Microbial Genetics Part III - Molecular Diagnosis
1/15
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
16 Terms

history and physical
biological specimen
phenotype-based diagnosis methods

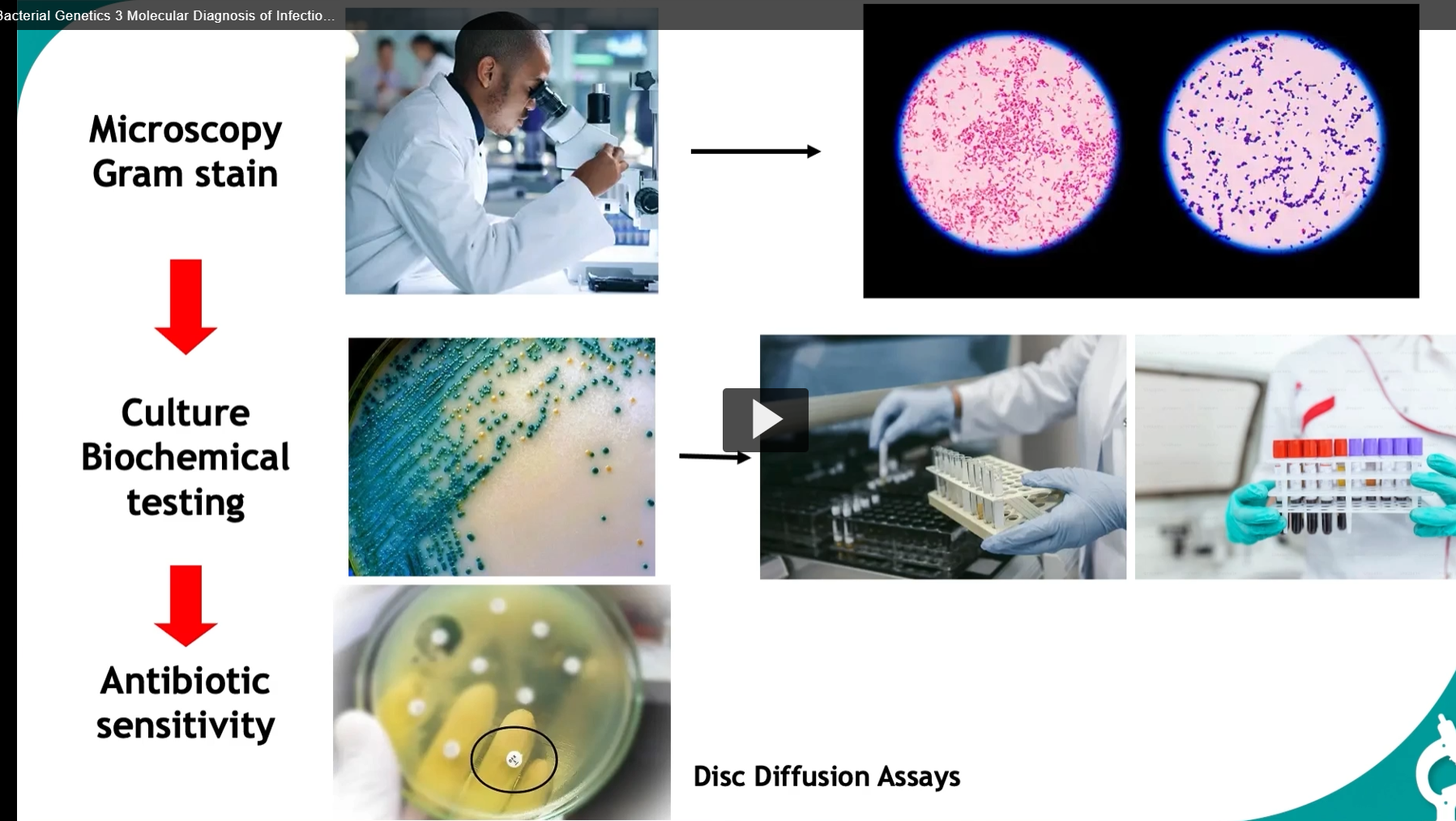
in diagnosing infections, we start with the gram stain:
microscopy gram stain
culture biochemical testing (agar or broth, help reach a definitive diagnosis)
antibiotic sensitivity (uses discs with various antibiotics in the agar plate to find the most effective treatment)
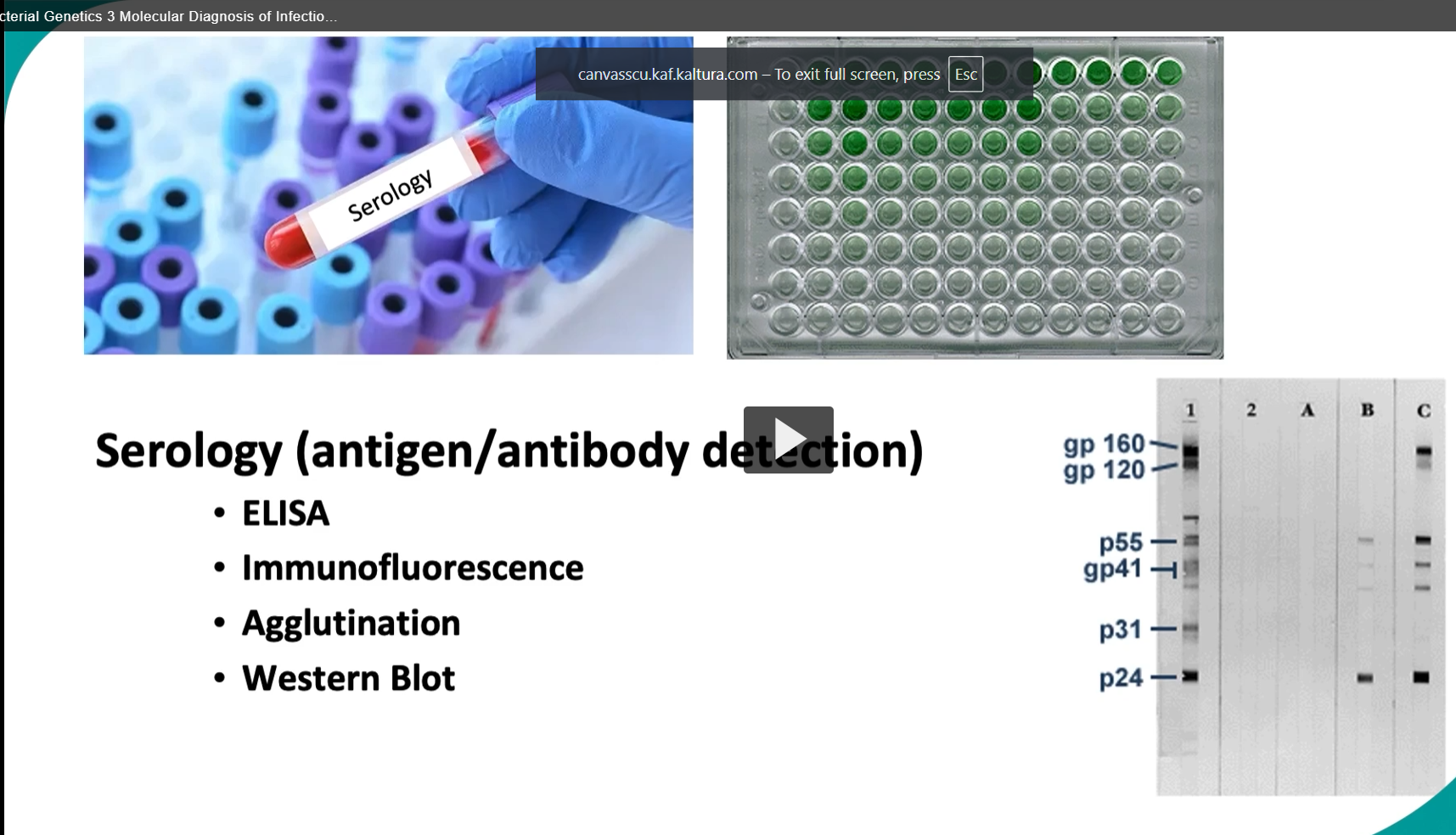
Serology (antigen/antibody detection)
-serology mechanisms for available for identifying infectious organisms.
This approach is based on the detection of antibodies produced in response to an infectious agent, or for the detection of epitopes from circulating antigens from the agent in the patients serum.
Eliza (useful for quantifying the presence of antibodies or antigens in a sample)
Immunofluorescence (can visually demonstrate the presence of specific antigens or antibodies by labeling them with a fluorescent dye.
Agglutination
Use of monoclonal and polyclonal antibodies in Western Blot (western blot is shown here and is particularly used for confirming the presence of proteins associated with certain infections).
In a western blot test, proteins are separated by gel electrophoresis and then transferred to a membrane where they are probed with antibodies that are specific to the target antigen. If the target antigen is present, it will bind to the specific antibody, which can then be detected often through a color change.
ELISA — Definition & Etymology
Definition:
ELISA stands for Enzyme-Linked Immunosorbent Assay.
It is a laboratory technique used to detect and quantify specific antigens or antibodies in a sample (e.g., blood, serum). It works by using antibody–antigen specificity and an enzyme-driven color change as a readout.
Etymology (break it down)
Enzyme
en- = “in”
zyme (Greek zymē) = “ferment, yeast”
→ A molecule that drives biochemical reactions
Linked
→ The enzyme is attached (linked) to an antibodyImmuno-
Latin immunis = “exempt, protected”
→ Refers to the immune system
Sorbent
Latin sorbere = “to absorb or suck in”
→ The assay uses a surface that binds (absorbs) molecules
Assay
Old French assai = “trial, test”
→ A test or analytical procedure
Put together: “A test where immune molecules are captured on a surface and detected using an enzyme-linked signal.”
