylomicrons
phospholipids breakdown
-phospholipases catalyzes hydrolysis of glycerophospholipids to form FFA
-phospholipases A1, A2, B, C, D attack different bonds in glycerophospholipid
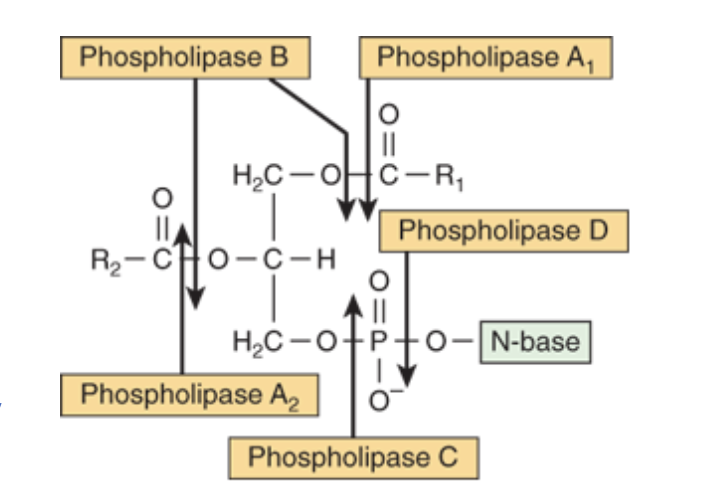
phospholipase A2
pancreatic fluid and snake venom
phospholipase C
one of major toxins secreted by bacteria
phospholipase D
known to be involved in mammalian signal transduction
sphingolipids
-carbs bind to lipids to form glycolipids
-glycerol backbone generally replaced by backbone of sphingosine (made from serine and 16-carbon fatty acid palmitate)
-can bind two other molecules with hydroxyl and amino groups from serine
-amino group always bound to another fatty acid to make ceramide
disease conditions associated with phospholipids and sphingolipids
-lung surfactants prevent alveoli from collapsing (ex: dipalmitoyl-phosphatidylcholine; composed mainly of lipid with some proteins and carbs)
-deficiency of lung surfactant in lungs of many preterm newborns gives rise to infant respiratory distress syndrome (IRDS)
-certain diseases characterized by abnormal quantities of lipids in tissues (often in nervous tissues)
-classified into 2 groups: true demyelinating diseases and sphingolipidoses
demyelinating disease
multiple sclerosis
-loss of both phospholipids (particularly ethanolamine and plasmalogen) and of sphingolipids from white matter
-lipid composition of white matter resembles that of gray matter
-cerebrospinal fluid shows raised phospholipid levels
sphingolipidoses
-lipid/lysosomal storage diseases = inherited
-caused by genetic defect in catabolism of lipids containing sphingosine
-part of larger group of lysosomal disorders
lysosomal storage diseases
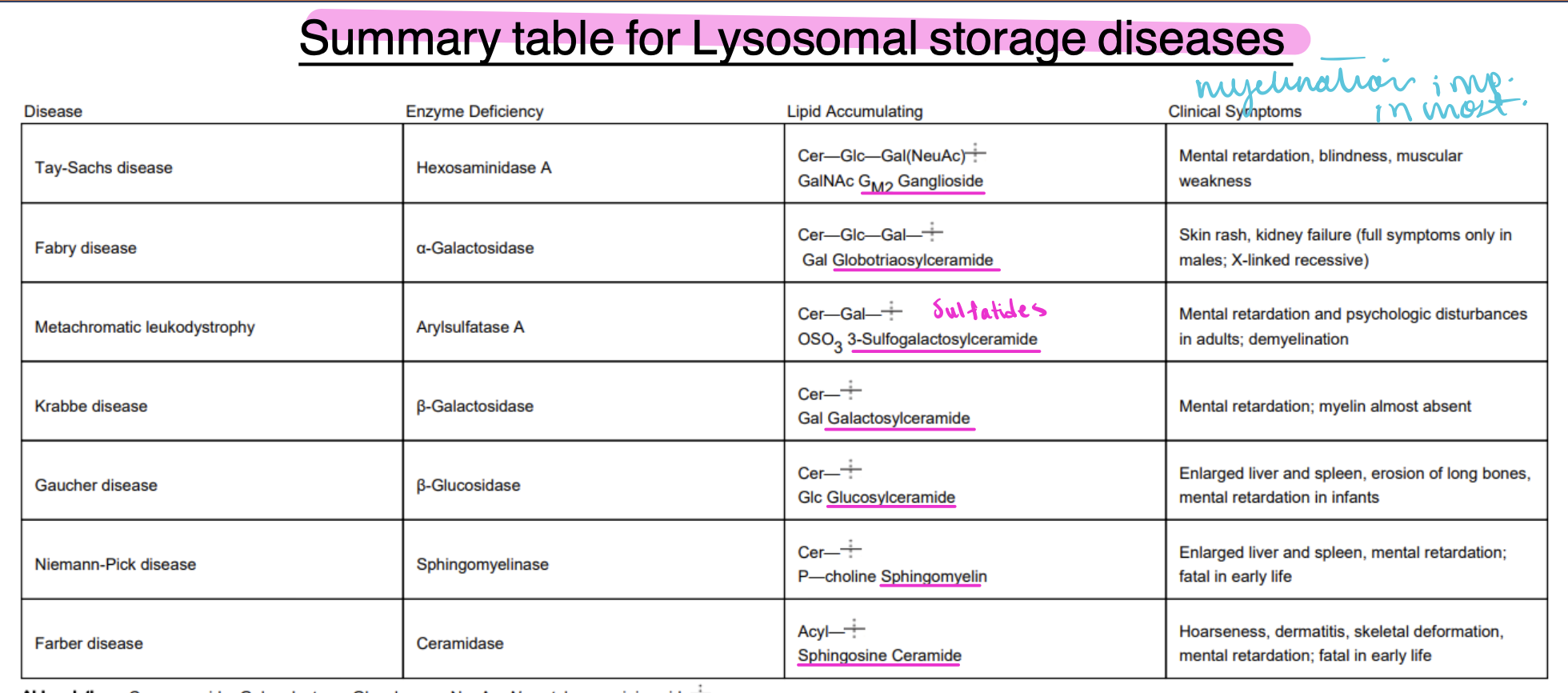
Farber's disease
Niemann-Pick disease
gaucher disease
krabbe disease
metachromatic leukodystrophy
fabry disease
tay-sachs disease
farber's disease
accumulation of sphingosine ceramides to due lack of ceramidase activity, disorders in many organs including CNS