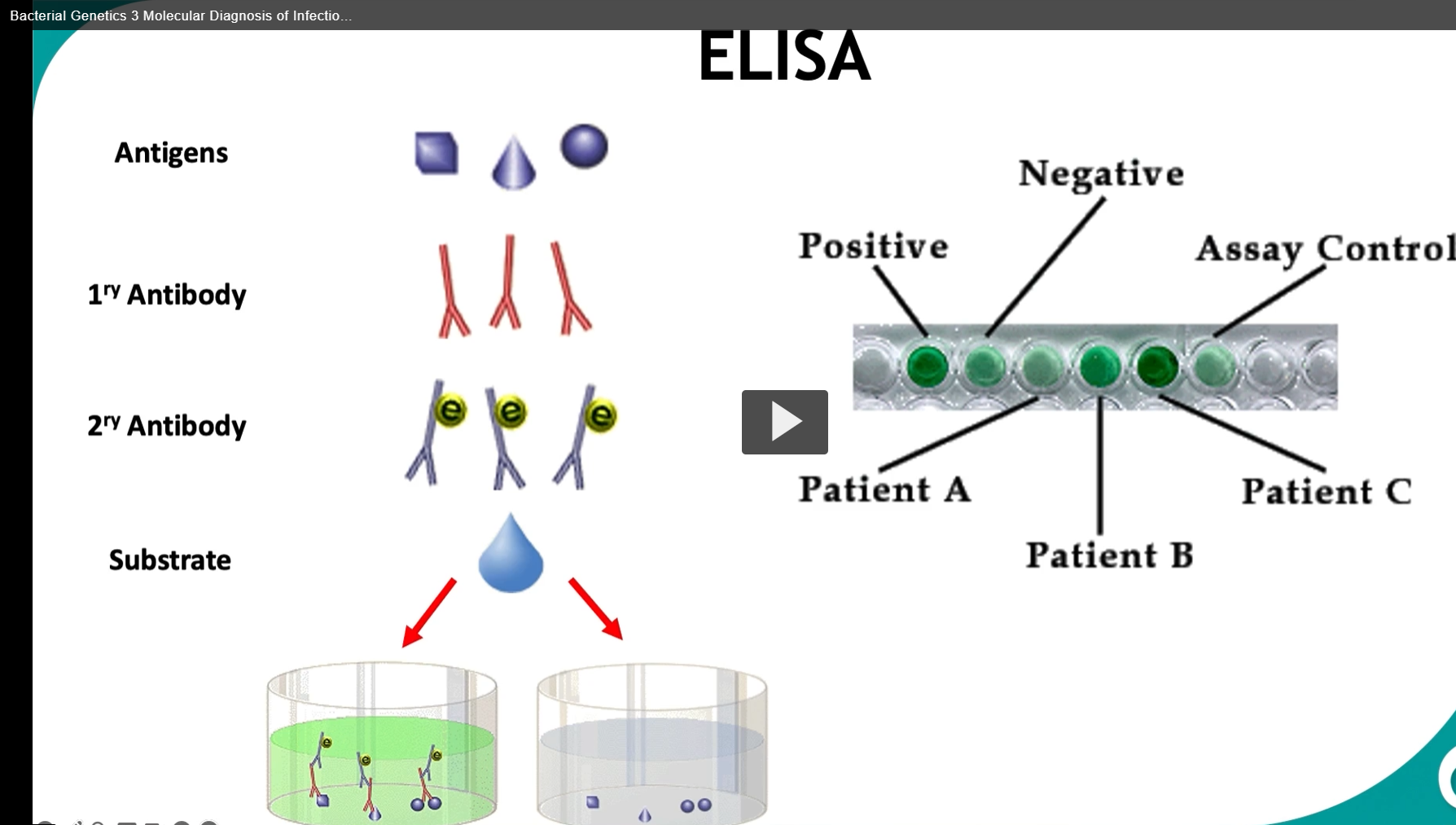
What’s happening in the diagram (step-by-step)
This slide shows a typical indirect ELISA workflow:
1. Antigen is present (top row)
Different shapes = different antigens (targets you’re trying to detect)
2. Primary antibody (1° antibody) binds antigen
The primary antibody is specific to the antigen
If the antigen is present → antibody binds
If not → nothing binds
3. Secondary antibody (2° antibody) binds the primary
The secondary antibody recognizes the primary antibody
It is enzyme-linked (the yellow “e” in the diagram)
This step amplifies the signal (more enzymes per antigen)
4. Substrate is added
A substrate (colorless chemical) is added
The enzyme converts it into a colored product
5. Color change = positive result
Green color → Positive
→ Antigen was present → full antibody chain formed → enzyme reaction occurredClear/no color → Negative
→ No antigen → no enzyme → no reaction
🧪 Interpreting the plate (right side)
Patient A → dark green → strong positive
Patient B → lighter green → weaker positive (lower concentration)
Patient C → no color → negative
Assay control → ensures the test is working properly
Conceptual takeaway (important for exams)
ELISA is essentially: “Convert invisible molecular binding into a visible color signal.”
Specificity → antibody–antigen binding
Sensitivity → enzyme amplification
Readout → color intensity ∝ amount of target
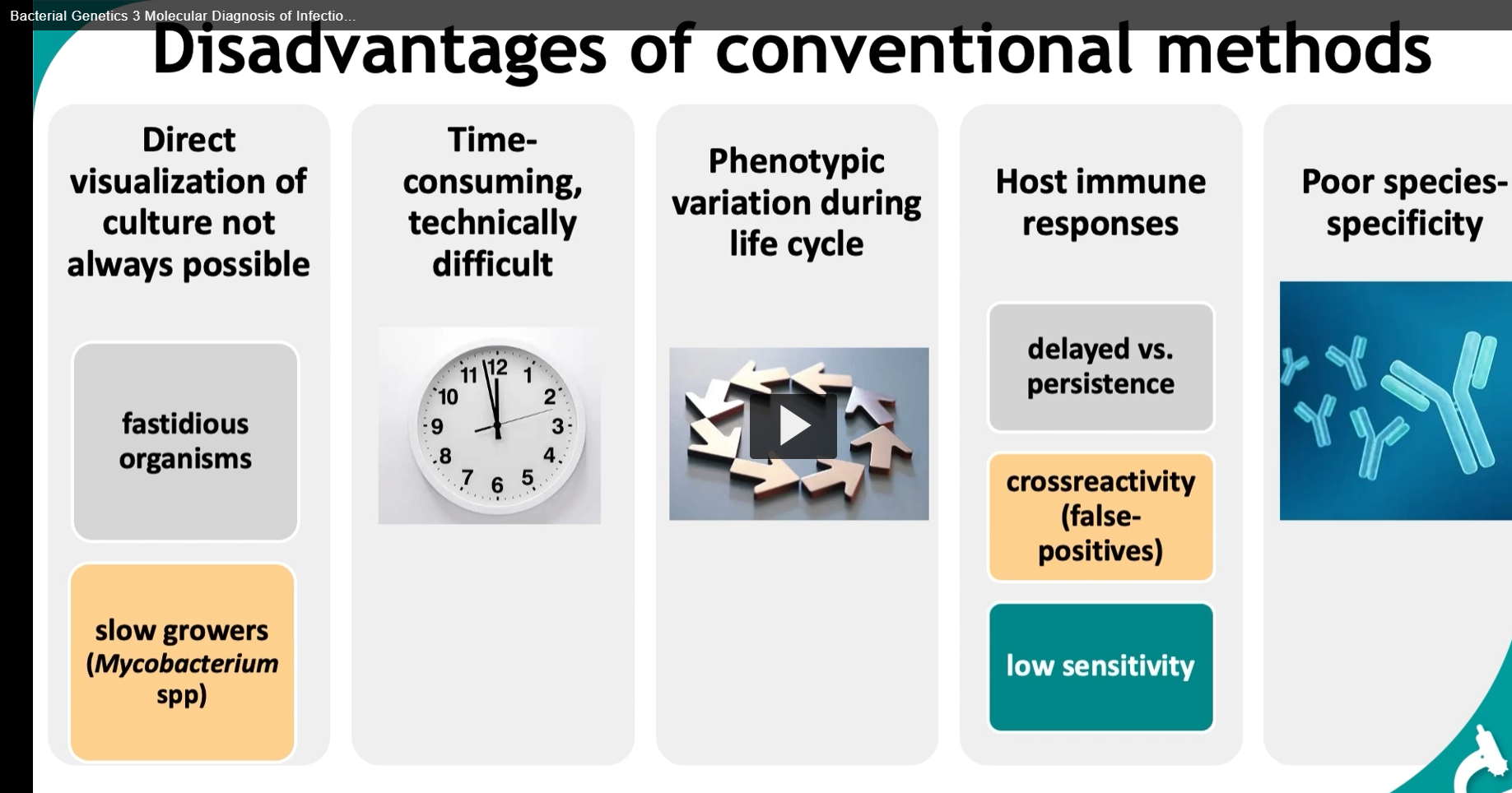
Disadvantages of conventional methods
Direct visualization of culture not always possible
fastidious organisms
slow growers (Mycobacterium spp)
Time-consuming, technically difficult
Phenotypic variation during life cycle
Host immune responses
delayed vs. persistence
crossreactivity (false-positives)
low sensitivity
Poor species-specificity
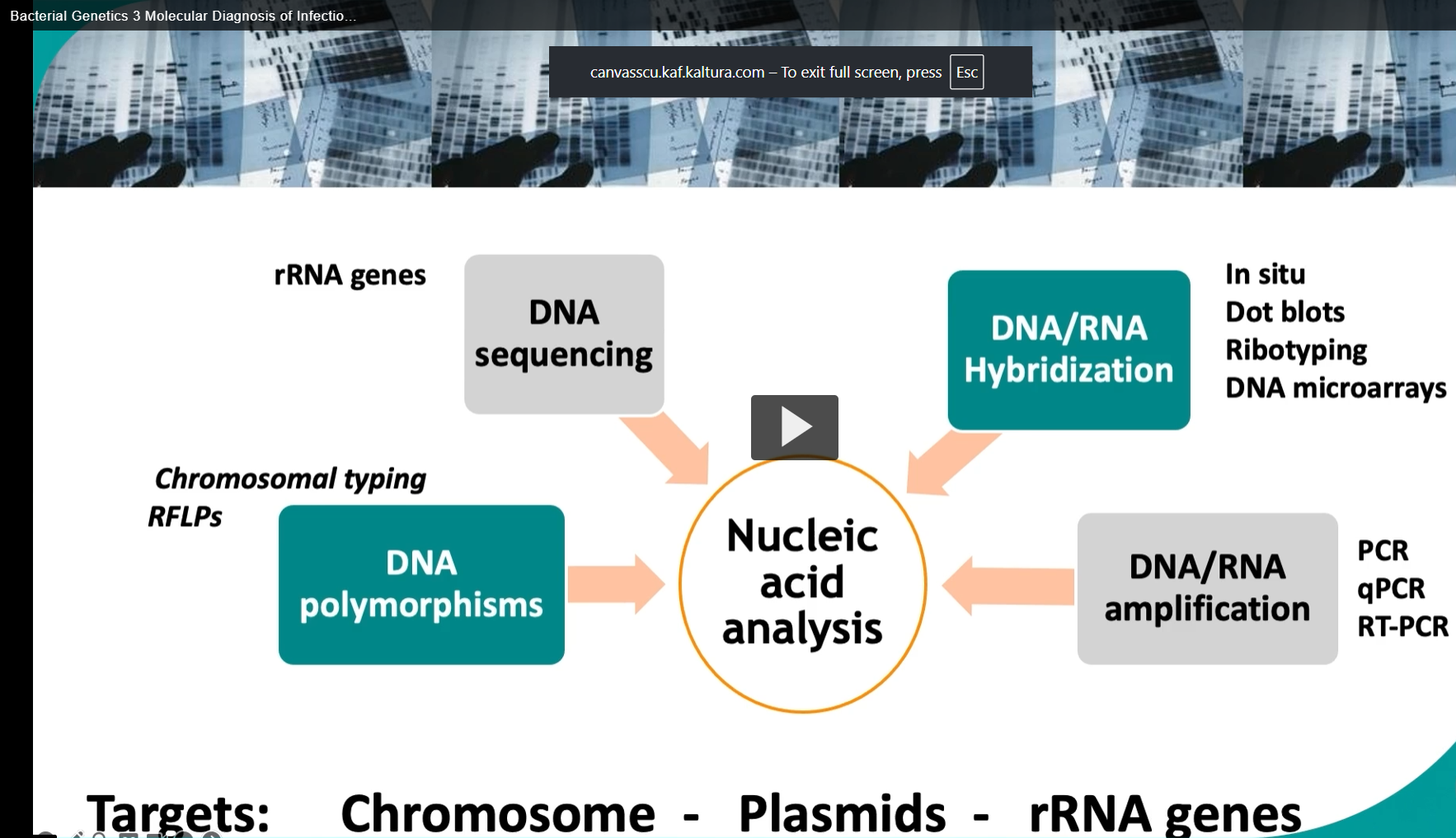
Genotype based tests that analyze DNA or RNA overcome traditional diagnostic limitations by identifying microbial pathogens through genetic variations using methods like restriction fragment length, polymorphism, RFLP, and specific hybridization probes.
DNA sequencing, especially of the 60S ribosomal RNA genes, offers precise organism identification.