-hoarseness, dermitis, skeletal deformation and mental retardation
-fatal in early life
niemann-pick disease
-type A=neurodegenerative disease
-type B=non-neurologic, visceral form
fatal in early life
accumulation of sphingomyelin due to lack of acid sphingomyelinase activity
enlarged liver and spleen, mental retardation
gaucher disease
glucosylceramides accumulate in spleen, liver, lungs, bone marrow, and brain due to deficiency of B-glucosidase
-enlarged liver and spleen, mental retardation in infants and erosion of long bones
krabbe disease
accumulation of galactosylceramides due to lack in activity of B-galactosidase (galactosylceramides)
-mental retardation, myelin almost absent
Metachromatic leukodystrophy
accumulation of sulfatides (sulfates galactosylceramides) due to deficiency of arylsulfatase A
-mental retardation and psychological disturbances in adults; demyelination
Fabry disease
X-linked recessive
accumulation of globotriaosylceramide due to lack of galactosidase activity
-skin rash and kidney failure (full symptoms only seen in males)
Tay-Sachs disease
accumulation of ganglioside GM2 due to lack of hexosaminidase A activity
-mental retardation, blindness and muscular weakness
summary of lysosomal storage diseases
constant features of sphingolipidoses
-complex lipids containing ceramide accumulate in cells, particularly neurons, causing neurodegeneration and shortening life span
-rate of synthesis of stored lipid is normal
-enzymatic defect in lysosomal degradation pathway of sphingolipids
-extent to which activity of affected enzyme decreased is similar in all tissues
possible treatment for sphingolipidos
-enzyme replacement therapy and bone marrow transplantation in treatment of Gaucher and Fabry diseases
-substrate deprivation therapy to inhibit synthesis of sphingolipids, chemical chaperone therapy, and gene therapy are currently under investigation