For samples with low nucleic acid levels, amplification techniques like PCR or RT PCR boost sensitivity and specificity, enabling the detection of minute quantities of genetic material. Potential targets for these techniques include genomic DNA of the bacterial chromosome, plasmids, such as R factors, and ribosomal RNA genes
By exploiting the unique characteristics of these genetic materials, laboratory professionals can accurately identify microorganisms, understand their resistance mechanisms, and guide the appropriate treatment strategies.
Genotype-based tests: Nucleic acid analysis
Applications
Diagnosis based on detection of specific sequences in microbial pathogens from the patient, environment, or culture samples.
Key applications: Detection of antibiotic resistance plasmids (vital for outbreak management in healthcare settings)
These techniques allow for precise Strain differentiation
Molecular epidemiology of outbreaks, helping trace infections, sources, and spread.
Provide insights of Disease pathogenesis and microbial evolution.
Taxonomy

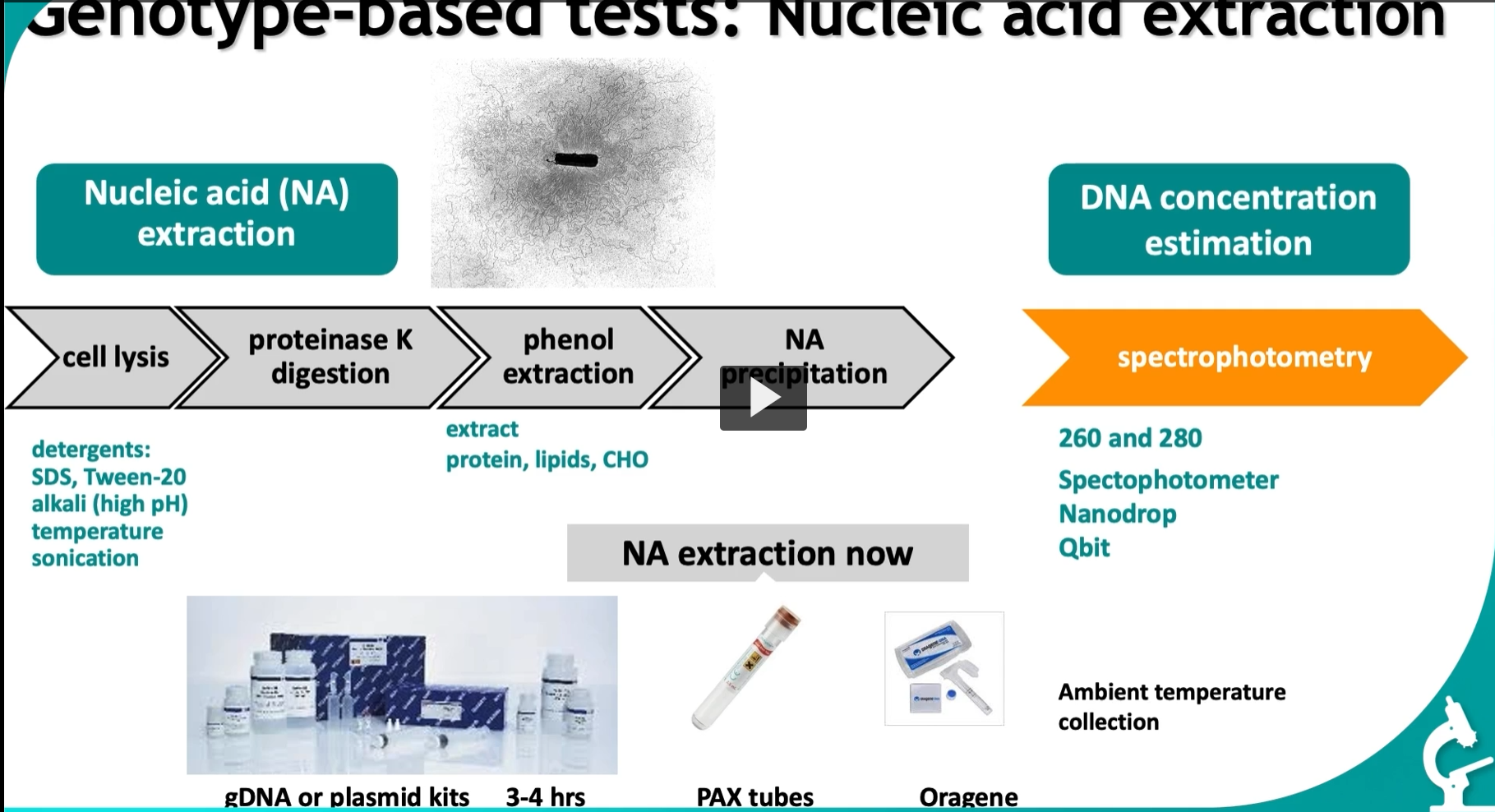
we extract nucleic acids (DNA/RNA) from cells and then measure how much we got.
Think of it as: “Break cells → isolate DNA → measure DNA quality & quantity.
1: Nucleic Acid Extraction (left side)
1. Cell lysis = breaking open the cell
Goal: release DNA from inside the cell
Methods listed:
Detergents (SDS, Tween-20) → dissolve cell membrane (lipids)
Alkali (high pH) → destabilizes membranes and proteins
Temperature → helps break structures
Sonication → sound waves physically disrupt cells
Concept: You’re destroying the “container” (cell membrane + nucleus) to access DNA.
2. Proteinase K digestion
Goal: remove proteins
Proteinase K is an enzyme that digests proteins (to remove them)
It removes:
histones (DNA-binding proteins)
enzymes that could degrade DNA
Concept: DNA is “wrapped in protein”—you must remove proteins to purify it.
3. Phenol extraction: separate DNA from other biomolecules
Phenol separates:
Proteins → organic layer
DNA → aqueous layer
Slide note:
“extract protein, lipids, CHO (carbohydrates)”
→ Concept: This is a chemical separation step—like oil and water layers.
4. Nucleic Acid (NA) precipitation: pull DNA out of solution
Add alcohol (ethanol/isopropanol) → DNA becomes insoluble
DNA forms a visible pellet
→ Concept: Turning dissolved DNA into a solid you can collect
Collected by centrifugation
the purity and concentration of DNA is assessed by spectrophotometry with absorbance ratios between 1.8 and 2.0 indicating HIGH PURITY.
Part 2: DNA Concentration Estimation (right side)
After extraction → you must check:
How much DNA?
How pure is it?
Spectrophotometry
Measures how much light DNA absorbs.
Key wavelengths:
260 nm → DNA absorbs here
280 nm → proteins absorb here
Ratio (important!):
260/280 ≈ 1.8 → pure DNA
Lower → protein contamination
AFTER, DNA extraction, the next step is assessing nucleic acid quality and fragment size.
Gel electrophoresis: using electricity (electrophoresis) to separate DNA based on size AND charge.
Why DNA moves
DNA has a negative charge (because of phosphate groups)
When electricity is applied:
DNA moves toward the positive electrode
What determines movement? 1. Size (most important)
Smaller fragments → move faster → travel farther
Larger fragments → move slower → stay near the top
That’s why: Distance traveled ∝ inverse of size
The gel = molecular sieve
Think of the gel like a mesh/net:
Small DNA → slips through easily
Large DNA → gets slowed down
different types of gels
Agarose gel (most common) Range:
Separates ~50 bp to 50 kb
Key concept:
Higher % agarose → tighter mesh → better for small DNA
Lower % agarose → looser mesh → better for large DNA
Easy memory: Tight gel= small DNA
Polyacrylamide gel
Used for:
Very small fragments
Can detect 1 nucleotide difference
→ Much higher resolution than agarose
Used in:
sequencing
SNP analysis
PFGE (Pulse-Field Gel Electrophoresis)
Very large DNA (>50 kb)
How it works:
Electric field changes direction periodically
Forces large DNA to reorient and separate
→ Used in: chromosomal typing


PFGE analysis of chromosomal DNA (from tracking infections)
DNA extraction (taken from isolates from various hospital areas)
↓
Restriction enzyme digestion
type II endonucleases (Cut DNA at specific sequences, Produce large DNA fragments
↓
Gel electrophoresis (This is NOT normal gel electrophoresis, (PFGE specifically)
↓
Pattern recognition (by comparing the resulting patterns, this is called a DNA fingerprint, we can identify the presence of the same organisms, crucial for understanding the presence of the same organism, crucial for understanding the spread of infections
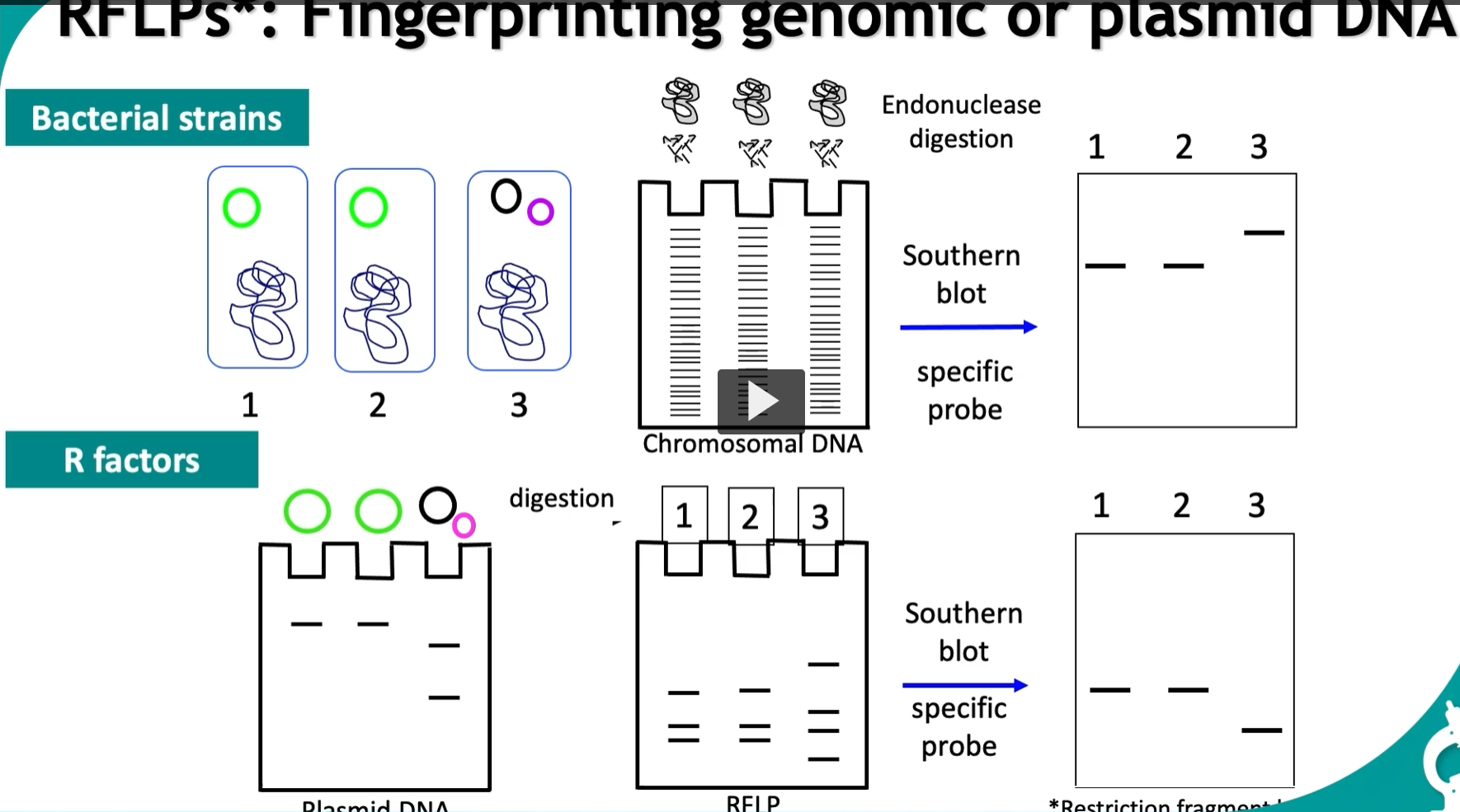
when DNA is digested with restriction enzymes, this process is known as RESTRICTION, FRAGMENT LENGTH POLYMORPHISM.
This technique targets specific DNA sequences that are unique to an organism, creating a distinctive fingerprint, useful for diagnostics.
The process involves:
isolating DNA
digesting it with restriction enzymes
separating the resulting fragments using agarose gel electrophoresis.
to enhance specificity, probes can hybridize conserved regions of the DNA, providing precise identification.
In this example, we use RFLP to differentiate between three bacterial strains,
we isolate
chromosomal DNA
digest it with type II endonucleases
and separate the fragments by gel electrophoresis.
Specificity is enhanced by hybridizing the specific probe that identifies the DNA fragment.
The results show that strains I and II are identical while strain III is a different bacterium. (strain III is at a different position)
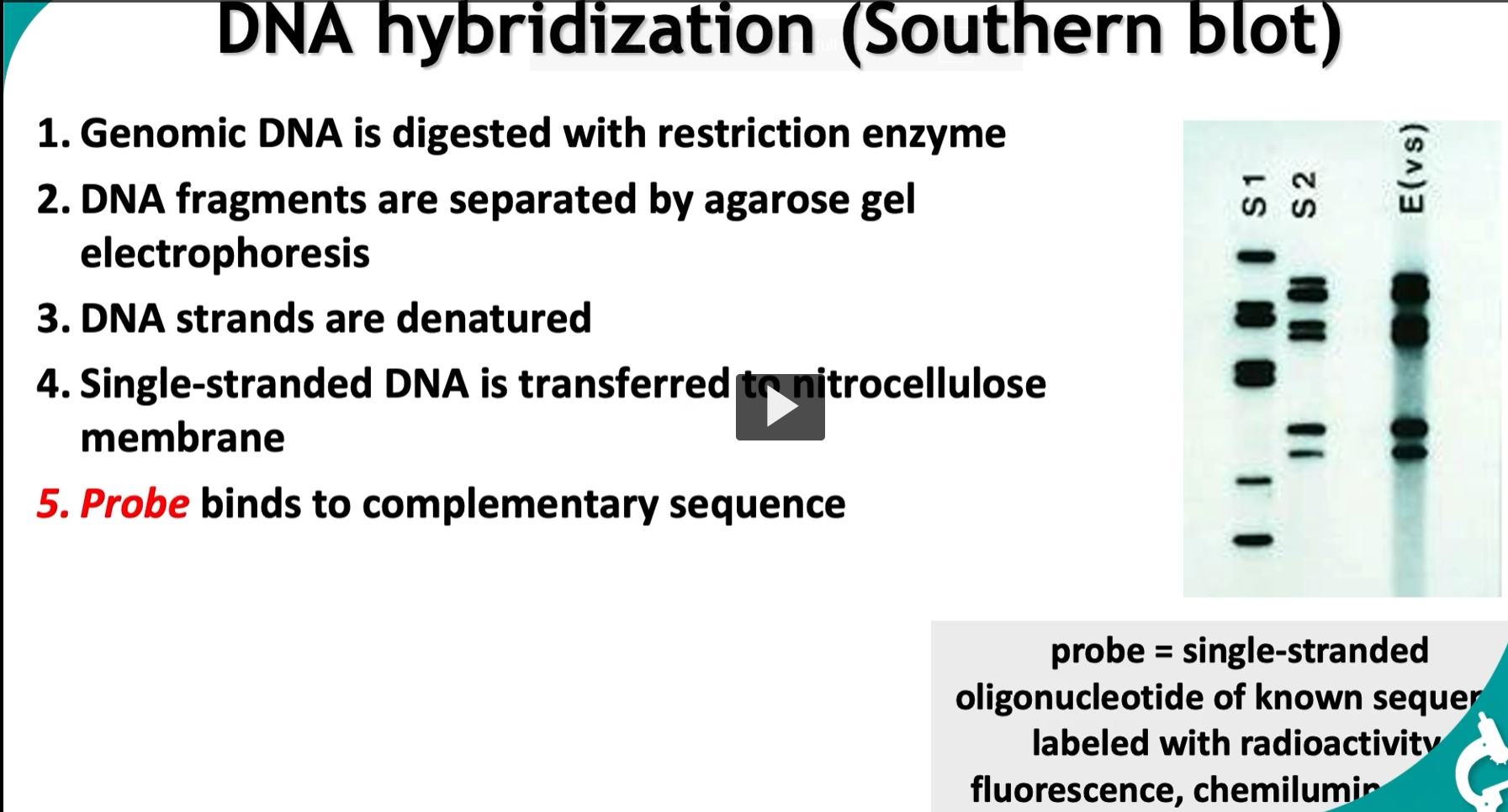
DNA hybridization (Southern blot)
Southern blot = find a specific DNA sequence by using a complementary labeled probe.
“Cut DNA → separate → transfer → detect with a probe.”
Genomic DNA is digested with restriction enzyme (These enzymes recognize specific sequences and cut DNA into fragments, A mixture of many DNA fragments of different sizes)
DNA fragments are separated by agarose gel electrophoresis (most common gel)
DNA strands are denatured
Single-stranded DNA is transferred to nitrocellulose membrane (The membrane is stable and accessible for probing, “Copy the gel pattern onto a durable sheet”)
Probe binds to complementary sequence (labeled (radioactive, fluorescent, or chemiluminescent), Probe binds ONLY where the matching sequence exists)
probe: single-stranded oligonucleotide of known sequence labeled with radioactivity, fluorescence, chemiluminescence
Applications of Southern blotting
South blotting allows Identification of microorganisms by hybridization to homologous NA sequences
genomic DNA, ribosomal RNA, plasmids
Southern blotting allows Rapid detection of infectious agents directly in clinical specimens
blood, urine, sputum, tissues
in situ hybridization
Southern blotting allows Gene expression studies
Northern blot (RNA)
Different HPV subtypes have different cancer risks, and molecular probes can detect which subtype is present.
👉 This lets us:
diagnose infection
predict cancer risk
🦠 Part 1: HPV and cancer What is HPV?
Human papillomavirus (HPV) = a DNA virus
Infects epithelial cells (skin, mucosa)
Diseases associated:
Cervical cancer
Anal cancer
Oral (oropharyngeal) cancer
Key concept:
There are ~70+ HPV subtypes
NOT all are dangerous
👉 Two categories:
Low-risk types (e.g., HPV 6, 11) → warts
High-risk types (e.g., HPV 16, 18) → cancer
🔴 Critical point on the slide
“Correlation between some viral subtypes and cancer risk”
👉 Meaning:
The specific DNA sequence (genotype) determines how dangerous the virus is
🧪 Part 2: How we detect this (dot blot)
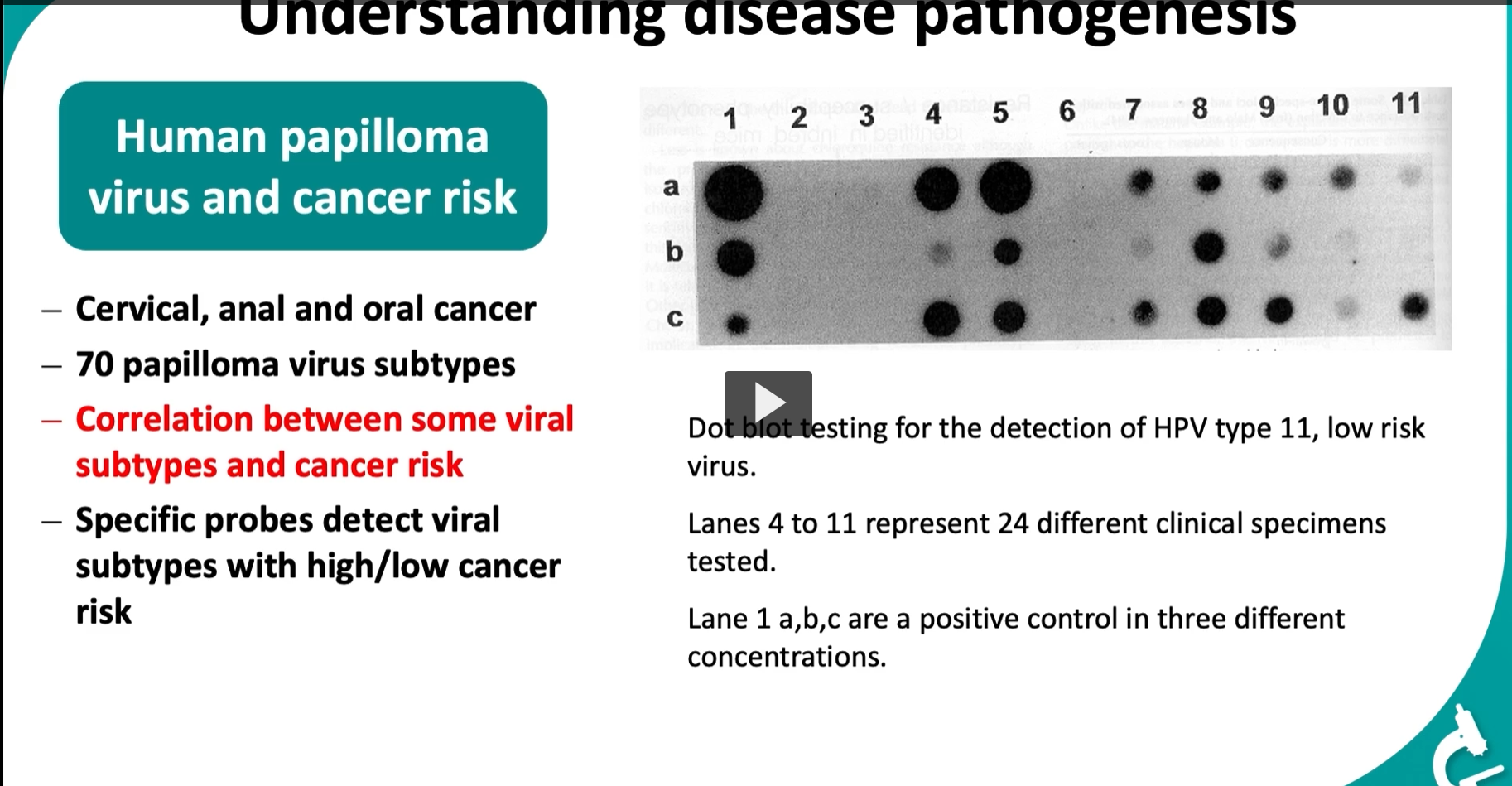
This slide shows a dot blot (a simpler version of Southern blot).
🔬 What is a dot blot?
DNA samples are placed as dots on a membrane
No size separation (unlike gel electrophoresis)
A probe is added
👉 If the target HPV DNA is present → dot becomes dark
📊 Interpreting the image Layout:
Columns labeled 1–11 = different samples
Rows a, b, c = different concentrations (controls)
Controls (Lane 1):
a, b, c = positive control
Show strong signals at different concentrations
👉 Confirms:
The test is working properly
Clinical samples (Lanes 4–11):
Each dot = one patient/sample
👉 Dark dot → HPV DNA present
👉 No dot → HPV not detected
Important detail:
This test is specifically detecting:
HPV type 11 (low-risk virus)
So:
Positive result → patient has low-risk HPV
Negative → no HPV 11 detected (could still have other types!)
🧠 Why probes matter
“Specific probes detect viral subtypes”
Each probe is designed for a specific HPV DNA sequence
You can:
distinguish HPV 11 vs HPV 16
classify low vs high cancer risk
🔥 Clinical significance
This is HUGE in medicine:
Determines:
Who is at risk for cancer
Who needs closer monitoring
Used in:
Pap smear follow-ups
HPV screening
🧩 High-yield summary
Molecular hybridization techniques (like dot blot) use sequence-specific probes to detect HPV subtypes, allowing identification of infections and assessment of cancer risk based on viral genotype.
This slide is about ribotyping, a molecular method used to identify and compare bacteria based on their rRNA genes. Here’s the logic step-by-step:
🧬 Big Idea
Ribotyping = using rRNA gene patterns as a “genetic fingerprint” to identify and compare organisms.
🧪 What are rRNA genes?
Bacteria have ribosomal RNA (rRNA) genes, which are essential for making ribosomes.
These include:
16S rRNA
23S rRNA
5S rRNA
👉 Together they form the rRNA operon
🔑 Why rRNA genes are special
Highly conserved → similar across many bacteria
BUT also have variable regions → allow differentiation
👉 This balance makes them perfect for:
identification + evolutionary comparison
🔬 How ribotyping works 1. DNA is cut (restriction enzymes)
Break genome into fragments
2. Fragments separated (gel electrophoresis)
Different sizes spread out
3. Probe hybridization
Use probes that bind specifically to rRNA genes
👉 Only fragments containing rRNA genes will be detected
4. Band pattern is generated
This pattern = ribotype
🧬 Key concept: “variable distribution”
Different bacteria have rRNA genes in different locations and numbers
So after cutting DNA:
rRNA-containing fragments differ in size and position
👉 Result:
Each species → unique banding pattern
📊 Species-specific patterns
Same species → similar pattern
Different species → different pattern
👉 This is like a:
barcode for bacterial identity
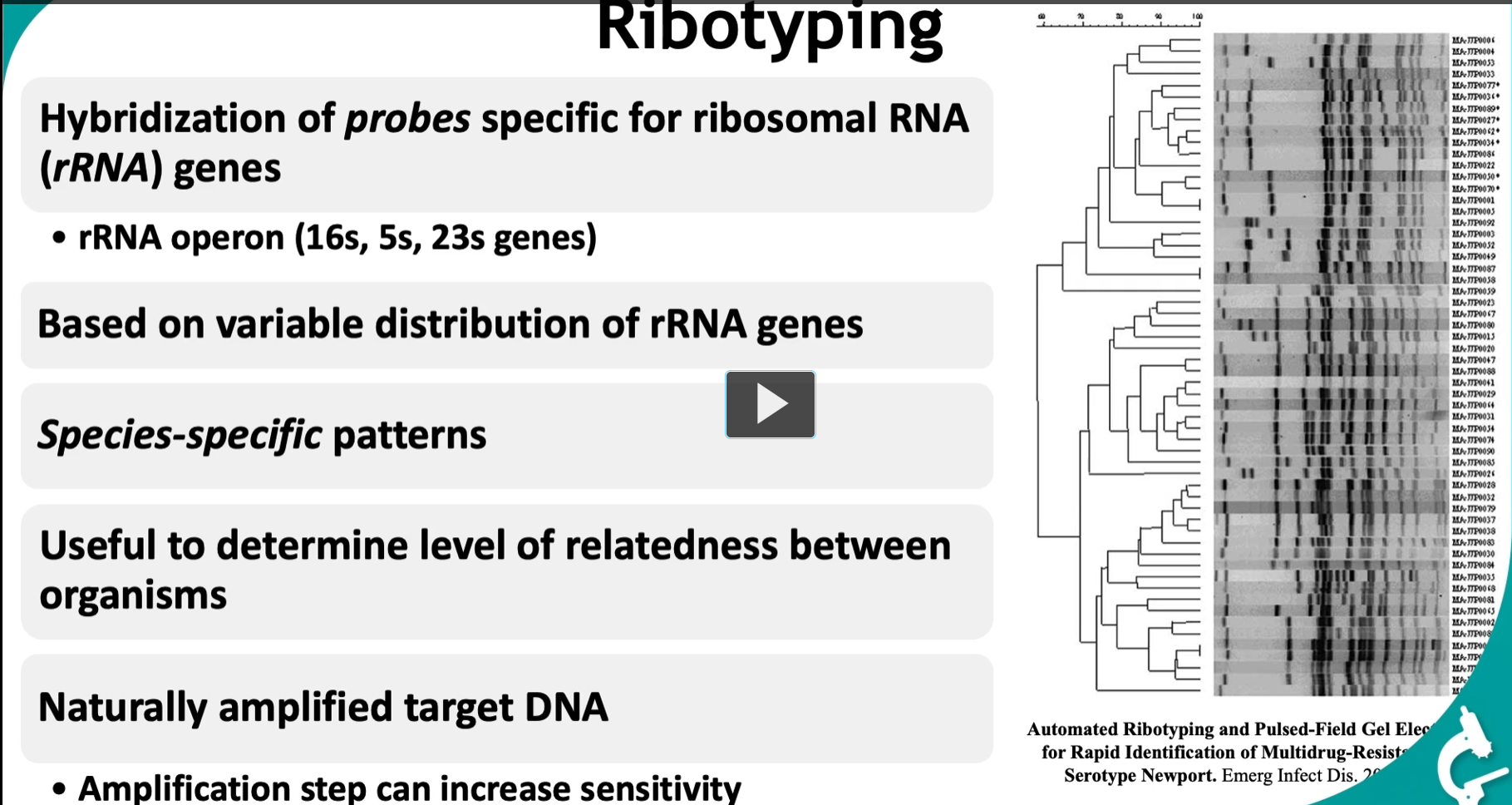
🌳 Determining relatedness
The tree (dendrogram on right side):
Groups organisms based on similarity of patterns
Closer branches → more closely related
👉 Used for:
taxonomy
evolutionary relationships
outbreak tracking
🔥 “Naturally amplified target DNA”
This is VERY important:
rRNA genes exist in multiple copies per genome
👉 Meaning:
Easier to detect
Stronger signal
Amplification (optional)
PCR can further increase sensitivity
Useful for low DNA samples
🧠 Why ribotyping is useful
Identify unknown bacteria
Compare strains
Study evolution
Track infections
🧩 High-yield summary
Ribotyping uses probes targeting rRNA genes to generate species-specific banding patterns, allowing identification and determination of genetic relatedness among organisms.
🧠 Simple mental model
rRNA genes = “landmarks” in the genome
Restriction enzymes = “cutting map”
Pattern = “genetic fingerprint”

This slide is showing RiboPrints®, which is basically an automated, standardized version of ribotyping used for bacterial identification.
🧬 Big Idea
RiboPrint = generate a DNA “barcode” (fingerprint) using rRNA genes and match it to a database to identify the organism.
👉 Think:
“Cut DNA → detect rRNA fragments → create pattern → match to database.”
🔬 Step-by-step (what’s happening) 1. Cells from a pure culture are lysed
Break open bacteria to release DNA
👉 “Pure culture” = only one organism present
→ important for accurate identification
2. DNA digestion (restriction enzyme)
DNA is cut at specific sequences
👉 Result:
Many fragments of different sizes
3. Gel electrophoresis
Fragments are separated by size
👉 Creates a band pattern
4. Transfer + hybridization (like Southern blot)
DNA is transferred to a membrane
A probe for rRNA genes is added
👉 Only fragments containing rRNA genes are detected
5. Chemiluminescent detection
A substrate is added
Bound probes produce light
👉 No color bands—this is detected as light emission
6. Camera detection → RiboPrint
A camera records the pattern
This pattern = RiboPrint (DNA fingerprint)
📊 What happens next (most important part)
The system compares your pattern to a large database
From the slide:
5,700+ patterns
180+ genera
1,200+ species
👉 Match = organism identified
🧠 Why rRNA genes are used
Present in all bacteria
Have:
conserved regions → probe can bind
variable distribution → different patterns
👉 Perfect for identification
📌 Right-side examples (what you’re seeing)
Different banding patterns correspond to different species:
Escherichia coli
Staphylococcus aureus
Pseudomonas aeruginosa
etc.
👉 Each species = unique barcode pattern
🔥 Why this is useful
Fast identification of bacteria
Standardized (automated system)
Useful in:
clinical microbiology
food safety
outbreak investigations
🧩 High-yield summary
RiboPrint is an automated ribotyping method that generates rRNA-based DNA fingerprints and compares them to a database to identify bacterial species.
🧠 Simple mental model
Restriction enzyme = scissors
rRNA probe = target detector
Pattern = barcode
Database = scanner system (like a grocery store)
This slide is showing how DNA sequencing (specifically 16S rRNA sequencing) is used to identify bacteria—this is the modern gold standard replacing many older methods like ribotyping.
🧬 Big Idea
Sequence the 16S rRNA gene → compare to a database → identify the organism.
👉 Think:
“Read the DNA code → match it like a fingerprint.”
🔬 Step-by-step explanation 1. DNA extraction from a pure isolate
Take a single bacterial species (pure culture)
Extract its DNA
👉 Important:
Pure isolate = ensures you’re sequencing ONE organism
2. Amplify the 16S rRNA gene (PCR)
Use PCR to copy the 16S rRNA gene
👉 Why 16S?
Present in all bacteria
Has:
conserved regions → primers can bind
variable regions → distinguish species
👉 This is the KEY:
Same gene in all bacteria, but slightly different sequence
3. Extension products are separated and sequenced
DNA is sequenced (often via Sanger sequencing)
👉 The graph on the right = chromatogram
Each peak = a nucleotide:
A, T, C, G (different colors)
👉 Output:
A string like: ATCGTTACG…
4. Match sequence to database
Compare your sequence to:
GenBank
BLAST
clinical databases
👉 Result:
Closest match = organism identity
🧠 Why this works (core concept)
The 16S rRNA gene is like a biological barcode.
Conserved → universal detection
Variable → species-level identification
🔥 Why this is powerful
Compared to older methods:
Method | What it uses | Limitation |
|---|---|---|
ELISA | proteins | indirect |
PFGE | patterns | complex |
Ribotyping | band patterns | lower resolution |
Sequencing | actual DNA sequence | most precise |
🧪 Clinical significance
Used for:
Identifying unknown bacteria
Detecting rare or unculturable organisms
Diagnosing infections
Microbiome studies
🧩 High-yield summary
16S rRNA sequencing identifies bacteria by amplifying and sequencing a conserved gene with variable regions and matching it to known sequences in databases.
🧠 Simple mental model
PCR = photocopier
Sequencing = reading the letters
Database = search engine
Match = organism ID


This slide is about DNA microarrays (“DNA chips”), which let you analyze thousands of genes at once. It’s a big step up from single-gene methods.
🧬 Big Idea
Microarrays = test many genes simultaneously by hybridization to known DNA probes on a chip.
👉 Think:
“Thousands of probes on a chip → your sample binds → glowing pattern tells the story.”
🔬 How the microarray works (step-by-step) 1. Chip contains known DNA probes
The chip has thousands of spots
Each spot = a known single-stranded DNA sequence (a gene or part of a gene)
👉 Like:
A grid of “questions” (each probe asks: is this gene present or expressed?)
2. Sample DNA or cDNA is added
From an unknown sample:
DNA → for gene presence
cDNA → for gene expression (most important use)
👉 cDNA is made from mRNA → reflects which genes are active
3. Hybridization occurs
If your sample contains a matching sequence → it binds (hybridizes) to that spot
👉 Specific binding = sequence match
4. Fluorescent signal detection
Sample is labeled with a fluorescent dye
Where binding occurs → spot glows
👉 Brightness = amount of gene expression (or abundance)
📊 What you get (the image on right)
A grid of colored dots:
Bright = high expression
Dim = low expression
No signal = not expressed
👉 This gives a global snapshot of gene activity
🧠 What makes this powerful 🔹 “One experiment = thousands of genes”
Instead of testing 1 gene → test entire genome patterns
🔹 “Whole picture”
You see how genes interact
Not just “on/off” → but patterns
🔬 Applications (from slide) 1. Gene expression analysis
Compare:
healthy vs diseased tissue
Example:
cancer vs normal cells
👉 Can reveal:
Which genes are turned ON or OFF in disease
2. Disease diagnosis
Detect:
infectious agents
genetic signatures
3. Cancer classification
Example from slide:
B-cell lymphoma → actually two different diseases based on gene expression
👉 Same appearance, different molecular behavior
4. Drug development
Identify:
disease-specific pathways
drug targets
Pharmacogenomics:
how patients respond differently to drugs
🧩 Key conceptual connection
Microarrays don’t just tell you what is there → they tell you what is active.
🔥 High-yield summary
DNA microarrays use thousands of immobilized DNA probes to detect gene presence or expression via fluorescent hybridization, enabling large-scale analysis of gene activity in a single experiment.
🧠 Simple mental model
Chip = thousands of locks
Sample DNA = keys
Binding = correct key fits lock
Fluorescence = light turns on when match happens
DNA Microarrays or “DNA chips”
The “Flu Chip” – a New Way to Diagnose the Flu
“By using the FluChip-55 microarray in conjunction with a well-established RNA amplification method, RNA from viruses of interest, including influenza viruses A/H1N1, A/H3N2, and A/H5N1 and influenza B virus, was typed and subtyped in 11 hours,” say the researchers.
Detection of multiple human herpes viruses by DNA microarray technology.
This approach can establish whether or not a handful of viral genes are present in a clinical sample: HSV-1, HSV-2, varicella zoster, Epstein Barr, CMV, HHV-6


This slide is one of the most important in molecular diagnostics—it explains PCR and RT-PCR, which are the backbone of modern testing (COVID tests, HIV viral load, etc.).
🧬 Big Idea
PCR = make millions of copies of a specific DNA sequence.
RT-PCR = convert RNA → DNA → then amplify it.
👉 Think:
“Find a tiny piece of genetic material → copy it → detect it.”
🔬 PART 1: PCR (Polymerase Chain Reaction) 🧪 What PCR does
Takes a small amount of DNA
Uses specific primers to target a sequence
Amplifies it → millions of copies
⚙ How it works (conceptually)
PCR cycles repeat:
Denaturation → DNA strands separate
Annealing → primers bind to target sequence
Extension → DNA polymerase copies DNA
👉 Repeat ~30 cycles → exponential amplification
🔑 Key feature:
Highly specific (because primers define the target)
🧪 Detection (from slide)
After PCR:
Run on gel electrophoresis
Or:
restriction enzyme digestion
probe hybridization
👉 Confirms identity of amplified DNA
🧬 PART 2: RT-PCR (for RNA)
Used when the target is RNA, not DNA
⚙ Steps: Step 1: Reverse transcription
Reverse transcriptase enzyme
Converts RNA → cDNA
Step 2: PCR
Amplify the cDNA using primers
🔑 Key idea:
RNA cannot be amplified directly → must first become DNA
🧪 “In situ PCR”
PCR done directly in tissue
Preserves location of DNA/RNA
👉 Shows:
where the pathogen or gene is inside tissue
⚡ Why PCR is powerful
Rapid → results in hours
Highly sensitive → detects tiny amounts
Highly specific → sequence-targeted
🧠 Applications (from slide) 1. Detect hard-to-grow organisms
Mycobacterium avium
Chlamydia trachomatis
Neisseria gonorrhea
👉 Important because:
Some bacteria are slow or difficult to culture
2. Measure viral load
HIV-1
HCV
👉 Quantifies:
How much virus is in the body
3. Monitor therapy
Is treatment working?
Viral load decreasing?
4. Prognosis
High viral load → worse outcome
Low viral load → better prognosis
🧪 Real-world example (on slide)
LightCycler SeptiFast Test (Roche)
Detects pathogens directly from blood
Used in sepsis diagnosis
🔥 High-yield summary
PCR amplifies specific DNA sequences using primers, while RT-PCR first converts RNA to cDNA before amplification, enabling rapid, sensitive detection and quantification of pathogens.
🧠 Simple mental model
PCR = DNA photocopier
RT-PCR = translate RNA → then copy it
Primers = address labels telling copier what to copy
⚠ Exam tip (very important)
PCR = DNA detection
RT-PCR = RNA detection (viruses like HIV, COVID)
This slide is about PCR-RFLP, a classic method used to detect genetic differences (mutations or strain variation) by combining PCR with restriction enzymes.
🧬 Big Idea
PCR-RFLP = amplify DNA → cut with restriction enzymes → analyze fragment pattern.
👉 If the DNA sequence changes → the cutting pattern changes → the band pattern changes.
🔬 Step-by-step (core workflow) 1. PCR amplification
Target a specific gene (e.g., HIV env gene, hemoglobin gene)
Make many copies
2. Restriction enzyme digestion
Add a restriction enzyme (e.g., AluI, HinfI)
Enzyme cuts DNA at specific sequences
👉 Important:
If a mutation changes the sequence → enzyme may cut differently or not at all
3. Gel electrophoresis
Separate fragments by size
Visualize band pattern
🧠 Key principle
DNA sequence → determines restriction sites → determines band pattern
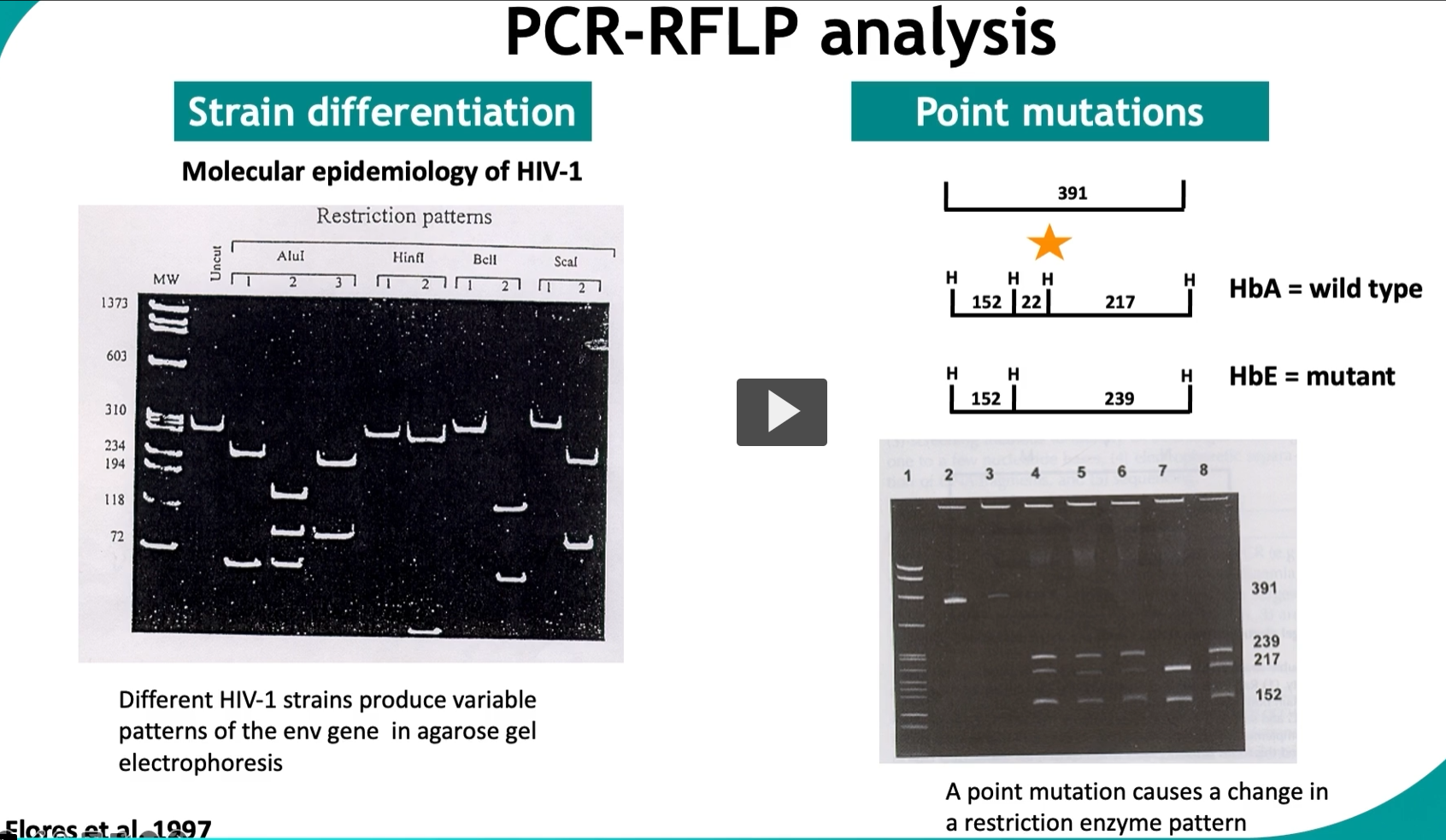
🧪 LEFT SIDE: Strain differentiation (HIV example) What’s happening:
Different HIV-1 strains have slightly different DNA sequences
When cut with enzymes → produce different fragment sizes
👉 Result:
Each strain → unique banding pattern
Why useful:
Track infection spread
Identify sources of outbreaks
Compare viral strains
👉 This is molecular epidemiology
🧬 RIGHT SIDE: Point mutation example (HbA vs HbE)
This is the most important concept.
🧪 Wild type (HbA)
Normal DNA sequence
Restriction enzyme recognizes its site → cuts
👉 Produces:
Multiple fragments (e.g., 152, 22, 217)
🧪 Mutant (HbE)
Single base change (point mutation)
Alters restriction site
👉 Result:
Enzyme can’t cut at that site anymore
Fragment sizes change (e.g., 152, 239)
📊 Gel interpretation
Different band sizes = different DNA sequence
Compare lanes:
Same pattern → same genotype
Different pattern → mutation present
🔥 Key concept (HIGH-YIELD)
A single nucleotide change can:
create a new restriction site
destroy an existing one
👉 → changes band pattern
🧠 Why this works
Restriction enzymes are:
sequence-specific “molecular scissors”
So even:
1 base change = different cutting = different pattern
🧩 High-yield summary
PCR-RFLP detects genetic variation by amplifying DNA, digesting it with restriction enzymes, and identifying sequence differences based on fragment size patterns.
🧠 Simple mental model
PCR = copy the sentence
Restriction enzyme = cut at specific words
Mutation = changes the word → cut changes
Gel = shows where cuts happened
⚠ Exam tip
Used for:
mutation detection
strain typing
Being replaced by:
sequencing (more precise)
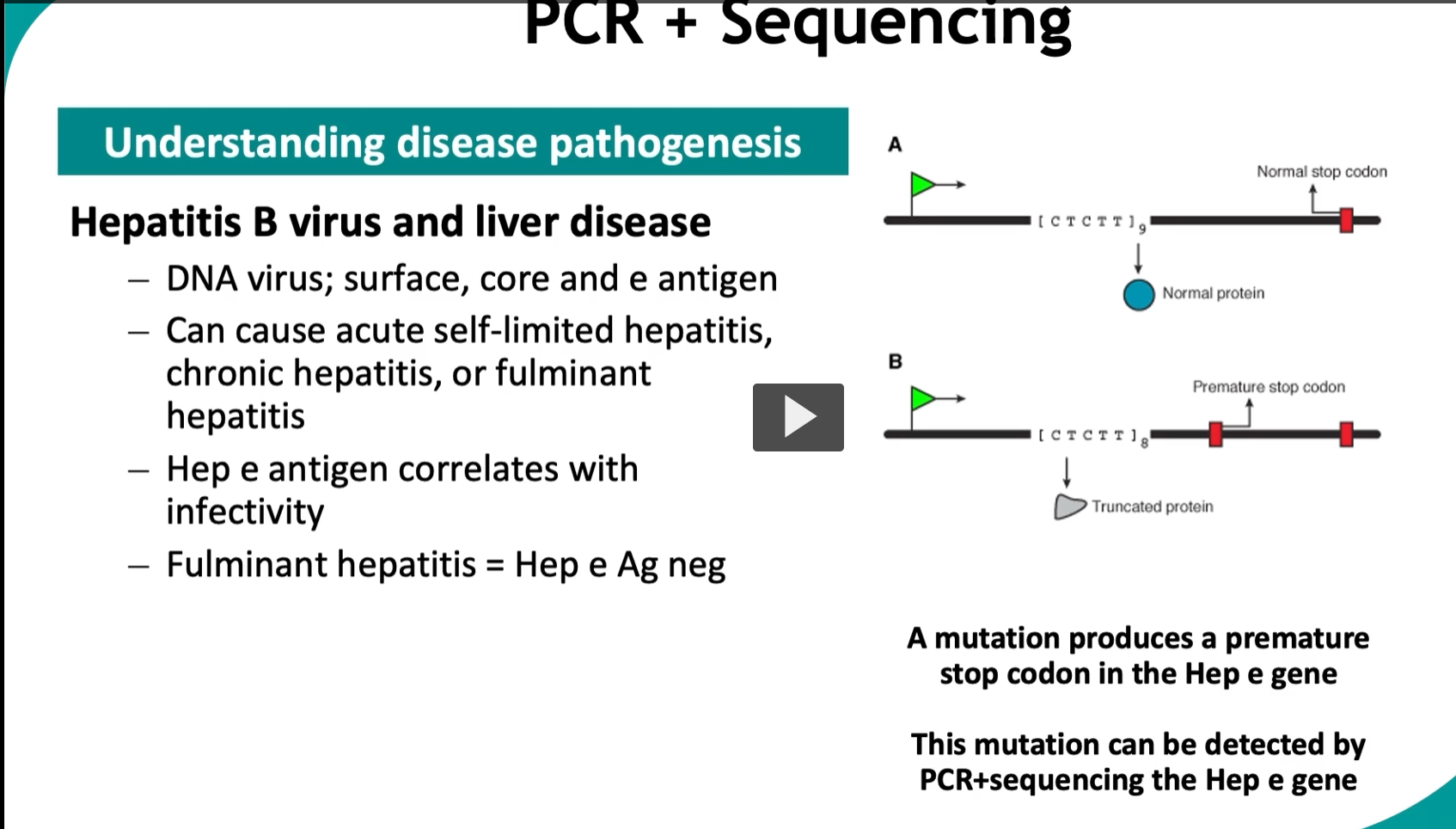
This slide shows how PCR + sequencing helps us understand disease mechanisms (pathogenesis)—using Hepatitis B virus (HBV) as the example.
🧬 Big Idea
A specific mutation in HBV changes a viral protein → changes disease behavior → detected by PCR + sequencing.
👉 This connects:
genetic mutation → protein change → clinical outcome
🦠 Part 1: Hepatitis B basics
HBV is a DNA virus
Produces key antigens:
Surface antigen (HBsAg)
Core antigen (HBcAg)
e antigen (HBeAg)
🔑 Important concept:
HBeAg = marker of infectivity
If HBeAg is present → high viral replication / high infectivity
If absent → usually lower infectivity… BUT not always (important!)
⚠ Disease outcomes
HBV infection can cause:
Acute hepatitis (self-limited)
Chronic hepatitis
Fulminant hepatitis (severe, rapid liver failure)
🧬 Part 2: The mutation (right side diagram) Normal case (A)
Gene has a normal stop codon at the correct position
Full protein is made → functional HBeAg
👉 Result:
Virus produces HBeAg
Infection is detectable via this marker
Mutant case (B)
A point mutation introduces a premature stop codon
👉 What happens:
Protein production stops early
Truncated (shortened) protein is made
HBeAg is NOT produced
🔥 Critical clinical insight
Fulminant hepatitis = HBeAg negative
BUT:
Virus is still active
Just not producing detectable HBeAg
👉 This can be misleading clinically
🧠 Why this matters
Patient appears:
HBeAg negative
But actually:
Still highly infected and dangerous
👉 This mutation explains:
Why some severe HBV cases lack HBeAg
🧪 How we detect this (PCR + sequencing) Step 1: PCR
Amplify the HBe gene
Step 2: Sequencing
Read the DNA sequence
Identify:
mutation causing premature stop codon
Result:
Detect hidden mutant virus
🧩 Conceptual connection
Level | What happens |
|---|---|
DNA | mutation (point mutation) |
Protein | truncated HBeAg |
Lab test | HBeAg negative |
Clinical effect | severe disease (fulminant hepatitis) |
🔥 High-yield summary
A mutation in the HBV Hep e gene creates a premature stop codon, preventing HBeAg production and leading to severe disease; this mutation can be detected by PCR and DNA sequencing.
🧠 Simple mental model
Normal gene → full protein → detectable marker
Mutated gene → early stop → no marker → hidden but dangerous infection
⚠ Exam tip (VERY important)
HBeAg negative does NOT always mean low infectivity
→ Could be a pre-core mutant HBV
This slide focuses specifically on RT-PCR for RNA viruses, especially how it’s used clinically to measure viral load and monitor disease.
🧬 Big Idea
RT-PCR measures how much viral RNA is present in a patient → tells you how active the infection is.
👉 Think:
“More RNA = more virus = worse disease (usually)”
🔬 What RT-PCR is doing (core concept) Step 1: Reverse transcription
Viral RNA → converted into cDNA using reverse transcriptase
Step 2: PCR amplification
cDNA is amplified → millions of copies
👉 Result:
Even tiny amounts of virus can be detected and quantified
📊 What makes this slide important: QUANTITATION
Unlike regular PCR:
RT-PCR can measure how much virus is present (viral load)
🧪 Application 1: Viral load measurement Used for:
HIV-1
Hepatitis C (HCV)
👉 Sample:
Blood plasma
What is “viral load”?
The amount of viral RNA in the blood
High viral load → lots of virus
Low viral load → less virus
💊 Application 2: Monitoring therapy
Example from slide:
Protease inhibitor treatment (HIV)
👉 After treatment:
Viral replication is blocked
RT-PCR shows decreasing RNA levels
Key concept:
RT-PCR = real-time feedback on treatment effectiveness
📉 Application 3: Prognostic value
Viral load predicts disease progression
From slide:
Lower RNA plasma levels → decreased progression
👉 Meaning:
Lower viral load → better outcome
Higher viral load → worse prognosis
🧠 Clinical interpretation
Viral Load | Meaning |
|---|---|
High | Active infection, poor control |
Decreasing | Treatment working |
Undetectable | Virus suppressed |
🔥 Why RT-PCR is so powerful
Extremely sensitive
Quantitative (not just yes/no)
Works for RNA viruses
🧩 High-yield summary
RT-PCR converts viral RNA to cDNA and amplifies it to quantify viral load, which is used to monitor treatment effectiveness and predict disease progression.
🧠 Simple mental model
RT step = translate RNA → DNA
PCR = copy it
Output = how much virus is present
⚠ Exam tip (very important)
RT-PCR is the gold standard for viral load monitoring (HIV, HCV)
NOT just detection—quantification

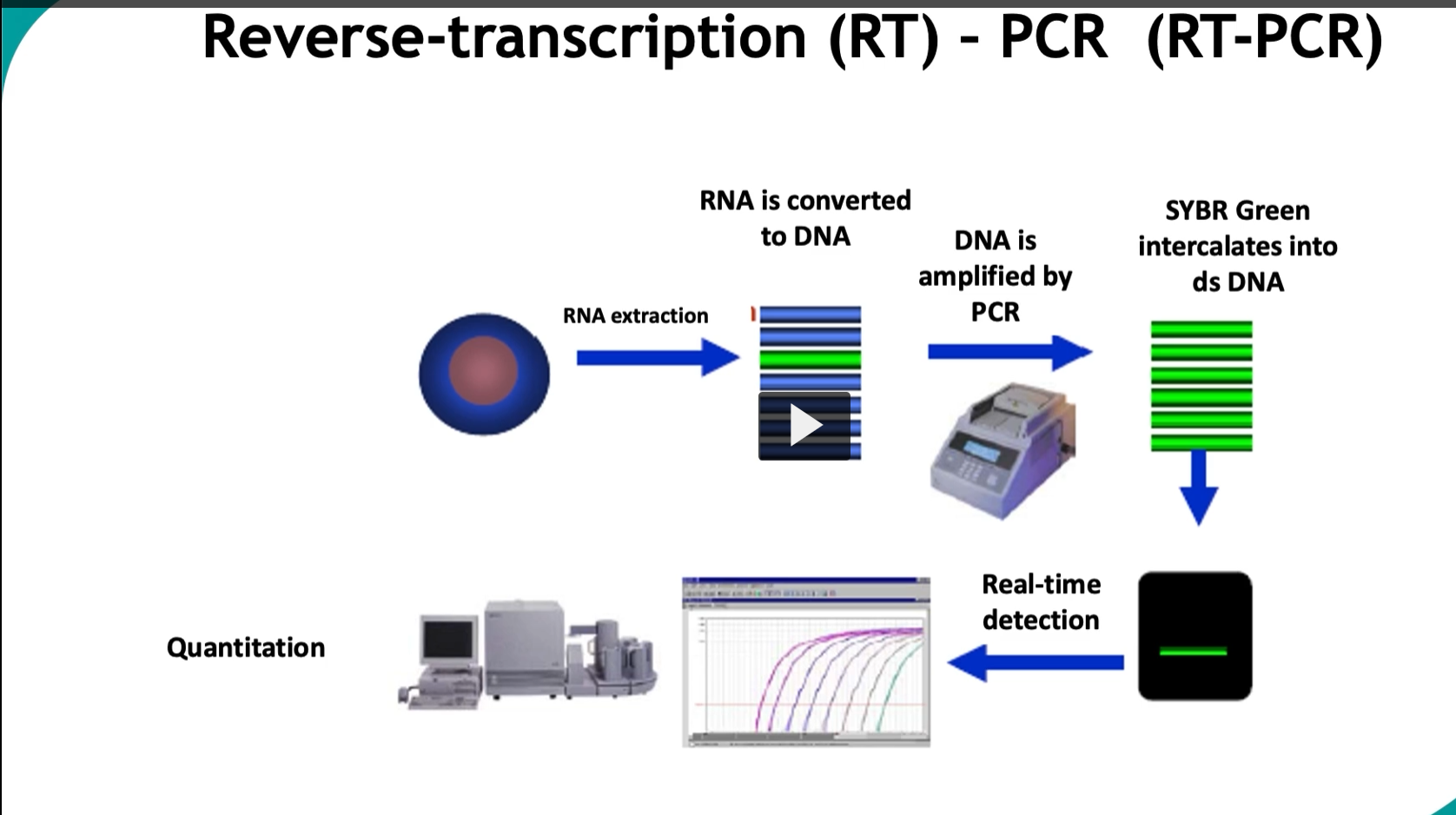
This slide is showing real-time RT-PCR (qRT-PCR)—the most advanced and clinically important version of PCR, used to detect AND quantify RNA in real time.
🧬 Big Idea
qRT-PCR = convert RNA → amplify DNA → measure fluorescence in real time → quantify how much RNA was present.
👉 Think:
“Copy it AND measure it while it’s happening.”
🔬 Step-by-step (what’s happening in the diagram)
1. RNA extraction
Start with a sample (blood, tissue, swab)
Extract RNA (e.g., viral RNA like HIV, SARS-CoV-2)
2. RNA → DNA (Reverse transcription)
Enzyme: reverse transcriptase
Converts RNA → cDNA
👉 Required because:
PCR only works on DNA
3. DNA amplification (PCR)
cDNA is amplified through cycles
Each cycle → doubles DNA amount
4. SYBR Green binds DNA
SYBR Green dye intercalates into double-stranded DNA
When bound → fluoresces (glows)
👉 More DNA → more fluorescence
5. Real-time detection
Machine measures fluorescence after each cycle
👉 Unlike regular PCR:
You don’t wait until the end
You monitor amplification live
6. Quantitation (most important)
The graph shows curves:
X-axis = cycle number
Y-axis = fluorescence
📊 Ct value (CRITICAL concept)
Ct (cycle threshold) = cycle at which fluorescence becomes detectable
Interpretation:
Ct Value | Meaning |
|---|---|
Low Ct (e.g., 15–20) | High starting RNA (lots of virus) |
High Ct (e.g., 30–40) | Low starting RNA |
No Ct | No detectable RNA |
🧠 Why this is powerful
Detects RNA viruses
Quantifies viral load
Extremely sensitive
Extremely fast
🧪 Clinical uses
HIV viral load
Hepatitis C monitoring
COVID-19 testing
Gene expression studies
🔥 Key difference from regular PCR
Regular PCR | qRT-PCR |
|---|---|
End-point detection | Real-time detection |
Qualitative (yes/no) | Quantitative |
Gel needed | No gel needed |
🧩 High-yield summary
Real-time RT-PCR converts RNA to cDNA, amplifies it, and uses fluorescent dyes like SYBR Green to measure DNA accumulation during each cycle, allowing quantification of the original RNA.
🧠 Simple mental model
Reverse transcription = translate RNA → DNA
PCR = copy it repeatedly
SYBR Green = glow when DNA increases
Machine = tracks glow over time
⚠ Exam tip
Lower Ct = higher viral load
This is one of the most tested concepts.
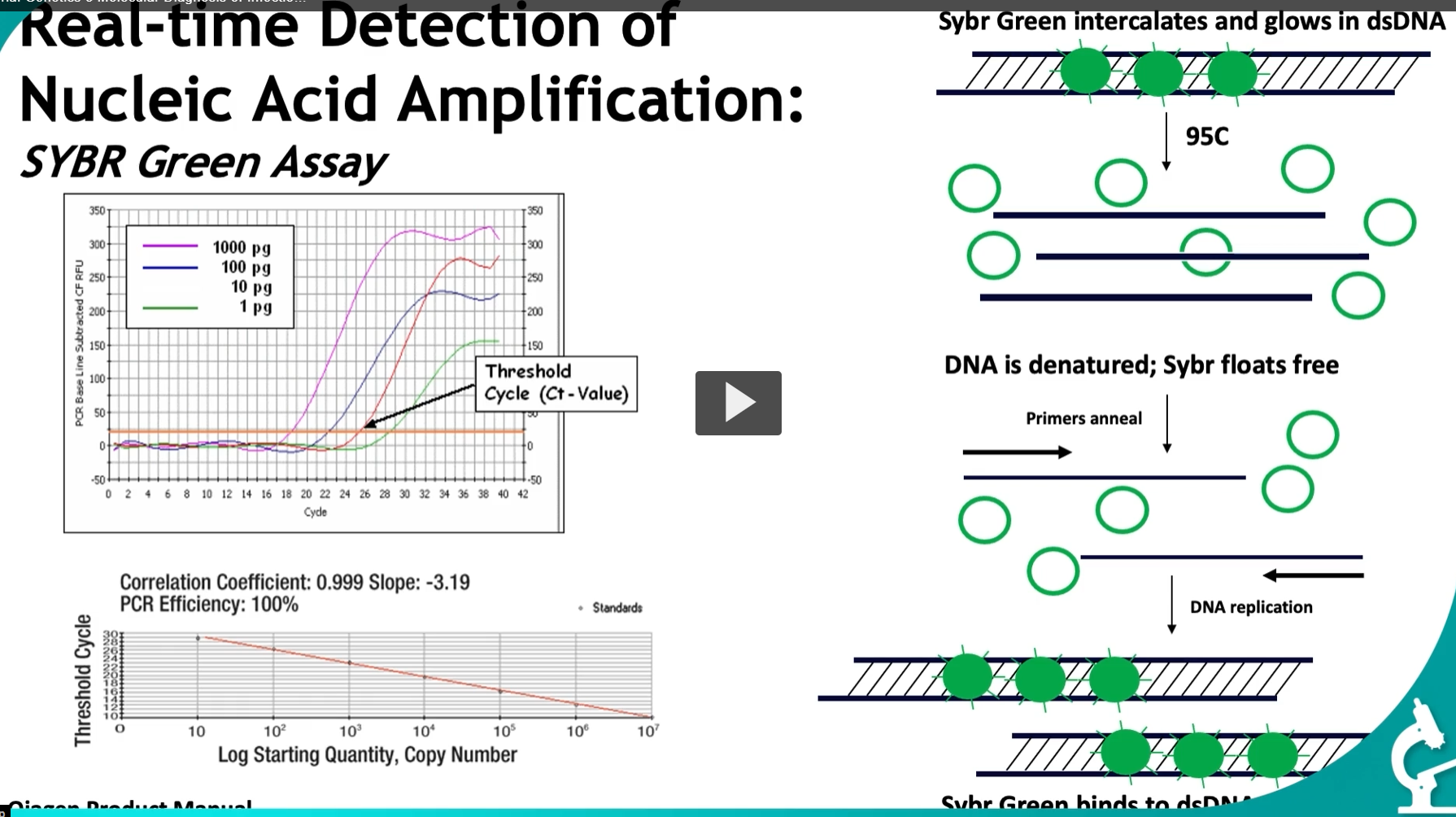
This slide explains how real-time PCR (qPCR) works using SYBR Green and how we quantify DNA using Ct values. This is a very high-yield concept.
🧬 Big Idea
SYBR Green glows when bound to double-stranded DNA → more DNA = more fluorescence → measure it each cycle to quantify starting material.
🔬 PART 1: What SYBR Green does 🧪 Key property:
SYBR Green binds (intercalates) into double-stranded DNA (dsDNA)
When bound → fluoresces (glows)
When not bound → no signal
🔁 What happens during each PCR cycle 🔥 1. Denaturation (95°C)
DNA strands separate → single-stranded DNA
SYBR Green falls off
👉 Result:
Low fluorescence
🧬 2. Annealing
Primers bind to DNA
⚙ 3. Extension (replication)
DNA polymerase makes new strands → dsDNA forms again
SYBR Green binds again
👉 Result:
Fluorescence increases
🔁 Each cycle:
More DNA → more SYBR binding → more fluorescence
📊 PART 2: The amplification curve
The graph shows:
X-axis = cycle number
Y-axis = fluorescence
Each line = different starting amount:
1000 pg → highest
1 pg → lowest
🧠 Key observation
More starting DNA → curve rises earlier
📍 Ct value (MOST IMPORTANT)
Ct (threshold cycle) = cycle where fluorescence crosses a set threshold
Interpretation:
Starting DNA | Ct value |
|---|---|
High (1000 pg) | Low Ct (early detection) |
Low (1 pg) | High Ct (late detection) |
👉 Inverse relationship:
Ct ↓ = more starting DNA
Ct ↑ = less starting DNA
📈 PART 3: Standard curve (bottom graph)
X-axis = log starting quantity
Y-axis = Ct
👉 Straight line relationship:
Used to calculate unknown samples
Values on slide:
Correlation coefficient: 0.999 → very accurate
Slope: -3.19 → ideal PCR efficiency
PCR efficiency: ~100%
👉 Meaning:
DNA is doubling perfectly each cycle
🧠 Why this matters
This allows:
Quantification of DNA/RNA
Viral load measurement
Gene expression analysis
🔥 High-yield summary
SYBR Green qPCR detects DNA amplification in real time by fluorescing when bound to double-stranded DNA, and Ct values are used to determine the initial amount of nucleic acid.
🧠 Simple mental model
DNA doubles → fluorescence doubles
Machine tracks glow → determines how much you started with
⚠ Important limitation (exam tip)
SYBR Green binds ANY dsDNA
→ includes non-specific products
→ less specific than probe-based methods
Ultimate takeaway
The earlier the signal appears (lower Ct), the more target DNA was present initially.

Molecular diagnosis = detecting DNA or RNA of a pathogen directly (instead of growing it in culture).
✅ Advantages (Why it’s so good)1. High sensitivity / specificity
Sensitivity → detects very small amounts of pathogen DNA/RNA
Specificity → primers/probes bind only to the exact organism
👉 You can detect infections early, even when pathogen levels are low.
2. Fast + automated
PCR results can come in hours (not days)
Machines run automatically
👉 Huge advantage over culture (which can take days–weeks)
3. Multiplexed
Can test multiple pathogens in one reaction
👉 Example: one test detects flu, RSV, COVID simultaneously
4. Crude extract is enough
Doesn’t require perfectly purified samples
Can use blood, saliva, sputum directly
👉 Easier sample prep
5. Pathogen doesn’t need to be alive
Detects DNA/RNA even if organism is dead
👉 Useful when:
Patient already took antibiotics
Pathogen is hard to culture
6. Target doesn’t need to be infectious
You’re detecting genetic material, not activity
👉 Good for viruses or latent infections
7. Works for many organisms
Bacteria
Viruses
Other pathogens (fungi, parasites)
👉 Very versatile technology
❌ Limitations (What’s the catch?)1. High sensitivity → contamination risk
Even tiny contamination = false positive
👉 Example:
A single DNA fragment from previous sample → wrong diagnosis
2. Expensive
Requires:
Specialized machines
Trained personnel
👉 Not always accessible everywhere
3. No culture = limited follow-up testing
Since you don’t grow the organism:
❌ Cannot easily test antibiotic sensitivity
❌ Harder to study strain variation
👉 Culture still needed for:
Drug susceptibility
Detailed microbiology
4. Quantification requires special methods
Standard PCR = just yes/no
To measure amount → need real-time PCR (qPCR)
👉 Important for:
Viral load (HIV, HCV)
Monitoring treatment
🧠 Key Concept to Remember
👉 Molecular tests answer:
“Is the genetic material present?”
BUT NOT always:
“Is the organism alive or clinically relevant?”
🔥 High-Yield Exam Summary
Pros: fast, sensitive, specific, works on non-viable organisms
Cons: contamination risk, expensive, no culture info
qPCR = quantification (viral load)